

The response to and repair of DNA damage are central to our understanding of the causes of breast cancer. DNA damage results from environmental exposure to genotoxic agents, lifestyle factors, and cellular metabolism. Among the most lethal forms of DNA damage are double-strand breaks. A failure to accurately repair double-strand breaks has catastrophic consequences for the cell, including aneuploidy and genomic instability. Double-strand breaks may be repaired by homologous recombination or by the error-prone process of nonhomologous end joining. Homologous recombination is an important factor in the susceptibility to breast cancer, because BRCA1 is directly involved in the repair of double-strand breaks; it functions as a coordinator of proteins that are important for the response to DNA damage and has influence over the activation of cell-cycle checkpoints, DNA damage recognition, and signaling.1 Through its binding partners, BRCA1 is able to act in sensing DNA damage as well as in facilitating the DNA-repair process (Fig. 1).
Figure 1. DNA-Repair Pathways.
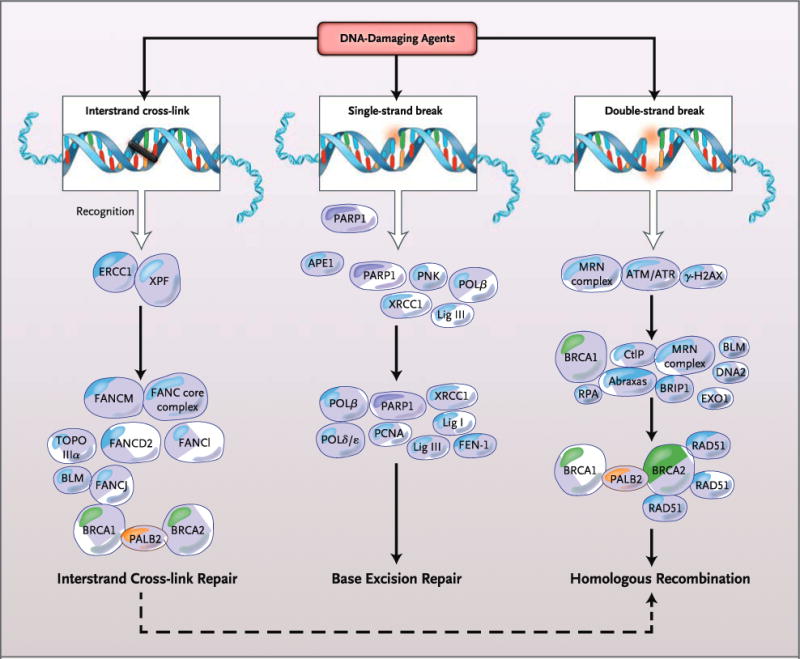
DNA damage from exogenous or endogenous sources may be repaired by a variety of DNA damage-response and damage-repair pathways, depending on the type of DNA damage induced. Interstrand cross-link repair, the single-strand-break repair subpathway of base excision repair, and homologous recombination address specific forms of damage with different, but in some cases common, proteins. The final step of interstrand cross-link repair involves homologous recombination. APE1 denotes apurinic-apirimidinic endonuclease 1; ATM ataxia telangiectasia mutated; ATR serine–threonine kinase; BLM Bloom’s syndrome, RecQ helicase–like; BRCA1 breast cancer 1, early onset; BRCA2 breast cancer 2, early onset; BRIP1 BRCA1-interacting protein C-terminal helicase 1; CtlP retinoblastoma binding protein 8; DNA2 DNA replication helicase/nuclease 2; ERCC1 excision repair cross-complementation group 1; EXO1 exonuclease 1; FANC core complex Fanconi’s anemia core complex; FANCM Fanconi’s anemia, complementation group M; FANCD2 Fanconi’s anemia, complementation group D2; FANCl Fanconi’s anemia, complementation group l; FANCJ Fanconi’s anemia, complementation group J; FEN-1 flap structure–specific endonuclease 1;·γ-H2AX phosphorylation of histone H2AX on serine 139; Lig I DNA ligase I; Lig III DNA ligase III; PALB2 partner and localizer of BRCA2; PARP1 poly(ADP-ribose) polymerase 1; PCNA proliferating-cell nuclear antigen; PNK polynucleotide kinase; Polδ/ɛ DNA polymerase δ/DNA polymerase ɛ; Polβ DNA polymerase β; RAD51 RAD51 recombinase; RPA replication protein A; TOPO IIIα DNA topoisomerase IIIα; XPF xeroderma pigmentosum, complementation group F; and XRCC1 x-ray repair complementing defective repair in Chinese hamster cells 1. The MRN complex is made up of Mre11, Rad50, and Nibrin.
