

Abstract
Objectives
Drug resistance confers a fitness advantage to parasites exposed to frequent drug pressure, yet these mutations also may incur a fitness cost. We assessed fitness advantages and costs of artemisinin resistance in Plasmodium falciparum in vitro to understand how drug resistance will spread and evolve in a competitive environment.
Methods
Genotyping of SNPs, drug susceptibility assays and copy number determination were used to assess the impact of artemisinin resistance on parasite fitness. An artemisinin-resistant clone (C9) selected in vitro from an isogenic parental clone (D6) was used to conduct competitive growth studies to assess fitness of artemisinin resistance. The resistant and susceptible clones were mixed or grown alone in the presence and absence of drug pressure (dihydroartemisinin or pyrimethamine) to quantify the rate at which artemisinin resistance was gained or lost.
Results
We experimentally demonstrate for the first time that artemisinin resistance provides a fitness advantage that is selected for with infrequent exposure to drug, but is lost in the absence of exposure to artemisinin drugs. The best correlations with artemisinin resistance were decreased in vitro drug susceptibility to artemisinin derivatives, increased copy number of Pf3D7_1030100 and an SNP in Pf3D7_0307600. An SNP conferring an E208K mutation in the kelch gene (Pf3D7_1343700) was not associated with resistance. Furthermore, we observed second-cycle ring-stage dormancy induced by pyrimethamine, suggesting that dormancy is a fitness trait that provides an advantage for survival from antimalarial drug stress.
Conclusions
Artemisinin-resistant P. falciparum have a fitness advantage to survive and predominate in the population even in the face of infrequent exposure to artemisinin drugs.
Introduction
Artemisinin-resistant Plasmodium falciparum appears to have emerged independently many times in south-east Asia.1 First described in Cambodia and along the Thai–Myanmar border as a prolonged parasite clearance phenotype following treatment with an artemisinin drug combination,2,3 artemisinin resistance appears to be spreading throughout south-east Asia.4 Multiple point mutations in the propeller domains of Pf3D7_1343700, the kelch gene on chromosome 13 (K13), have been associated with the delayed parasite clearance phenotype in vivo and have provided a molecular marker for tracking the spread of artemisinin resistance.5,6 While evidence suggests the independent emergence of artemisinin resistance across geographic locations in Asia, emergence appears to be dependent on the genetic background of preexisting mutations in the parasite.7 Parasites in south-east Asia have historically been associated with multidrug resistance, and given that artemisinin combination therapy (ACT) has been the primary treatment option globally for more than a decade, parasites have most likely evolved mechanisms to resist multiple classes of antimalarial drugs.
Selection of antimalarial drug resistance often occurs early after introduction to clinical use and can be conferred by a single point mutation.8,9 In contrast, a complex series of mutations may be required as compensation for fitness costs or to enhance higher levels of resistance. For instance, antifolate resistance is associated with a sequential accumulation of point mutations in dhfr or dhps that are associated with reduced susceptibility to antifolate drugs.10 Resistance arose from a single point mutation that was followed by multiple compensatory point mutations that increased resistance levels. Similarly, for chloroquine resistance at least four mutations in Pfcrt are necessary for resistance, with the pivotal mutation being K76T. Once these resistance-associated mutations are present in a population, they can spread rapidly due to fitness advantages over WT parasites in the presence of drug pressure. For both chloroquine and pyrimethamine, resistance-selective sweeps occurred that rapidly spread across malaria-endemic regions.11–13
Although drug resistance confers a fitness advantage for parasites exposed to frequent drug pressure, these mutations also may incur a fitness cost.8 For example, after chloroquine efficacy fell to low levels in Malawi, chloroquine use was discontinued and sulfadoxine/pyrimethamine was used as the primary therapy. Remarkably, chloroquine susceptibility in Malawi returned a decade later14 and this was attributed to re-expansion of susceptible parasites when chloroquine drug pressure was removed.15
The selection process for artemisinin resistance appears to be more complicated than was observed with chloroquine and antifolate resistance; therefore, it is not well understood how resistance to ACTs will spread and how long it will take for resistance to emerge in Africa. Natural selection is likely to be the determinant of combination therapy resistance due to the parasite's requirement to survive two antimalarials simultaneously, including a long-lasting partner drug.16 Therefore, the subsequent spread of resistance to ACTs must include mutations beneficial to resist multiple antimalarials and therefore may be associated with a significant fitness cost. These fitness costs are hypothetical at present since fitness associated with artemisinin resistance has not been determined experimentally.
Fitness costs of drug resistance can be measured in vitro to understand how drug resistance will spread and evolve in a competitive environment.8 In vitro models allow for more precise control and higher throughput to predict the spread of resistance in the field.17 In vitro models of fitness can more easily project the spread of antimalarial drug resistance due to the selective nature of antimalarial drug resistance and the major role that intraerythrocytic-stage parasites have in the spread of drug resistance.18,19 While host and environmental factors play a role in the dissemination of antimalarial drug resistance in the field, in vitro models provide significant insights into the relative advantages of parasite resistance versus fitness costs.
In this study we used an artemisinin-resistant clone selected from an isogenic parental clone to conduct fitness experiments in the presence and absence of drug pressure and to quantify the rate at which artemisinin resistance can be gained or lost in a competitive environment. By using a combination of pyrosequence analysis of multiple SNPs, copy number determination and in vitro drug susceptibility assays, we experimentally demonstrate for the first time that artemisinin resistance incurs a fitness cost that is selected for with infrequent exposure to drug, but is lost in the absence of artemisinin drug pressure.
