

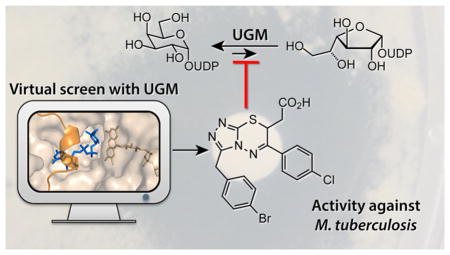
Abstract
Galactofuranose (Galf) is present in glycans critical for the virulence and viability of several pathogenic microbes, including Mycobacterium tuberculosis, yet the monosaccharide is absent from mammalian glycans. Uridine 5′-diphosphate-galactopyranose mutase (UGM) catalyzes the formation of UDP-Galf, which is required to produce Galf-containing glycoconjugates. Inhibitors of UGM have therefore been sought, both as antimicrobial leads and as tools to delineate the roles of Galf in cells. Obtaining cell permeable UGM probes by either design or high throughput screens has been difficult, as has elucidating how UGM binds small molecule, noncarbohydrate inhibitors. To address these issues, we employed structure-based virtual screening to uncover new inhibitor chemotypes, including a triazolothiadiazine series. These compounds are among the most potent antimycobacterial UGM inhibitors described. They also facilitated determination of a UGM–small molecule inhibitor structure, which can guide optimization. A comparison of results from the computational screen and a high-throughput fluorescence polarization (FP) screen indicated that the scaffold hits from the former had been evaluated in the FP screen but missed. By focusing on promising compounds, the virtual screen rescued false negatives, providing a blueprint for generating new UGM probes and therapeutic leads.
The monosaccharide D-galactofuranose (Galf) is found in cell surface glycoconjugates of many human pathogens, but it is absent from the mammalian glycome.1 Glycans containing Galf residues in human pathogens can be required for virulence as with K. pneumoniae2 or viability as in M. tuberculosis.3 Enzymes involved in Galf metabolism are therefore potential targets for the treatment of human disease. Uridine 5′-diphosphate (UDP)-galactopyranose mutase (Glf, also known as UGM) generates the biosynthetic precursor for Galf incorporation. UGM possesses a flavin adenine dinucleotide (FAD) cofactor that it uses to catalyze ring contraction of UDP-galactopyranose (UDP-Galp) to form UDP-Galf4–6 (Figure 1a). All organisms that generate Galf-containing glycans encode a UGM homologue. As a result, the gene encoding UGM has been linked to the virulence and viability of many infectious microbes, such as Mycobacterium tuberculosis,3,7 Klebsiella pneumoniae,1,8 Leishmania major, Aspergillus fumigatus,9 and parasitic nematodes.10,11
Figure 1.

UGM catalyzes the formation of UDP-Galf from UDP-Galp. (a) The isomerization reaction catalyzed by UGM. (b) A view of the active site from the crystal structure of K. pneumoniae UGM (wheat; PDB 3INT:B) in complex with UDP-Galp (blue) and the FAD cofactor. The structure represents the active conformation, with FAD reduced and the flexible 166–178 loop (orange) closed over the substrate.
The rise of multi-drug-resistant and extremely drug-resistant strains of M. tuberculosis12 has prompted a search for new tuberculosis drug candidates and new druggable targets. UGM is essential,3,7 and the gene encoding it is highly conserved between strains.13 UGM is required to generate the essential Galf polysaccharide known as the galactan, which is found within the thick and hydrophobic mycobacterial cell envelope.14,15 These features render the enzyme an attractive potential target. Moreover, the presence of UGM homologues in a variety of pathogens suggests that UGM inhibitors could be useful against multiple classes of microbes.10,16–23 We set out to devise cell-permeable UGM inhibitors that could be used as therapeutic leads and probes of the roles of Galf-containing glycans.
