

Abstract
Motivation: Detection of allelic imbalances in ChIP-Seq reads is a powerful approach to identify functional non-coding single nucleotide variants (SNVs), either polymorphisms or mutations, which modulate the affinity of transcription factors for chromatin. We present ABC, a computational tool that identifies allele-specific binding of transcription factors from aligned ChIP-Seq reads at heterozygous SNVs. ABC controls for potential false positives resulting from biases introduced by the use of short sequencing reads in ChIP-Seq and can efficiently process a large number of heterozygous SNVs.
Results: ABC successfully identifies previously characterized functional SNVs, such as the rs4784227 breast cancer risk associated SNP that modulates the affinity of FOXA1 for the chromatin.
Availability and implementation: The code is open-source under an Artistic-2.0 license and versioned on GitHub (https://github.com/mlupien/ABC/). ABC is written in PERL and can be run on any platform with both PERL (≥5.18.1) and R (≥3.1.1) installed. The script requires the PERL Statistics::R module.
Contact: mlupien@uhnres.utoronto.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide association studies (GWAS) have identified thousands of single nucleotide variants (SNV) representing genomic loci associated with human traits and disease (Hindorff et al., 2009). The causal variant(s) underlying each association are hard to identify because most of the associated loci fall outside of annotated genes (Schaub et al., 2012). ChIP-Seq allows for the identification of transcription factor (TF) binding sites across the genome (Furey, 2012). Functional studies have shown that many trait/disease-associated loci modulate the affinity of TFs for chromatin (Zhang et al., 2014). Noncoding somatic mutations found in tumors can also modulate TF binding to promote tumor growth and progression (Horn et al., 2013; Huang et al., 2013). The generation of ChIP-seq data for TFs in normal cells and tumors (Dunham et al., 2012; Ross-Innes et al., 2012) provides an opportunity to directly assess the functional effect of risk-variants and somatic mutations on TF binding. This creates a need for a computational tool to systematically identify SNVs imparting an allelic imbalance in TF binding to the chromatin.
To eliminate alignment biases existing tools (Reddy et al., 2012; Rozowsky et al., 2011; Younesy et al., 2014) incorporate the alignment of the ChIP-Seq reads to two separate genomes. The use of two representative parental genomes assumes a diploid set of chromosomes. Therefore, the applicability of these tools to cancer samples may be limited. Tumor samples harbor numerous chromosomal abnormalities and exhibit clonal heterogeneity, which implies that some inherited SNVs and somatic mutations, in particular, are unlikely to be present at an equal ratio within tumors. In addition, assigning the alleles of multiple somatic SNVs to the correct haplotype in tumor samples may prove to be challenging. We addressed this issue by designing the allele-specific binding from ChIP-Seq (ABC) tool. Our approach does not require phased haploid genomes and can readily serve to call allele-specific TF binding to the chromatin from normal or cancer samples where genotype information is available.
2 Methods
The number of ChIP-Seq reads is proportional to the binding intensity of the profiled TF. Each ChIP-Seq read spanning a SNV will contain the allele that was sequenced. A deviation from the expected proportion of reads mapping to each allele of heterozygous SNVs indicates an allelic imbalance in TF binding. The null expectation within ChIP-Seq reads should be equal to the genomic allele ratio (gAR) or the number of reads mapping to each allele in the genomic DNA. Accounting for the gAR adequately controls for biases caused by differences in copy number (CNVs and aneuploidy), clonal heterogeneity or mapping to a reference genome (Degner et al., 2009), assuming similar read lengths, since these biases should affect both the genomic and ChIP-Seq reads equally. ABC applies a binomial probability test to call an allele-specific bias in the ChIP-Seq reads using the observed gAR as the expected occurrence of the two alleles. ABC requires aligned reads from a ChIP-Seq experiment in Sequence Alignment/Map (SAM) format (Li et al., 2009) and a file containing the position, strand, observed alleles and the gAR of heterozygous SNVs. Ideally the gAR is calculated from genomic sequencing reads of similar length aligned to the same reference genome.
Unprocessed ChIP-Seq reads result in two strand-specific read pile-ups (peaks) surrounding the location of a TF-binding site (Fig. 1A). Binding site(s) can be inferred by extending the mapped reads in a strand-specific manner by an estimated fragment size (Park, 2009). Thus, the detection of an allelic imbalance using only aligned reads has the most power on the edges of a binding site and not within the centre. The available read information for a SNV is also limited to twice the read length used (Fig. 1B).
Fig. 1.