Materials and methods
Artemisinin-resistant P. falciparum culture in vitro
P. falciparum clone D6 (Sierra Leone) and an in vitro-selected artemisinin-resistant clone derived from D6 were cultivated using previously described methods.20 The artemisinin-resistant progeny used in this study were derived as previously reported. In brief the resistant progeny from D6 were initially selected with small increments of artelinic acid (AL) up to 80 ng/mL (D6.AL80).21 D6.AL80 then was subjected to artemisinin pressure up to 2400 ng/mL four times and cloned by limiting dilution.22 A single clone, C9, was exposed one additional time with 2400 ng/mL artemisinin and maintained in culture in the absence of drug pressure for over 400 days. C9 maintained a stable in vitro resistance phenotype in the absence of drug pressure as described by Tucker et al.22
Fitness study design
Asynchronous D6 and C9 were co-cultured in a 1: 1 (D6/C9) ratio and also maintained as monocultures. Parasite cultures were maintained between 2% and 5% parasitaemia at 4% haematocrit in complete medium supplemented with 10% heat-inactivated AB+ human plasma. D6/C9, D6 and C9 cultures at 2% parasitaemia each were exposed to 700 nM dihydroartemisinin or 10 μM pyrimethamine or were sham treated (0.25% DMSO) for 48 h. Following treatment, cultures were washed three times with RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) and observed daily for growth. Every 24 h following 48 h of drug exposure, medium was changed and blood smears were made. When parasite cultures recovered and parasitaemia exceeded 5%, cultures were diluted to 2% parasitaemia. The remaining parasitized blood pellet was frozen at −20°C for genomic DNA extraction and analysis. Parasites were exposed to drug four times with a 21 day grow-out period between each exposure; between exposure two and three there was a 42 day grow-out period. As noted above, parasitaemia was maintained between 2% and 5% during the grow-out period.
Recrudescence assay following drug exposure
Following a drug-exposure cycle, parasitaemia was determined every 24 h using microscopy. The ratio of ring, trophozoite and schizont-infected red blood cells/total red blood cells was used to determine parasitaemia for each timepoint. Dormant and dead parasites were not included in the calculation.
Drug susceptibility assay using radioisotope addition at T0
Susceptibility to artemisinin derivatives was determined using a modified [3H]hypoxanthine incorporation assay.22 [3H]hypoxanthine was added to cultures at the start of the assay when drug was added and allowed to incorporate for 48 h. Parasites were harvested at 48 h to determine IC50. For pyrimethamine, IC50 was determined traditionally, by adding [3H]hypoxanthine 24 h after drug exposure.23
Genomic DNA extraction
Parasitized red blood cells were frozen at −20°C for at least 24 h and treated with 0.5% saponin to free parasites from red blood cells. Parasite pellets were frozen for 12–24 h in Tris EDTA buffer. Thawed pellets were treated with 4% SDS and 20 mg/mL RNase (Life Technologies, Grand Island, NY, USA). Equal volumes of phenol/chloroform/isoamylalcohol (25: 24: 1) were added and extracts were spun at 14 000 rpm for 5 min at room temperature and then the upper aqueous phase was transferred to a new tube. This separation step was repeated with chloroform/isoamylalcohol (24: 1) and chloroform, respectively. The final aqueous transfer was treated with 3 M potassium acetate and 100% ethanol. Pellets were washed with 100% ethanol and suspended in Tris EDTA buffer. Genomic DNA was quantified using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Real-time quantitative PCR to determine gene copy number
Previously C9 was found to have an amplified region of chromosome 10, whereas the parent clone has only one copy of genes in this region.24 Pf3D7_1030100 is in the amplicon and C9 has two copies, whereas parental D6 maintains one copy. Therefore in this study we used real-time quantitative PCR to assess the copy number of D6, C9 and the D6/C9 mixed culture following the second and fourth drug-exposure cycles. We used the primer sequences in Table S1 (available as Supplementary data at JAC Online). In brief, Brilliant II QPCR SYBR Green 2× mastermix (Stratagene, La Jolla, CA, USA) and 30 nM ROX were used to amplify Pf3D7_1030100 at a final primer concentration of 200 nM. The relative copy number was determined by the standard curve method comparing threshold cycle values of gene of interest to ldh using MX3000P real-time quantitative PCR system (Stratagene). Average copy number was calculated from three replicates with a confidence interval >95% (GraphPad Prism).
Determination of SNP frequency by pyrosequencing
PCR and sequencing primers were designed using Biotage Pyrosequencing Assay Design software (Charlottesville, VA, USA) to amplify SNP-containing genes. Primer sequences and SNP details are listed in Table S1. A 5′ biotin label was added to the PCR primer running in the opposite direction to the sequencing primer. Each 50 μL PCR mixture contained 31.5 μL of sterile water, 10 μL of 5× HF buffer, 1 μL of 10 mM dNTPs, 2 μL each of forward and reverse primer (10 μM), 0.5 μL of Phusion Hot Start II DNA polymerase (Fisher Scientific, Pittsburg, PA, USA) and 2 μL of template DNA. Thermal cycling conditions were 98°C for 30 s (initial denaturation), followed by 40 cycles of denaturation at 98°C for 10 s, annealing at the temperature indicated in Table S1 for 30 s and extension at 72°C for 50 s, followed by a final extension at 72°C for 7 min, and a final hold at 4°C. PCR products (10 μL) were visualized using QiAxcel (Qiagen, Valencia, CA, USA).
Sequencing primer (0.6 μM) was diluted in annealing buffer (Biotage, Charlottesville, VA, USA) and 40 μL of the mixture was aliquotted into a 96-well PSQ plate. The binding reaction mixture consisted of 4 μL of streptavidin-coated sepharose beads (GE Healthcare) and 51 μL of binding buffer. In each well of a round-bottom PCR plate, 55 μL of the binding mixture was added to 25 μL of PCR product and mixed at 14 000 rpm for 10 min. Using the Vacuum Prep Workstation (Biotage, Charlottesville, VA, USA) the Sepharose-bound PCR products were captured with the Vacuum Prep Tool immediately after being mixed and washed as follows: 70% ethanol for 10 s; denaturing buffer (0.2 M NaOH) for 10 s; and washing buffer (10 mM Tris-acetate, pH 7.6) for 15 s. The vacuum was released and the Sepharose-bound PCR products were released into the PSQ plate containing sequencing primer. The plate was incubated at 80°C for 2 min and allowed to cool to room temperature. Plates were read on a Pyromark Q96 ID pyrosequencer (Qiagen) in AQ mode using a Pyromark Gold Q96 reagent kit. Assays were completed three times for each DNA template and the average percentage of WT allele was plotted over time. In addition standard curves were established for each SNP using ratios of D6/C9 gDNA (Figure S1).