A number of researchers have sought UGM inhibitors.16,18–20,22,24–30 Though inhibitors have emerged,18–20,31 few have been shown to function against microbial pathogens. We previously developed a fluorescence polarization (FP) assay to identify competitive UGM inhibitors.17,19 When implemented in a high-throughout screen (HTS) of 16 000 compounds, it afforded a series of thiazolidinones with an overall “hit-rate” of 0.4%.19,20 The thiazolidinone leads, however, are susceptible to conjugate addition by biological thiols20 and are classified as pan assay interference compounds (PAINS).32 Scaffold hopping was used to find 2-aminothiazole inhibitors,18 the most potent of which had an IC50 of 7.2 μM and 37 μM against K. pneumoniae UGM and M. tuberculosis UGM enzymes, respectively.16,17 Despite their promising antimycobacterial activity, they suffer from low solubility and mammalian cell toxicity at levels near their minimum inhibitory concentrations (MICs).16 We postulate that their off-target effects are a consequence of their modest potency. With no structures of UGM-small molecule inhibitor complexes to optimize potency, we sought new scaffolds by screening 320 000 compounds through the Molecular Libraries Small Molecule Repository (MLSMR). The screen had a low hit rate (0.06%) and afforded but one lead inhibitor (IC50 > 250 μM) of the M. tuberculosis UGM (PubChem AID 504439).
The issues we encountered are emblematic of those impeding pharmacological targeting and inhibitor discovery for nucleotide-sugar-binding enzymes. Few assays amenable to HTS have been developed. The available assays often use binding as a proxy for catalysis,19,33 and screens of this type can have high false negative rates. The scarcity of inhibitors for enzymes that act on nucleotide-sugar substrates is also problematic as it is unclear what scaffolds might give rise to leads. In principle, structure-based virtual screens can address these challenges.34 Docking can be used to evaluate large libraries of molecules to find those that physically fit a protein site. The world’s available compounds may be sampled, via libraries such as ZINC,35,36 and screens of 3–6 million molecules can be routinely implemented that can sample chemotypes not represented in small HTS libraries. Because only a limited number of prioritized molecules from the library are ultimately tested, one can use more sensitive low-throughput assays to evaluate candidate ligands. Nevertheless, docking screens have their own risks: testing so few molecules leads to docking false-negatives,37 and the technique remains plagued by false positives. The methodological problems of approximate scoring functions and under-sampling of molecular degrees-of-freedom are still unresolved.38,39 Indeed, others have struggled to find productive inhibitors of UGM using virtual screening.40
Despite the above caveats, the lack of progress in applying experimental HTS strategies motivated us to use DOCK3.641,42 to virtually screen a database of 4.6 million commercially available compounds.35,36 Three compounds that inhibit UGM in vitro were identified. Structure-based optimization produced a class of competitive triazolothiadiazine UGM inhibitors. Notably, members of this class exhibit antimycobacterial activity in cell culture against M. tuberculosis and are some of the most potent UGM inhibitors described to date. They also inhibit eukaryotic UGMs, a class of enzymes for which no high throughput assays are available. Moreover, a compound from this new series facilitated the determination of the first X-ray structure of a UGM in complex with a nonsubstrate inhibitor. We anticipate that this structure can be used to further optimize the lead compounds.
RESULTS AND DISCUSSION
Docking Screen
As a starting point for the screen, we examined the structures of UGM:UDP-Galp substrate complexes available in the Protein Data Bank. Structures of UGM homologues from two eukaryotic species (Aspergillus fumigatus43 and Trypanosoma cruzi44) and five different bacterial species6,21,45–48 have been determined. Because we were seeking antimycobacterial agents, we focused on structures of UGMs in the active conformation from bacterial species. We therefore used the structure of the substrate-bound, flavin-reduced K. pneumoniae UGM (PDB ID: 3INT) as our starting point.46 In this form of the enzyme, a flexible loop (residues 166–178) is closed over the active site (Figure 1b). To evaluate docking sampling parameters, we created 1700 property-matched decoys49 for 25 2-aminothiazole18 and 13 thiazolidi-none20 UGM inhibitors (Figure S1). A property-matched decoy is a molecule with the physical properties of a known ligand (here the 38 inhibitors) but with topological features that render it unlikely to bind the enzyme. A well-performing retrospective docking calculation will highly rank the true ligands and deprioritize the decoy molecules —this strategy has long been used to calibrate docking calculations.49 To quantify how well true ligands were separated from decoy molecules, we calculated an “adjusted log AUC” score,42 a metric that emphasizes early enrichment of ligands ranked highly and corrects for what one would expect at random. For context, a random ranking of docked compounds yields an adjusted log AUC of zero, while values greater than 20 indicate good performance. The optimal docking configuration led to a log AUC of 34.5 overall. Though thiazolidinones, members of the PAINS, can act as UGM inhibitors,20 they scored poorly by docking. In contrast, ligands from the 2-aminothiazole series (e.g., Figure 2, ED103), had a log AUC of 54.8 (Figure S2a), indicating high early enrichment (i.e., more than 50% of the known 2-aminothiazole ligands ranked within the top 1% of the library).