PALB2 (partner and localizer of BRCA2) was previously identified as a moderate-risk gene in breast cancer.2 Monoallelic mutations result in cancers, whereas biallelic mutations lead to Fanconi’s anemia, complementation group N, as explained below. PALB2 encodes a protein that functions as a tumor suppressor by binding and colocalizing with BRCA2 in nuclear foci. PALB2 permits localization of BRCA2 in the nucleus and provides the molecular scaffold for the BRCA1–PALB2–BRCA2 complex. PALB2 not only works with BRCA2 to prevent cells from accumulating DNA damage but also interacts with BRCA2 to replace replication protein A with RAD51 on the processed single-stranded DNA end. Homologous recombination is also important as the final step in the interstrand cross-link repair pathway, which is deficient in Fanconi’s anemia. BRCA1, BRCA2 (FANCD1), and PALB2 (FANCN), along with FANCJ (BRIP1), facilitate strand invasion to complete interstrand cross-link repair.3
In this issue of the Journal, Antoniou and colleagues4 examine the lifetime risk of breast cancer resulting from germline loss-of-function PALB2 mutations. Their analysis suggests that the risk for PALB2 mutation carriers is as high as the risk borne by BRCA2 mutation carriers. Using a modification of the complex-segregation-analysis approach, these authors examined PALB2 truncating, splice, and deletion mutations and assessed the age-specific risk of breast cancer in families. The family members who carried germline PALB2 mutations had a risk of breast cancer that was increased by a factor of 9.5, as compared with U.K. incidence statistics. The mean cumulative risk of breast cancer by 70 years of age was estimated to be 35%. The age-specific relative risk for mutation carriers was highest among those younger than 40 years of age (relative risk, 8 to 9), with slight decrements in risk among those 40 to 60 years of age (relative risk, 6 to 8) and those older than 60 years of age (relative risk, approximately 5). The increased relative risk for PALB2 mutation carriers extends to men as well, who have a risk of breast cancer that is 8.3 times as high as the risk in the male general population.
The study assessed the residual familial component contributing to risk and showed that the risks associated with PALB2 mutations synergize with unknown environmental, lifestyle, or additional genetic factors. The authors confirm that the mean risk estimates are markedly increased when risk is evaluated within the context of genotype and family history, which highlights the need to use genetic information within the context of family history and lifestyle factors when assessing risk.
Because of the racial homogeneity of this study population, future studies will be required to provide a wider examination of the role of PALB2 in breast cancer in other populations. Although black American women have a lower incidence of breast cancer than white American women, the disease among black Americans tends to occur earlier, to have more unfavorable prognostic signs, and to be associated with higher mortality. Small cohort studies of PALB2 mutation status in cohorts with African ancestry have shown a modestly increased risk but perhaps different mutational spectra.5–7
The findings of Antoniou et al. regarding PALB2 highlight the emerging opportunities to treat breast cancer by pursuing the synthetic lethality of cancer therapy. Synthetic lethality, first described by Calvin Bridges in the 1920s, occurs when there is a loss of function of two related genes simultaneously that results in cell death, whereas a loss of function of only one gene is not lethal.8 Cells that have mutant BRCA1 or BRCA2 are exquisitely sensitive to inhibition of the base excision repair enzyme poly(adenosine diphosphate–ribose) polymerase (PARP) 1.9 In cells with defective homologous recombination due to BRCA1 or BRCA2 loss of heterozygosity, exposure to PARP inhibitors results in the persistence of single-strand and double-strand breaks, leading to genomic instability and ultimately to cell death. Loss of heterozygosity at the PALB2 locus is likely to result in increased sensitivity to cell death with PARP inhibition as well, given that synthetic lethality is seen with other proteins that are involved in homologous recombination.10 The promise of synthetic lethality in breast-cancer treatment through genetic and pharmacologic targeting of the Fanconi’s anemia–breast cancer pathway will lead the way to the examination and exploitation of defective DNA-repair mechanisms in other cancers.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–9. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stecklein SR, Jensen RA. Identifying and exploiting defects in the Fanconi anemia/BRCA pathway in oncology. Transl Res. 2012;160:178–97. doi: 10.1016/j.trsl.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng Y, Zhang J, Niu Q, Huo D, Olopade OI. Novel germline PALB2 truncating mutations in African American breast cancer patients. Cancer. 2012;118:1362–70. doi: 10.1002/cncr.26388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding YC, Steele L, Chu LH, et al. Germline mutations in PALB2 in African-American breast cancer cases. Breast Cancer Res Treat. 2011;126:227–30. doi: 10.1007/s10549-010-1271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71:2222–9. doi: 10.1158/0008-5472.CAN-10-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bridges CB. The origin of variations in sexual and sex-limited characters. Am Nat. 1922;56:51–63. [Google Scholar]
- 9.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 10.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]