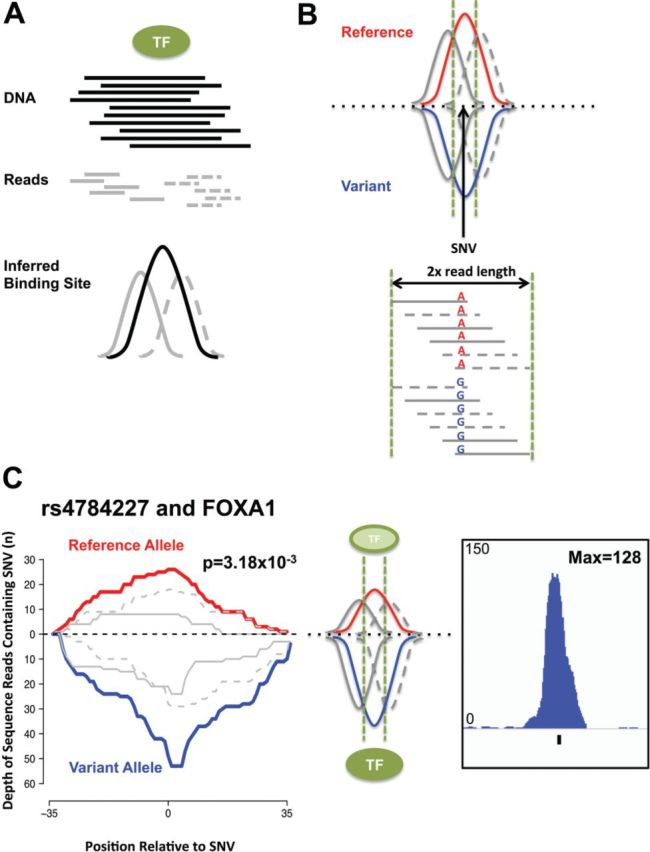
The ABCs of ChIP-Seq. (A) DNA fragments (black) are sequenced from both ends in ChIP-Seq assays. The reads (grey) are short and less than the DNA fragment size selected for sequencing. Reads map to the positive (solid grey) and negative (dashed grey) strands. The TF binding site (black) is inferred by extending the reads in a strand-specific manner. (B) The SNV’s location within the TF binding site can be inferred from the aligned reads. The distribution of total, positive and negative strand reads should be equal between alleles. The coverage window of a SNV is limited to twice the read length used (dashed green lines). (C) Allele-specific binding of FOXA1 caused by the rs4784227 SNV. The reads containing the reference (red) and variant (blue) alleles, as well as the distribution of the positive (solid grey) and negative (dashed grey) reads are shown (left). The interpretation of the results is illustrated (middle) indicating the preference of the TF (green) for the variant allele. The processed signal, peak, is shown (right)
However, a SNV’s position can be inferred by assessing the strand distribution of the reads containing each allele. Reads containing a SNV that map in both orientations identify SNVs closer to the centre of a binding site. ABC performs a Fisher’s exact test to determine that the strand distribution is similar for both alleles. Unlike genomic DNA sequencing the expectation of equal coverage of a SNV by reads in both orientations is not held for reads derived from ChIP-Seq assays (Fig. 1A). A position bias where the alleles of a SNV are not equally distributed along the length of the reads spanning it can be used to identify potential false-positive allelic imbalances. ABC applies a Mann-Whitney U test to assess a potential read position bias observed between the alleles. Confidence in the allele-specific binding called can also be gained by accounting for the maximum signal intensity found within the window surrounding the SNV. Instructions and a tutorial on how to run ABC can be found in the Supplementary Information.
3 Results
The rs4784227 SNP imposes allele-specific binding of FOXA1 in MCF7 cells based on ChIP-qPCR (Cowper-Sal lari et al., 2012). Using ABC against FOXA1 ChIP-Seq data from MCF7 cells (Hurtado et al., 2011) reports increased binding intensity of FOXA1 to the variant versus reference allele of the rs4784227 SNP. Specifically, 53 aligned reads from the FOXA1 ChIP-Seq data map to the variant allele while 26 map to the reference allele (P = 3.18 × 10−3) (Fig. 1C). No strand or position biases are observed (P > 0.05) and a high processed signal is reported at this site (normalized read depth = 128). In addition, we applied ABC genome-wide to identify SNVs modulating binding affinity of the PU.1 (or SPI1) (Supplementary Information) and ZNF143(Bailey et al., 2015) in GM12878 cells.
ABC provides the ability to identify allelic imbalance in TF binding, similar to a traditional allele-specific ChIP-qPCR assay and to what is reported by Ni and colleagues (Ni et al., 2012). However, we do not attempt to call SNVs from the ChIP-Seq data and we control for potential biases in the alignment across alleles. ABC can be used to help understand the role of SNVs associated with traits or disease by directly assessing their capacity to modulate TF binding to the chromatin.
Funding
The NCI/NIH (R01CA155004 to M.L.), the PMCF (M.L) and the Gattuso-Slaight Personalized Cancer Medicine Fund/PMCF (B.H-K.) supported this research. M.L. holds a young investigator award from the OICR, a new investigator salary award from the CIHR and a Movember Rising Star award from PCC (RS2014-04). S.D.B. is a CIHR Postdoctoral Fellowship recipient.
Conflict of Interest: none declared.
Supplementary Material
References
- Bailey S.D., et al. (2015) ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat. Commun., 2, 6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowper-Sal lari R., et al. (2012) Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet., 44, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner J.F., et al. (2009) Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics, 25, 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I., et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey T.S. (2012) ChIP-seq and beyond: new and improved methodologies to detect and characterize protein–DNA interactions. Nat. Rev. Genet., 13, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff L.A., et al. (2009) Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl Acad. Sci. USA, 106, 9362–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S., et al. (2013) TERT promoter mutations in familial and sporadic melanoma. Science, 339, 959–961. [DOI] [PubMed] [Google Scholar]
- Huang F.W., et al. (2013) Highly recurrent TERT promoter mutations in human melanoma. Science, 339, 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A., et al. (2011) FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet., 43, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y., et al. (2012) Simultaneous SNP identification and assessment of allele-specific bias from ChIP-seq data. BMC Genet., 13, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P.J. (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet., 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T.E., et al. (2012) Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res., 22, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes C.S., et al. (2012) Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature, 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J., et al. (2011) AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol., 7, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub M.A., et al. (2012) Linking disease associations with regulatory information in the human genome. Genome Res., 22, 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younesy H., et al. (2014) ALEA: a toolbox for allele-specific epigenomics analysis. Bioinformatics, 30, 1172–1174. [DOI] [PubMed] [Google Scholar]
- Zhang X., et al. (2014) Laying a solid foundation for Manhattan—‘setting the functional basis for the post-GWAS era'. Trends Genet., 30, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.