Figure 2.

Normalized inhibitory activity of predicted UGM ligands against the UGM from K. pneumoniae. (a) Thirteen commercially available, predicted inhibitors were tested for inhibition in an enzyme activity assay.52 Error bars show standard deviation (n = 2). Limited solubility precluded the testing of compound 9 under these conditions. (b) Structures of compounds that displayed dose-dependent inhibition. Docking rank (out of 4.6 million) from the virtual screen is indicated in parentheses. See Figure S3 for the structures of all tested molecules.
The configuration generated for known UGM binders was used to dock a library of 4.6 million “lead-like” 50 molecules (250 < molecular weight <350; xLogP ≤ 3.5; number of rotatable bonds ≤ 735,50). On average, each molecule in the library was docked in 2163 conformations and in 246 000 orientations, or 6.39 × 108 configurations per molecule. Over the entire screen, 3.24 × 1015 complexes were sampled. This calculation took 2595 CPU days, or about 104 real-time hours on our cluster. This analysis does not include the substantial prescreening calculations that led to parameter optimization. The 2-aminothiazoles showed good enrichment in this larger ligand set as well, with a log AUC of 46.3 (40% of the known 2-aminothiazole ligands ranked in the top 0.1% of the screened library; Figure S2b).
Experimental Evaluation of Docking
A small collection of compounds representing novel, previously untested scaffolds was selected from the most highly ranked compounds for in vitro testing. Many sources of error can exist in large-scale automatic docking screens, from approximate and incomplete scoring functions to misrepresentation of compounds in databases. These and other factors are not considered in the scoring function and are more effectively evaluated by a trained investigator. High-ranking molecules were rejected due to the following criteria: unlikely ionization state at physiological pH values, no commercial source docked in a high-energy conformation or with unsatisfied polar interactions. They were also prioritized for their potential to interact with either of the two critical active site arginines51 and a complementary fit to the binding site. Using these criteria, 13 compounds were chosen to test as UGM inhibitors (Figure S3). These compounds had docking scores that ranked among the top 0.01% of the library.
We first tested compounds against the K. pneumoniae UGM using an HPLC activity assay.52 We employed this enzyme homologue as its structure was used in the virtual screen. Of the six compounds blocking more than 25% of UGM activity at 100 μM, five exhibited dose-dependent inhibition (15–33% at 50 μM). At concentrations in this range, nonspecific inhibition can occur if compounds undergo colloidal aggregation.53 To eliminate small molecules that act via this mechanism, the assay was repeated in the presence of detergent (0.025% Tween 80), and compounds (3, 11, 12) whose activity was severely diminished when detergent was present were not analyzed further (Figure S4). Three compounds (6, 8, and 10; Figure 2b) showed dose-dependent UGM inhibition and insensitivity to detergent, giving an overall hit rate of 23% (3 out of 13).
We further characterized the features of compounds 6 and 10 relevant for their inhibitory activity. When dynamic light scattering (DLS) was used to directly assess their propensity to aggregate, neither 6 nor 10 formed colloids at concentrations of 100 μM (Figure S5). This finding is consistent with their inhibitory activity in the presence of detergent. Additionally, at 100 μM, neither compound inhibits AmpC β-lactamase, an enzyme effective in counter-screens for promiscuous aggregation.54,55 The FP assay indicated that compound 6 binds to K. pneumoniae UGM with Kd = 43 ± 20 μM and compound 10 with Kd = 6 ± 3 μM (Figure S6).