Results
Competitive fitness of artemisinin-resistant and -susceptible clones
Understanding fitness costs associated with artemisinin resistance is of extreme importance due to the potential spread of resistance globally. An in vitro model to determine fitness cost associated with artemisinin resistance has not been previously described yet could provide essential information to elucidate how efficiently artemisinin resistance will spread. Therefore in these studies we assessed the fitness of artemisinin resistance in vitro in competitive growth experiments. We used a stable artemisinin-resistant clone (C9) derived from a cloned parent (D6) following repetitive pulse exposures to artemisinin drugs in vitro.22 C9 is artemisinin-resistant as evidenced by its ability to survive repeated exposure to 2400 ng/mL artemisinin and reduced susceptibility to multiple artemisinin derivatives in vitro. An advantage of using these cloned parasites is that they are derived from an isogenic background8 and, following resistance selection, several molecular markers were identified that allowed us to discriminate between the resistant and susceptible clones in mixed population growth studies. In preliminary studies we identified several SNPs (see Table S1) and a copy number variant (CNV) on chromosome 10 that were present in the C9 (resistant) clone.24 By using these markers, we were able to quantify the phenotypic responses associated with the prevalence of C9 genotypes in a heterogeneous population.
We first assessed fitness in a competitive growth experiment in which an asynchronous parent clone (D6) and its artemisinin-resistant progeny (C9) were mixed equally (1: 1) at the beginning of the study. In direct comparison with both susceptible and resistant clones grown in monocultures, we exposed the cultures to four cycles of drug pressure (21 or 42 days apart) with dihydroartemisinin or pyrimethamine; as a control we also included a sham treatment with 0.25% DMSO. Following the first pulse exposure to dihydroartemisinin, the resistant C9 clone recovered from ring-stage drug-induced dormancy earlier than D6 (Figure 1), as reported previously.22 Conversely D6 and mixed cultures recrudesced a day later, although the mixed culture recovered to higher parasitaemia faster than D6. Following the first exposure to pyrimethamine there was no difference in recovery between the susceptible, the resistant and the mixed parasite populations (Figure 1). The resistant C9 clone recovered earlier than D6 following dihydroartemisinin pressure in all but the third drug-exposure cycle. Importantly, the parasites were not synchronized at any time of the experiment; therefore, mixed asexual stages were present and these ratios may be different between cultures at the time of drug exposure. Interestingly, the C9-resistant clone recovered from dormancy earlier than susceptible parasites only following exposure to dihydroartemisinin; these results suggest enhanced survival of resistant parasites and the potential of resistance to spread when exposed to artemisinin drugs. In contrast, when exposed to a drug that acts through a different target (e.g. pyrimethamine), artemisinin-resistant parasites recover from dormancy similarly to susceptible parasites. In all sham-treated cultures parasites grew normally without any evidence of the dramatic reductions in parasitaemia seen with dihydroartemisinin or pyrimethamine exposure (data not shown).
Figure 1.
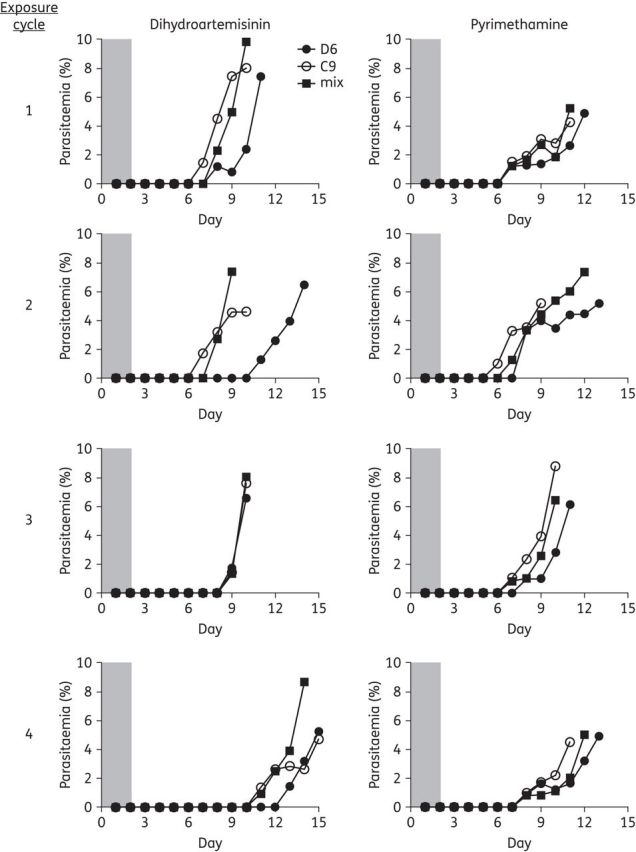
Recrudescence following antimalarial drug exposure. Recrudescence following four exposures to dihydroartemisinin or pyrimethamine was assessed by microscopy of Giemsa-stained blood smears. Asynchronous artemisinin-resistant (C9) and parent D6 ring-stage parasites were co-cultured 1: 1 and exposed to 700 nM dihydroartemisinin or 10 μM pyrimethamine for 48 h; data for sham exposure with 0.25% DMSO are not shown. C9 and the mixed culture recovered up to 2–3 days earlier than D6 when exposed to dihydroartemisinin. When exposed to pyrimethamine mixed culture, recrudescence was similar to that of drug-susceptible D6 in monoculture.