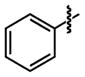
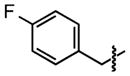
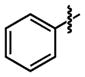
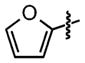
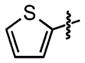
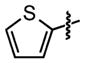
To elucidate the structure–activity relationships (SARs) of compounds 6 and 10, commercially available analogs were purchased and tested (Figure 3). One analog of 10 showed improved K. pneumoniae UGM inhibition (Figure S7), but limited availability of additional analogs and the solubility of 10 deterred us from further pursuing this series. A focus on compound 6, however, proved productive. Lineweaver–Burk analysis indicated that compound 6 is a competitive inhibitor of K. pneumoniae UGM with a Ki of 78 ± 31 μM (Figure S8). Of the 11 analogs of 6 sourced for testing, nine showed better inhibition of K. pneumoniae UGM at 100 μM than the parent compound (Table 1), and five had better Kd values. The most active compounds, 19, 22, and 24, exhibited Kd values of 18 ± 11 μM, 9 ± 5 μM, and 9 ± 4 μM, respectively (Figure 3a and Figure S9). Although these results were promising, previous studies have shown that series that begin as nonaggregators can drift into this mechanism upon optimization.56 Analysis by DLS indicated that 19 and 24 form particles at 100 μM, but compound 22 did not (Figure S5). Lineweaver–Burk analysis indicated that 22 is a competitive inhibitor of K. pneumoniae UGM (Figure 3c).
Figure 3.

Evaluation of compounds 22 and 30 for competitive K. pneumoniae UGM inhibition and M. smegmatis cell killing. (a,b). First-generation (22) and second-generation (30) compounds were tested with K. pneumoniae in a fluorescence poloarization assay. Kd values determined using this assay are 9 ± 5 μM for 22 and 5 ± 1.5 μM for 30 (error bars represent standard deviation; n = 3). (c,d) Lineweaver–Burk analysis of UGM inhibition by 22 and 30 (Ki,22 = 12 ± 5 μM and Ki,30 = 1.1 ± 0.2 μM; error bars represent standard deviation; n = 3). (e) Nonlinear regression plot of K. pneumoniae UGM inhibition by 30 is shown for comparison to double reciprocal analysis. (f) M. smegmatis viability in the presence of the compounds was assessed in liquid culture using Alamar Blue. MICs were determined after growth (46 h) in the presence of UGM inhibitors (error bars represent standard deviation; n = 2). MIC of 30: 20 μM.
Table 1.
Commercially Available Analogs of Compound 6 Evaluated as K. pneumoniae UGM Inhibitors
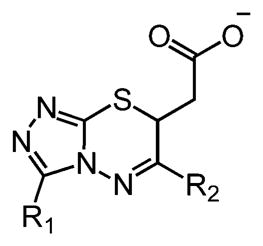
| ||||
|---|---|---|---|---|
| # | R1 | R2 | Kd (μM) | % inhibition (100 μM) |
| 6 |
|
|
41 ±20 | 34 ±3 |
| 16 |
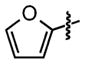
|
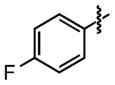
|
>100 | 38 ±2 |
| 17 |
|
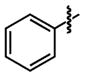
|
75 ±36 | 8.9 ±0.4 |
| 18 |
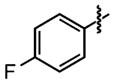
|
|
59 ±27 | 74 ±3 |
| 19 |
|
|
18 ± 11 | 92 ± 0.0 |
| 20 |
|
|
N/A | 46 ±0.5 |
| 21 | H− |
|
64 ±24 | 72 ±1 |
| 22 |
|
|
9±5 | 95 ± 0.0 |
| 23 |
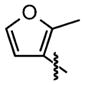
|
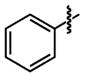
|
92 ±42 | 25 ±10 |
| 24 |
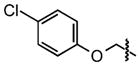
|
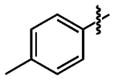
|
9±4 | 97 ± 0.6 |
| 25 |
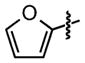
|
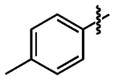
|
33 ±9 | 62 ±1 |
| 26 |
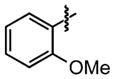
|
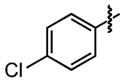
|
24 ±9 | 91 ±0.8 |
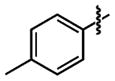
| 27 |
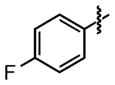
|
|
15 ±4 | 96 ±0.3 |
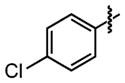
| 28 |
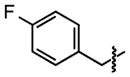
|
|
9.9 ±3 | 96 ±0.1 |
| 29 |
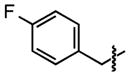
|
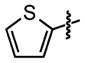
|
59 ±27 | 92 ± 0.2 |
| 30 |
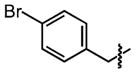
|
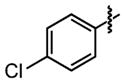
|
5.0 ±1 | 100 ±0.0 |
| 31 |
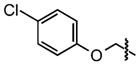
|
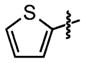
|
13 ±3 | 95 ± 0.7 |
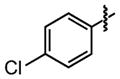
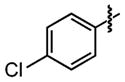
Inhibitors Act on Multiple Species
We next compared the selectivity of the new inhibitors by evaluating them with UGMs from other pathogens. The UGMs from M. tuberculosis and C. diphtheriae share about 43% sequence identity with that from K. pneumoniae but share 76% identity with each other (ClustalO; Figure S10). Eukaryotic UGMs from organisms such as Caenorhabditis elegans have low sequence identity with bacterial UGMs.10 Structural studies of the bacterial and eukaryotic enzymes reveal some differences in substrate binding,43 yet the general features of the UGM active site are highly conserved across species.57 Consistent with this conservation, compound 22 inhibited all the UGM homologues tested. It was less effective for the C. elegans UGM (Ki = 25 ± 8 μM), M. tuberculosis UGM (Ki = 31 ± 18 μM), and C. diphtheriae UGM (Ki = 77 ± 37 μM) than for the K. pneumoniae UGM (Ki = 8 ± 3 μM; Figure 4). This trend was preserved other analogs from this series (Figure 4). Higher activity against the K. pneumoniae homologue may reflect the use of a K. pneumoniae UGM structure for the docking screen.
Figure 4.

Comparison of inhibitory activity with UGM homologues. Data for compounds (a) 22 and (b) 30 were fit with one-site IC50 nonlinear regression curves, and Ki values were determined using the Cheng–Prusoff equation.69 Error bars show standard deviation of the mean (n = 2).
The ability of the triazolothiadiazines to block a nematode UGM is noteworthy, as the FP assay used in HTS to find prokaryotic UGM inhibitors is not applicable to eukaryotic UGMs. Specifically, the fluorescent UDP derivative used in the FP assay binds too weakly to eukaryotic UGMs to be useful in screening. Recent structural studies of the A. fumigatus UGM reveal that the substrate uracil adopts a conformation in the binding site43 that differs from that observed with the prokaryotic enzymes. These distinctions may be important for FP probe binding and catalysis, but the use of the prokaryotic enzyme in the docking screen yielded inhibitors that act against both prokaryotic and eukaryotic UGM homologues.
X-ray Crystallography of a UGM–22 Complex
A major barrier to optimizing previously identified UGM inhibitors has been the lack of a UGM–inhibitor structure. We therefore used X-ray crystallography to determine a structure of UGM in complex with small molecule 22. Although we obtained crystals of the K. pneumoniae UGM in the presence of 22 that diffracted, lattice-translocation disorder complicated structure determination. Fortunately, the structure of C. diphtheriae UGM with inhibitor 22 was solved at 2.65 Å resolution (see Table S1 for data collection and refinement statistics). Its modest resolution notwithstanding, the structure provides insight into the binding orientation and interactions of 22.