Pyrimethamine induces second-cycle ring-stage dormancy
The ring stages of P. falciparum exposed to artemisinin arrest development and enter a state of dormancy characterized by a prolonged period of no proliferation and reduced metabolic activity followed by recrudescence in vitro.22,25,26 The artemisinin-induced dormant ring stages express a characteristic phenotype (condensed nuclei and reduced cytoplasm) and these dormant rings have been shown experimentally to be the source of recrudescence in vitro.22,25,26 In this study, exposure to dihydroartemisinin induced ring-stage dormancy as expected (Figure 2). In contrast, following pyrimethamine exposure, the ring-stage parasites continued development into late schizogony, where the drug effect was maximal. Interestingly, pyrimethamine exposure also reduced parasitaemia in the cultures similarly to dihydroartemisinin, and this effect was followed by subsequent recrudescent growth (Figure 1). Recrudescence following pyrimethamine exposure has been previously described,27,28 yet the mechanism by which the parasite survives pyrimethamine exposure is not known. By examining the artemisinin-resistant (C9) or -susceptible (D6) clone morphology following drug exposure, we discovered that pyrimethamine induced ring-stage dormancy in the subsequent cycle of development after drug exposure. That is, the parasites were not arrested in the first ring stage when drug was applied, but following development to mature schizonts, some schizonts successfully produced merozoites that invaded new erythrocytes and then arrested as dormant ring stages (Figure 2). The morphologies of pyrimethamine- and dihydroartemisinin-induced dormant rings were similar, as was the period of dormancy induced by both drugs following 48 h of exposure. This is the first demonstration of drug-induced second-cycle dormancy by any antimalarial drug other than artemisinin derivatives.
Figure 2.
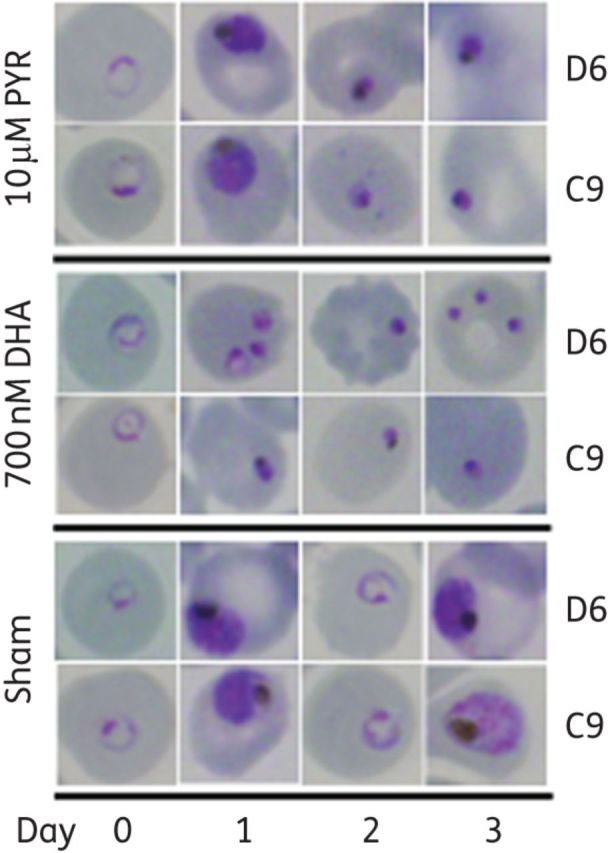
Pyrimethamine induces second-cycle ring-stage dormancy. P. falciparum exposed to pyrimethamine enter ring-stage dormancy in the subsequent life cycle of development. Late-stage parasites were observed 24 h following 10 μM pyrimethamine exposure to ring-stage parasites progressing the same as untreated parasites. Ring-stage dormant parasites were observed 48 h after starting pyrimethamine treatment. This is in contrast to ring-stage-induced dormancy by dihydroartemisinin in which dormant parasites are observed 24 h following 700 nM dihydroartemisinin exposure to ring-stage parasites. PYR, pyrimethamine; DHA, dihydroartemisinin.
Drug susceptibility assays confirm resistance phenotypes in competitive growth studies
The artemisinin-resistance phenotype in vitro was measured using a modified assay that includes addition of [3H]hypoxanthine at the start of the assay when the drug is added22; pyrimethamine susceptibility was measured by traditional drug-susceptibility methods in which [3H]hypoxanthine was added 24 h after the drug. By using these drug susceptibility methods we were able to assess the prevalence of artemisinin-resistance phenotypes in the fitness competitive growth studies. We found that the mixed population was equally resistant to artemisinin derivatives to the C9 resistant clone by the end of the study after exposure to four pulse cycles of dihydroartemisinin (Figure 3). Conversely, resistance was lost and the mixed population was equally susceptible to D6 by the end of study in the absence of dihydroartemisinin pressure or following pyrimethamine exposures. Pure C9 resistant clones also maintained resistance to artemisinin derivatives in vitro in the presence or absence of artemisinin drug pressure as expected (Figure 3).22 Interestingly, the degree of C9 resistance to artemisinin derivatives in vitro was similar to that of K13 propeller mutants from Cambodia and Thailand.29 Sensitivity to pyrimethamine was maintained in this experiment for all parasite cultures.
Figure 3.

Artemisinin-resistant phenotypes correlate with prevalence of resistant genotypes in mixed populations. In vitro drug susceptibility assays (n = 2) were performed on the clones before the experiment (pretreatment) and on the D6, C9 and mixed cultures following recovery from the fourth drug or sham exposure. Reduced susceptibility of C9 to AL, artemisinin and dihydroartemisinin was observed in the absence of dihydroartemisinin exposure. The mixed culture also demonstrated reduced susceptibility to artemisinin derivatives in the dihydroartemisinin-exposed groups, but not in the sham group or the pyrimethamine-exposed group. PYR, pyrimethamine; QHS, artemisinin; DHA, dihydroartemisinin.
Prevalence of SNPs in competitive growth studies
Next we assessed the relative proportions of resistant and susceptible genotypes in the parasite cultures following each drug or sham exposure. Resistant and susceptible parasites in co-culture were quantified by pyrosequencing SNPs previously identified in artemisinin-resistant C9 (Figure S1). In this study we quantified SNPs on chromosomes 3 (Pf3D7_0307600; conserved protein with unknown function), 7 (Pf3D7_0700900; RESA-like protein, pseudogene) and 13 (Pf3D7_1343700, K13) to discriminate between C9 and D6 genotypes in the competitive growth fitness studies.
The Pf3D7_0307600 SNP allele frequency of mixed culture reached almost 80% following recovery from dihydroartemisinin exposure (Figure 4). Conversely in the absence of dihydroartemisinin pressure, the percentage of the WT allele became increasingly abundant over time. This was most likely due to artemisinin-resistant parasites recovering from dihydroartemisinin-induced dormancy faster than susceptible parasites22,25; however, due to apparent fitness costs associated with artemisinin resistance, WT parasites outgrew resistant parasites in the absence of dihydroartemisinin exposure. C9 produces fewer merozoites than D6,22 thus allowing D6 to outgrow C9 in the absence of artemisinin pressure. These results suggest a positive, although not causal, association of the Pf3D7_0307600 SNP with artemisinin resistance.