The UGM complex crystallized in the open, oxidized conformation. Co-crystals of C. diphtheriae UGM and 22 in the reduced, closed form of the enzyme were not obtained, but our structure shows some additional density proximal to the ligand that likely corresponds to the closed lid (Figure S11). In both monomers (chain A and chain B), compound 22 binds in the active site (Figure 5a; Figure S12). In both chains, the R1 thiophene of 22 occupies a hydrophobic pocket formed by Tyr326, Tyr364, Trp162, and Pro327, while the R2 chlorophenyl stacks on face-to-face with Tyr326 and edge-to-face with Trp162. In chain A, there is a salt bridge between the carboxylate group of the inhibitor and Arg288, a residue essential for UGM activity.51 In chain B, however, density for the ligand carboxylate is more ambiguous. Therefore, despite its marginally higher average temperature factors (106 versus 97 Å2) and slightly lower occupancy (0.81 versus 0.87) (Table S1), we focused on the ligand in chain A in our structural analysis.
Figure 5.

Structure of C. diphtheriae UGM in complex with 22 (PDB ID: 4XGK). (a) The complex of compound 22 (black) and oxidized C. diphtheriae UGM (white) determined by X-ray crystallography to 2.65 Å. The active site of chain A is shown. Green mesh depicts the Fo–Fc omit map (with the ligand removed and structure rerefined) at 2σ. (b) The ligand omit map in chain B of the UGM crystal dimer. (c) Comparison of the docking pose predicted for 6 binding to reduced UGM (thicker bonds, carbons in light blue) and the crystallized ligand in chain A. The two ligand poses differ by 5.1 Å over the triazolothiadiazine core. For detailed analysis of the closed UGM ligand docking pose generated and the open, oxidized crystallographically determined structure of the complex, see Figure S12.
Comparison of X-ray Structure and Docking Pose
The structure provided a means to compare the docked ligand pose to that determined experimentally. The C. diphtheriae UGM–ligand complex determined by X-ray crystallography is in the open form rather than in the closed loop conformation of the K. pneumoniae UGM structure used for the docking screen. The difference is not surprising as X-ray structures of UGM bound to substrates or substrate analogs have captured the enzyme in both open and closed forms.29 The key catalytic Arg174 residue is located on the flexible loop of UGM and thus moves into the substrate-binding pocket in the active form of the enzyme.46,51 Overlay of the X-ray structure of the UGM–inhibitor complex and the K. pneumoniae UGM structure employed in docking indicates that some reorientation of the Arg174-containing loop or of compound 22 would need to occur for 22 to bind the closed form of the enzyme (Figures 5c and S13). The enzyme’s flexibility leaves open the issue of whether compound 22 can bind to the closed conformation of the enzyme. This flexibility was also manifested when we examined the predicted docking pose of high-ranking hit 6: the ligand occupies the binding pocket present in the X-ray structure. The salt bridge between the inhibitor scaffold carboxylate and Arg288 (C. diphtheriae UGM numbering; Figure S12), seen in chain A also is predicted. Similarly, when 22 was computationally docked into C. diphtheriae UGM in the open conformation, the ligand pose matched that experimentally determined for chain A (Figure S14), suggesting that docking can capture the important features of binding to this conformation of the protein.
Though the orientation of compound 6 docked in the K. pneumoniae UGM active site differs somewhat from that in the X-ray structure of C. diphtheriae UGM, there are reasons to suspect that both of these are relevant to the activity of the series.58 In both, the ligand carboxylate participates in an ionic interaction with Arg288 and interactions that occur with many of the conserved aromatic residues in the UGM–22 complex are observed in the docked model. Opposing conformations of the flexible loop (Figure S13), sequence differences between K. pneumoniae UGM and C. diphtheriae UGM distal to the binding site, and the small variation in the ligand (6 versus 22) itself may contribute to discrepancies between the poses. Still, it seems likely that the hydrophobic subpockets of the binding site can support a large range of substitutions and/or binding modes. Interestingly, both ligand orientations are consistent with the observed SAR, as they indicate larger R1 side-chains are favorable and that bulky R2 para substitutions are preferred (Figure S12). We anticipate that both the crystal structure and docking pose can guide inhibitor optimization.
The structure of C. diphtheriae UGM bound to compound 22 adds to the growing number of UGM structures from human pathogens.6,21,44–46,59,60 Most significantly, however, it serves as the founding structure of a UGM bound to a small molecule, nonsubstrate inhibitor. Though the first UGM inhibitors were identified more than 10 years ago23 and several small molecule inhibitors have been reported subsequently,18–20,31 no structures of a complex have been disclosed. Our success in obtaining a complex with the triazolothiadiazines provides guidance for augmenting UGM inhibitor potency.
Structure–Activity Relationship Analyses
The initial activities and ligand efficiency (LE = ΔG/Number of heavy atoms) values of 0.361 provides impetus for triazolothiadiazine optimization. We therefore analyzed the structure determined by X-ray crystallography and the predicted docking pose to optimize potency within this series (Table 1; Figure S12). We anticipated that ligands with larger or more polarizable para substituents on the R2 phenyl ring would be more effective, and the SAR we had obtained to date are consistent with this analysis (i.e., pCl-Phe 26 > pMe-Phe 25 > pF-Phe 16). The parachlorophenyl substituent was expected to be especially favorable given the activities of 26 and 22. With regard to the R1 substituent, we anticipated that larger groups would give rise to enhanced inhibitor potency, as we had observed (i.e., compound 19 > 18 > 17 and compound 22 > 21). We therefore tested five additional commercially available “second generation” analogs (compounds 27–31, Table 1). All second-generation analogs afforded more than 90% inhibition at 100 μM, with three of the five showing greater than 90% inhibition at 50 μM. It is interesting to note that both ligand orientations (docking and X-ray structure) are consistent with the observed SAR, as they indicate that larger R1 side-chains are favorable and that bulky R2 para substitutions are preferred (Figure S12).
The best inhibitor (30) fully blocked K. pneumoniae UGM activity at 50 μM. We therefore characterized its binding to and inhibition of UGM (Figure 3b,d). Compound 30 is a competitive inhibitor of K. pneumoniae UGM (Ki = 1.1 ± 0.2 μM. The ability of 30 to block different UGM homologues followed the same relative order as observed with 22: Ki values of 28 ± 15 μM, 29 ± 8 μM, and 7 ± 3 μM were obtained for the M. tuberculosis, C. diphtheriae, and C. elegans UGM, respectively (Figure 4). It is notable that compound 30 is substantially more active any of its analogs tested against all UGM homologues. We detected no colloidal aggregates (Figure S5) nor nonspecific inhibition of malate dehydrogenase (Figure S14) at relevant compound concentrations. Additionally, detergent had no effect on competitive binding of 30 to UGM (Figure S15). Together, these data indicate that the inhibitory activity of 30 is specific.
Antimycobacterial activity of UGM inhibitors
For screens focused either on enzyme binding or inhibition in solution, a concern often raised is that inhibitors will fail to function in cells. To test whether the virtual screen yielded cell-permeable inhibitors, we evaluated our compounds against Mycobacterium smegmatis in broth microdilution assays. We focused on this organism, which is often used as a model for pathogenic mycobacteria, because UGM is nonessential in K. pneumoniae62 and because of the need for new antimycobacterial agents.
Growth curves for M. smegmatis were generated in liquid media in the presence of the most promising UGM inhibitors. Trends in growth inhibition aligned well with each compound’s in vitro activities (Figures 3 and 6). Notably, the MIC of 30 for M. smegmatis was 20 μM (9.7 μg/mL). Compound 30 showed no significant killing of bacterial species lacking a UGM ortholog, such as B. subtilis (MIC = 500 μM) and B strain E. coli (MIC > 500 μM; data not shown). Cytotoxicity of 30 with HEK293 cells (Figure S16) was observed only at the high concentrations (LD50 ~ 100 μM or 47.5 μg/mL) at which the compound aggregates.
Figure 6.

A disk diffusion assay for antibacterial activity of UGM inhibitors. Compounds 22, 27, 28, and 30 (400 nmol) were dissolved in DMSO and transferred onto a disk centered on an agar plate containing bacteria of the strain denoted. The activity of a previously described UGM inhibitor ED10318 is shown for comparison. The radius of the growth inhibition zone (measured from outer edge of paper disk to the border of cell growth) is indicated in millimeters in the upper left corner. The reported error represents the standard deviation of four independent measurements.