Figure 4.

Artemisinin-resistant allele frequency. Allele frequency of SNPs was assessed by pyrosequence analysis and is plotted as the percentage of WT (D6) SNP found at each timepoint in the competitive growth experiment. Drug or sham exposures were for 48 h and are denoted as bars. The SNPs quantified were on chromosomes 3 (Pf3D7_0307600; conserved protein with unknown function), 7 (Pf3D7_0700900; RESA-like protein, pseudogene) and 13 (Pf3D7_1343700, K13). The Pf3D7_0700900 and Pf3D7_1343700 SNPs were lost in C9 and the mixed culture in the absence of dihydroartemisinin pressure. Conversely, the Pf3D7_0307600 SNP persisted throughout the study in C9 and the mixed culture exposed to dihydroartemisinin. The K13 SNP was maintained in artemisinin-resistant C9 over four consecutive treatments plotted as proportion of WT allele. DHA, dihydroartemisinin; PYR, pyrimethamine.
The apparent fitness costs of artemisinin resistance were further evident in D6 and C9 monocultures treated with dihydroartemisinin compared with pyrimethamine. C9 recovered from drug-induced dormancy earlier than D6 only when exposed to dihydroartemisinin. Furthermore, this was evident in the Pf3D7_0307600 SNP frequency of the mixed culture immediately following recovery from pyrimethamine-induced dormancy (Figure 4). There were 60% WT alleles in co-cultures immediately following recovery, reaching 95% when grown in the absence of drug pressure prior to the next pyrimethamine exposure cycle. Artemisinin-resistant and -susceptible parasites recovered similarly from pyrimethamine-induced ring-stage dormancy. In addition Pf3D7_0307600 SNP frequency increased in the mixed population remarkably quickly following a dihydroartemisinin exposure cycle, yet diminished to nearly 100% WT (D6) after just one exposure to drug.
We also assessed the prevalence of an SNP in Pf3D7_0700900 that was identified in C9. The prevalence of the Pf3D7_0700900 SNP was significantly reduced in the mixed culture by the end of the study, with most parasites having WT sequence regardless of drug exposure.
The prevalence of an SNP in K13 (Pf3D7_1343700) is of interest given that previous studies demonstrated a correlation between mutations in the propeller domains (≥440 amino acids) of K13 with clinically relevant resistance that is expressed as a slow parasite clearance phenotype in vivo. The SNP identified in the artemisinin-resistant C9 clone results in a non-synonymous change at amino acid 208 of K13 (E208K) that has not previously been observed in the field and is outside the propeller domains associated with artemisinin-resistance mutations. Interestingly, the prevalence of Pf3D7_1343700 SNP in C9 and the mixed cultures was similar to that observed for Pf3D7_0700900 (Figure 4). That is, the WT allele appeared to increase in C9 or mixed cultures after dihydroartemisinin exposures, and by the end of the study the K13 SNP was no longer prevalent in any of the cultures exposed to dihydroartemisinin. These results suggest that neither the Pf3D7_1343700 nor Pf3D7_0700900 SNPs are strongly associated with the artemisinin-resistance phenotype in vitro. It is important to note that the SNP discrimination for the K13 SNP in mixtures of D6 and C9 was not as good as that for the other SNPs (see Figure S1), thus the SNP calls were binary (D6 or C9).
Pf3D7_1030100 copy number and artemisinin resistance
The artemisinin-resistant C9 clone has two copies of a multigene amplicon on chromosome 1024; therefore we also assessed the relative proportion of resistant and susceptible clones by copy number analysis of Pf3D7_1030100, a pre-mRNA-splicing factor ATP-dependent RNA helicase located within the amplified region of chromosome 10. In this study, the artemisinin-resistant C9 clone maintained two copies of Pf3D7_1030100 in the presence or absence of dihydroartemisinin or pyrimethamine drug pressure over the course of the study (Figure 5). Once recovered from the second dihydroartemisinin exposure, the mixed culture had two copies of Pf3D7_1030100, but only one copy when exposed to pyrimethamine or no drug pressure. In addition following the fourth drug-exposure cycle, two copies of Pf3D7_1030100 were found in mixed culture only when exposed to dihydroartemisinin. These results suggest a positive association of increased Pf3D7_1030100 copy number and the artemisinin-resistance phenotype assessed by drug-susceptibility assays.
Figure 5.
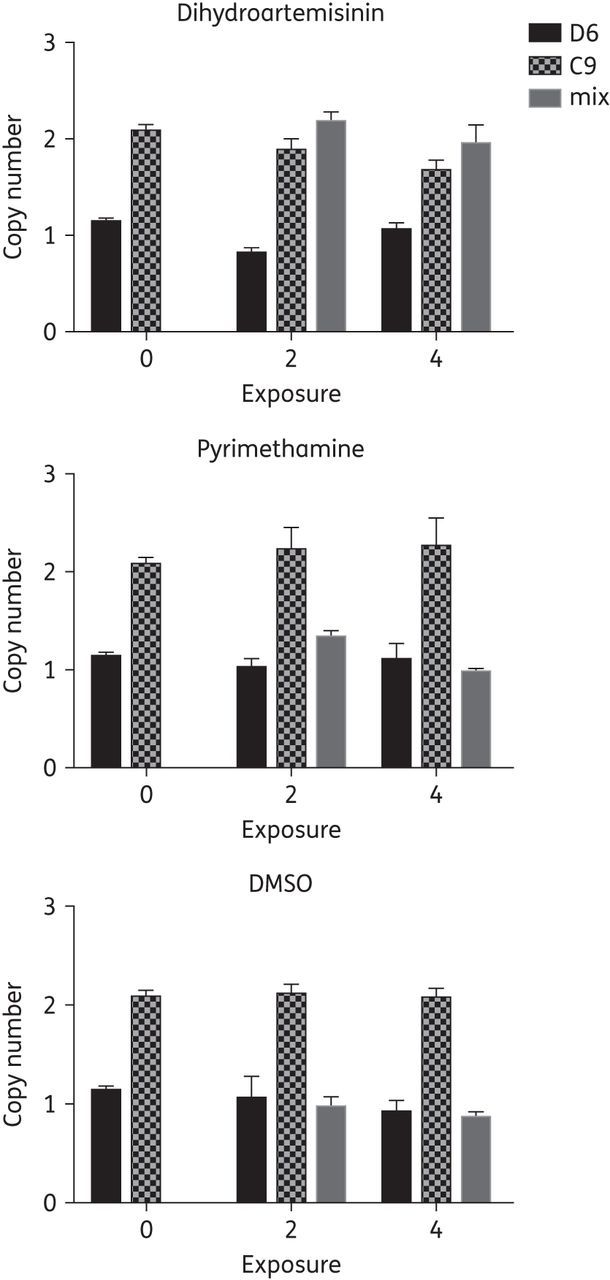
Increased copy number of Pf3D7_1030100 is associated with artemisinin resistance in P. falciparum. The artemisinin-resistant clone (C9) has an amplified region on chromosome 10 that includes Pf3D7_1030100, a pre-mRNA-splicing factor ATP-dependent RNA helicase; therefore, the copy number of Pf3D7_1030100 was analysed by real-time PCR (n = 3) following the second and fourth drug- or sham-exposure cycles in the competitive fitness experiment. After recovery from the second exposure, one copy of Pf3D7_1030100 was observed in the absence of dihydroartemisinin exposure in mixed cultures. Artemisinin-resistant C9 maintained two copies of Pf3D7_1030100 in the absence of artemisinin exposure. Co-cultured parasites maintained two copies of Pf3D7_1030100 only when treated with dihydroartemisinin.
Discussion
Antimalarial drug exposure ultimately leads to the emergence of resistant parasites. All too often resistance is selected quickly due to SNPs in target genes or transporters or via amplification of regions of the genome that confer an advantage for parasites exposed to drugs. The ease and speed of resistance development varies with the target, the specific chemotype of drug used, the presence of preexisting cross-resistance and the duration of exposure to the drug in vitro and in vivo. As resistant organisms emerge, they have a competitive advantage over susceptible organisms when exposed to the drug, yet often the genetic alterations that confer resistance come at a cost to the parasite in the absence of drug pressure. Therefore without drug pressure, the less-fit resistant genotypes can be outcompeted by WT parasites. In areas of low endemicity and high drug pressure (e.g. south-east Asia), even the less-fit resistant genotypes can be selected for at such a rate as to reach fixation in a population. An example of this is stable chloroquine and pyrimethamine resistance in Thailand where these drugs have not been used for primary treatment of falciparum malaria in decades. Conversely, in high transmission areas resistance may emerge multiple times, yet the relative absence of drug pressure plus the massive genetic diversity of susceptible parasites mean that they outcompete the resistant parasites. Obviously this is a complex, yet critically important issue, specifically for the future utility of artemisinin drugs. Resistance has emerged in south-east Asia and some data suggest that K13 mutations have emerged independently in both Asia and Africa, although K13 mutations associated with slow clearance in vivo have only been reported in Asia thus far.1,30 Clearly the relative fitness of artemisinin-resistance traits and the potential for even soft selective sweeps of artemisinin-resistant P. falciparum require urgent attention.
In this study we have assessed the fitness of an in vitro-selected artemisinin-resistant clone in competitive growth experiments with its isogenic parent (D6). The clear advantage of this approach is that the clones are genetically homogenous with the exception of SNPs and CNVs that arose during selection of resistance. In addition we were able to use three SNPs and a CNV on chromosome 10 to quantify the resistant C9 genotype in relation to the susceptible D6 parental clone. Finally we had a drug resistance phenotype that allowed us to assess the relative shifts in population structure and to correlate with the molecular markers. The results of the competitive growth studies demonstrate a fitness advantage of the resistant parasite with infrequent pulse exposures to dihydroartemisinin. The period between drug exposures was either 21 or 42 days, which is equivalent to ∼11 or 22 asexual parasite generations. Conversely, the artemisinin-resistant parasites incurred fitness costs and were outcompeted in the absence of drug pressure or with a drug that has a different mechanism of action and resistance (pyrimethamine). These studies demonstrate for the first time the relative fitness of artemisinin-resistant P. falciparum and provide contextual information about the potential for this resistance to spread.
There is no gold standard method for assessing antimalarial drug fitness. The competitive growth experiments offer perhaps the most direct comparison of fitness of resistant and susceptible organisms at the level of the parasite.8 This approach has been used successfully to demonstrate fitness of cytochrome b and pfmdr1 mutants using similar approaches9,17 and in some cases to correlate with in vivo fitness.8,17 Despite the advantages of our approach to assess fitness, there are some limitations that must be considered. First the resistance mechanism(s) selected in C9 may differ from artemisinin-resistant genotypes that have emerged in south-east Asia. The artemisinin-resistant parasites in Asia have mutations in the propeller regions of K13 that are associated with slow clearance in vivo; the in vitro-selected clone has a K13 SNP, but it is not in the propeller region. Despite this difference, the in vitro-selected clone (C9) expresses the same degree of cross-resistance to several artemisinin derivatives in vitro as artemisinin-resistant clones with K13 propeller SNPs.29 These results suggest a similar mechanism of resistance that is not associated with K13 SNPs. A direct comparison of resistant and susceptible clones from Asia would provide an interesting comparison with our results, but the inherently different growth rates between isolates and the fact that some artemisinin-resistant P. falciparum from Cambodia have altered asexual life cycles29,31 will make competitive growth studies difficult to interpret. More importantly the ultimate goal is to understand the relative fitness of artemisinin resistance in the clinic and how often and how fast these resistant genotypes can emerge and spread. Modelling studies of artemisinin use in south-east Asia suggest that mostly effective drug coverage in this hypoendemic region is rapidly selecting for the most resistant parasites and that the “last man standing” will be the genotypes most resistant to ACTs.32 Our data support this contention, especially since relatively infrequent drug exposures are enough to give a fitness advantage to the artemisinin-resistant parasite.
Interestingly, the best correlation of artemisinin resistance in our study was with three measured phenotypes or genotypes: in vitro drug susceptibility in a modified assay format; a CNV on chromosome 10; and an SNP in Pf3D7_0307600. In comparison, SNPs in two other genes, including K13 in C9, were lost during the competitive growth studies and appear to be unrelated to the artemisinin-resistance phenotype. The C9 resistant clone was selected from the D6 parent by numerous pulses of AL and artemisinin, followed by recovery and growth before another drug-exposure cycle. During this selection process a limited number of mutations were observed in C9 versus D6.22,24 These included the three SNPs and the chromosome 10 CNV used to monitor resistant genotypes in our study. It is important to note that SNPs or CNVs can arise in long-term culture even in the absence of drug pressure33; therefore, the molecular markers we used could be neutral and not linked to the resistance phenotype. That appears to be the case for the SNPs in Pf3D7_1343700 and Pf3D7_0700900. The prevalence of these SNPs decreased over time following exposure to dihydroartemisinin, even though the artemisinin resistance phenotype persisted. In contrast, the Pf3D7_0307600 SNP was linked to the artemisinin-resistance phenotype and the CNV on chromosome 10. Further studies are required to determine whether the SNP in Pf3D7_0307600 is causally associated with artemisinin resistance.
Conversely, we identified a strong positive association of artemisinin resistance with a duplicated region on chromosome 10, as exhibited by quantitative analysis of Pf3D7_1030100 copy number. The C9 clone maintained two copies of Pf3D7_1030100 throughout the study, in either the absence or the presence of dihydroartemisinin or pyrimethamine drug pressure. In addition, the increased copy number of Pf3D7_1030100 was selected for in the mixed culture exposed to infrequent pulses of dihydroartemisinin. Furthermore, this molecular marker provided the best correlation with resistance as assessed by in vitro drug susceptibility to four different artemisinin drugs (Figure 3). The association of this CNV and the artemisinin resistance phenotype is intriguing, yet we do not know if any of the genes in the amplicon have a direct role in conferring artemisinin resistance. In a previous study of clonal variation in transcriptional responses, one stock of 3D7 had a 56 kb amplicon on chromosome 10 that covered from Pf3D7_1028700 to Pf3D7_1030300 and increased expression 2-fold of five of six genes analysed within the duplicated region.34 This observation is interesting given that, in a different study, 3D7 was identified as having reduced susceptibility to artemisinin drugs in vitro,35 although it is not known if that stock of 3D7 had a CNV on chromosome 10. The potential role of chromosome 10 CNVs in artemisinin resistance and dormancy certainly deserves further study.
Exposure of ring stages of P. falciparum to artemisinin derivatives induces a cell-cycle arrest that is time of exposure and dose dependent. The drug-induced ring stages are known as dormant rings given their ability to persist for days to weeks in a non-replicating state before recovery and normal asexual growth resumes. Previously we have confirmed artemisinin-induced dormancy in both drug-resistant and drug-susceptible P. falciparum.22,25 These dormant ring-stages have distinct physiological differences from normal rings, as shown by up-regulation of fatty acid and pyruvate metabolism26 and maintenance of mitochondrial membrane potential that is linked to the viability of dormant rings.36 In this study we observed ring-stage dormancy with subsequent recrudescence following exposure of resistant, susceptible or mixed cultures to both dihydroartemisinin and pyrimethamine. Although recrudescence following pyrimethamine drug pressure has been observed previously,27,28 the mechanism for parasite survival was not known. By observing parasite morphology following pyrimethamine drug exposure, we observed for the first time induction of second-cycle ring-stage dormancy. The difference in induction of dormancy between dihydroartemisinin and pyrimethamine is that only artemisinin drugs induce dormancy immediately upon exposure to ring stages. Conversely, under pyrimethamine pressure the rings progressed through the asexual cycle until late schizogony (segmenters) when development was arrested in most parasites. An unknown proportion of these segmenters produced viable merozoites that invaded new erythrocytes and then arrested as dormant rings. Although our data suggest that a similar state of ring-stage dormancy is induced by dihydroartemisinin and pyrimethamine, additional studies are required to fully characterize the pyrimethamine-induced dormant rings. Regardless, these observations may explain the results of previous studies with pyrimethamine27,28 and suggest a more broadly adaptive fitness advantage of dormancy for parasite survival. We hypothesize that ring-stage dormancy is a fitness trait that provides an advantage to P. falciparum survival to stress, especially from antimalarial drugs. The potential role of second-cycle dormancy and recrudescence in resistance development should be investigated with additional antimalarial drugs.
The relative fitness associated with artemisinin resistance is of critical importance, given our current reliance on ACTs as the first-line therapy for malaria worldwide. Although new promising drugs are in clinical trials, there are no new therapies that can immediately replace the loss of ACTs; therefore, fitness and its role in the emergence and spread of this resistance trait need additional scrutiny. What our data show is the fitness advantage of artemisinin-resistant genotypes to survive and predominate the population even in the face of infrequent drug exposure. What we could not measure in this study are other important traits that could enhance the spread of these genotypes. In particular, the transmission potential of artemisinin-resistant P. falciparum is of critical importance. Previous studies suggest enhanced transmission potential of resistant parasites by earlier commitment to produce gametocytes as well a competitive advantage of resistant parasites to survive drug exposure long enough to transmit the traits to the next generation. Clearly, if artemisinin-resistant parasites are more fit to transmit to mosquitos, the effects we observed will be amplified in the field. These are intriguing biological questions that more importantly have immediate relevance to the malaria elimination and eradication efforts. Data from these competitive fitness studies offer an opportunity to model the fitness of artemisinin resistance and to begin to assess these fitness advantages and costs in vivo.
Funding
This study was supported by grants (R01AI058973 and R01AI79709) from the National Institute of Allergy and Infectious Diseases of the US National Institutes of Health. The funder played no part in study design, conduct of the study or analysis of the results.
Transparency declarations
None to declare.
Supplementary data
Acknowledgements
We thank Drs Alberto van Olphen and Theresa Trindade for advice, assistance and access to equipment for pyrosequence analysis.
References
- 1.Takala-Harrison S, Jacob CG, Arze C, et al. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J Inf Dis 2015; 211: 670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dondorp AM, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. New Engl J Med 2009; 361: 455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noedl H, Se Y, Schaecher K, et al. Evidence of artemisinin-resistant malaria in western Cambodia. New Engl J Med 2008; 359: 2619–20. [DOI] [PubMed] [Google Scholar]
- 4.Ashley EA, Dhorda M, Fairhurst RM, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. New Engl J Med 2014; 371: 411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Straimer J, Gnadig NF, Witkowski B, et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015; 347: 428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ariey F, Witkowski B, Amaratunga C, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014; 505: 50–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miotto O, Amato R, Ashley EA, et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 2015; 47: 226–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenthal PJ. The interplay between drug resistance and fitness in malaria parasites. Mol Microbiol 2013; 89: 1025–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters JM, Chen N, Gatton M, et al. Mutations in cytochrome b resulting in atovaquone resistance are associated with loss of fitness in Plasmodium falciparum. Antimicrob Agents Chemother 2002; 46: 2435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregson A, Plowe CV. Mechanisms of resistance of malaria parasites to antifolates. Pharmacol Rev 2005; 57: 117–45. [DOI] [PubMed] [Google Scholar]
- 11.Roper C, Pearce R, Bredenkamp B, et al. Antifolate antimalarial resistance in southeast Africa: a population-based analysis. Lancet 2003; 361: 1174–81. [DOI] [PubMed] [Google Scholar]
- 12.Wootton JC, Feng X, Ferdig MT, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 2002; 418: 320–3. [DOI] [PubMed] [Google Scholar]
- 13.Nair S, Williams JT, Brockman A, et al. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol 2003; 20: 1526–36. [DOI] [PubMed] [Google Scholar]
- 14.Laufer MK, Thesing PC, Eddington ND, et al. Return of chloroquine antimalarial efficacy in Malawi. New Engl J Med 2006; 355: 1959–66. [DOI] [PubMed] [Google Scholar]
- 15.Laufer MK, Takala-Harrison S, Dzinjalamala FK, et al. Return of chloroquine-susceptible falciparum malaria in Malawi was a reexpansion of diverse susceptible parasites. J Inf Dis 2010; 202: 801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hastings IM, Donnelly MJ. The impact of antimalarial drug resistance mutations on parasite fitness, and its implications for the evolution of resistance. Drug Res Updates 2005; 8: 43–50. [DOI] [PubMed] [Google Scholar]
- 17.Froberg G, Ferreira PE, Martensson A, et al. Assessing the cost-benefit effect of a Plasmodium falciparum drug resistance mutation on parasite growth in vitro. Antimicrob Agents Chemother 2013; 57: 887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pongtavornpinyo W, Hastings IM, Dondorp A, et al. Probability of emergence of antimalarial resistance in different stages of the parasite life cycle. Evol Appl 2009; 2: 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White NJ, Pongtavornpinyo W. The de novo selection of drug-resistant malaria parasites. Proc Biol Sci R Soc 2003; 270: 545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science 1976; 193: 673–5. [DOI] [PubMed] [Google Scholar]
- 21.Chavchich M, Gerena L, Peters J, et al. Role of pfmdr1 amplification and expression in induction of resistance to artemisinin derivatives in Plasmodium falciparum. Antimicrob Agents Chemother 2010; 54: 2455–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tucker MS, Mutka T, Sparks K, et al. Phenotypic and genotypic analysis of in vitro-selected artemisinin-resistant progeny of Plasmodium falciparum. Antimicrob Agents Chemother 2012; 56: 302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desjardins RE, Canfield CJ, Haynes JD, et al. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 1979; 16: 710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tucker MS. Phenotypic and genotypic analysis of in vitro selected artemisinin resistant Plasmodium falciparum. (Doctoral Dissertation) University of South Florida, 2010. Ann Arbor: ProQuest/UMI; (Publication No. 822142847). [Google Scholar]
- 25.Teuscher F, Chen N, Kyle DE, et al. Phenotypic changes in artemisinin-resistant Plasmodium falciparum lines in vitro: evidence for decreased sensitivity to dormancy and growth inhibition. Antimicrob Agents Chemother 2012; 56: 428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen N, LaCrue AN, Teuscher F, et al. Fatty acid synthesis and pyruvate metabolism pathways remain active in dihydroartemisinin-induced dormant ring stages of Plasmodium falciparum. Antimicrob Agents Chemother 2014; 58: 4773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakazawa S, Kanbara H, Aikawa M. Plasmodium falciparum: recrudescence of parasites in culture. Exp Parasitol 1995; 81: 556–63. [DOI] [PubMed] [Google Scholar]
- 28.Nakazawa S, Maoka T, Uemura H, et al. Malaria parasites giving rise to recrudescence in vitro. Antimicrob Agents Chemother 2002; 46: 958–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hott A, Casandra D, Sparks KN, et al. Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes. Antimicrob Agents Chemother 2015; 59: 3156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor SM, Parobek CM, DeConti DK, et al. Absence of putative artemisinin resistance mutations among Plasmodium falciparum in sub-Saharan Africa: a molecular epidemiologic study. J Inf Dis 2015; 211: 680–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mok S, Ashley EA, Ferreira PE, et al. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science 2015; 347: 431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maude RJ, Pontavornpinyo W, Saralamba S, et al. The last man standing is the most resistant: eliminating artemisinin-resistant malaria in Cambodia. Malaria J 2009; 8: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bopp SE, Manary MJ, Bright AT, et al. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Gen 2013; 9: e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rovira-Graells N, Gupta AP, Planet E, et al. Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Res 2012; 22: 925–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klonis N, Xie SC, McCaw JM, et al. Altered temporal response of malaria parasites determines differential sensitivity to artemisinin. Proc Natl Acad Sci USA 2013; 110: 5157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peatey CL, Chavchich M, Chen N, et al. Mitochondrial membrane potential in a small subset of artemisinin-induced dormant Plasmodium falciparum parasites in vitro. J Infect Dis 2015; doi:10.1093/infdis/jiv048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.