We next evaluated compounds for activity against pathogenic M. tuberculosis H37Rv. We employed a disk diffusion assay to compare control bacterial strains to mycobacteria (Figure 6). Compound 30 out-performed others tested in the series, significantly inhibiting growth of virulent M. tuberculosis. No zone of inhibition for BL21(DE3) E. coli was observed. These results are consistent with the lack of UGM expression in B strain E. coli. They further indicate that, compared to the previous lead ED103, compound 30 is much more potent against M. tuberculosis (Figure 6). The finding that triazolothiadiazine 30 exhibited enzyme-inhibitory activity in solution and also antimycobacterial activity in cells is notable. The data add to the growing evidence that UGM is a novel antimycobacterial drug target, and they suggest that triazolothiadiazine inhibitors can serve as leads. The challenges of penetrating the mycobacterial cell wall are substantial,7 so the efficacy of the identified compounds against M. smegmatis and M. tuberculosis is especially notable.
Comparing Docking and HTS Libraries
To understand why virtual screening gave rise to more productive UGM leads than our MLSMR screen, we assessed the chemical similarity of compounds in the docking and MLSMR libraries. Of the many options available for measuring chemical similarity, we selected an ECFP4-based Tanimoto-coefficient (Tc), wherein 1 indicates identical molecules and values lower than 0.35 typically mean the two molecules are of distinct chemotypes.63 Of the 4.6 million molecules docked, only 560 000 (12.3%) compounds had a Tc < 0.5 from the closest molecule in the MLSMR library. By these criteria, there was structural overlap in the two libraries, though there were many more molecules, and therefore finer granularity, in the docked library. The chemotypes can also be compared by clustering. Clustering MLSMR alone at 60% similarity resulted in 42 683 clusters, while clustering the union of the two libraries resulted in 175 501 clusters. The new 133 000 clusters represent chemo-types present only in the docking library; therefore, they provide a measure of the increased chemical space afforded by the structure-based approach.
Although docking hits 6, 8, and 10 themselves were not directly assayed in HTS, close analogs of these compounds were present in the screened MLSMR library (Table 2). When we retested these MLSMR analogs in the assays used to evaluate the primary docking hits; the analogs had comparable levels of UGM inhibition as their virtual screening counterparts (Figure 7). As these molecules were not identified as hits in the MLSMR screen, they represent HTS false negatives. These results suggest that suitable chemotypes for inhibition of nucleotide-sugar-dependent enzymes are not necessarily absent from screening libraries. Rather, the stringency that is built into HTS assays to make them reproducible and rapid and to limit promiscuous activities can lead researchers to overlook the weaker binding hits that may be evolved to useful leads.
Table 2.
Closest Analogs for Docking Hits in the Empirical HTS Screen Library
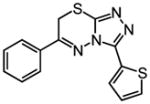
| Docking Hit | Closest MLSMRa | Tcb | Closest MLSMR Anionicc | Tcb |
|---|---|---|---|---|
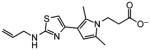
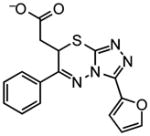 6 |
|
0.66 |
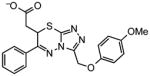 ~6 |
0.7 |
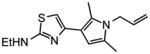
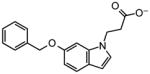 8 |
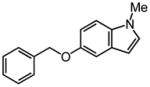
|
0.59 |
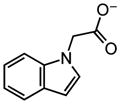 ~8 |
0.55 |
 10 |
|
0.65 |
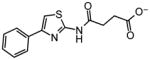 ~10 |
0.50 |
Closest analog in the entire MLSMR (May 2011) library.
Tanimoto coefficient (Tc) between docking hit and analog using ECFP4 fingerprints as implemented in Pipeline Pilot (version 6.1; SciTegic Inc., San Diego, CA). This is a common chemical similarity measure ranging from 0 to 1, where 1 indicates identical molecules, and values lower than 0.35 typically mean the two molecules are of distinct chemotypes.
Closest analog out of the subset of anionic compounds in the MLSMR library.
Figure 7.
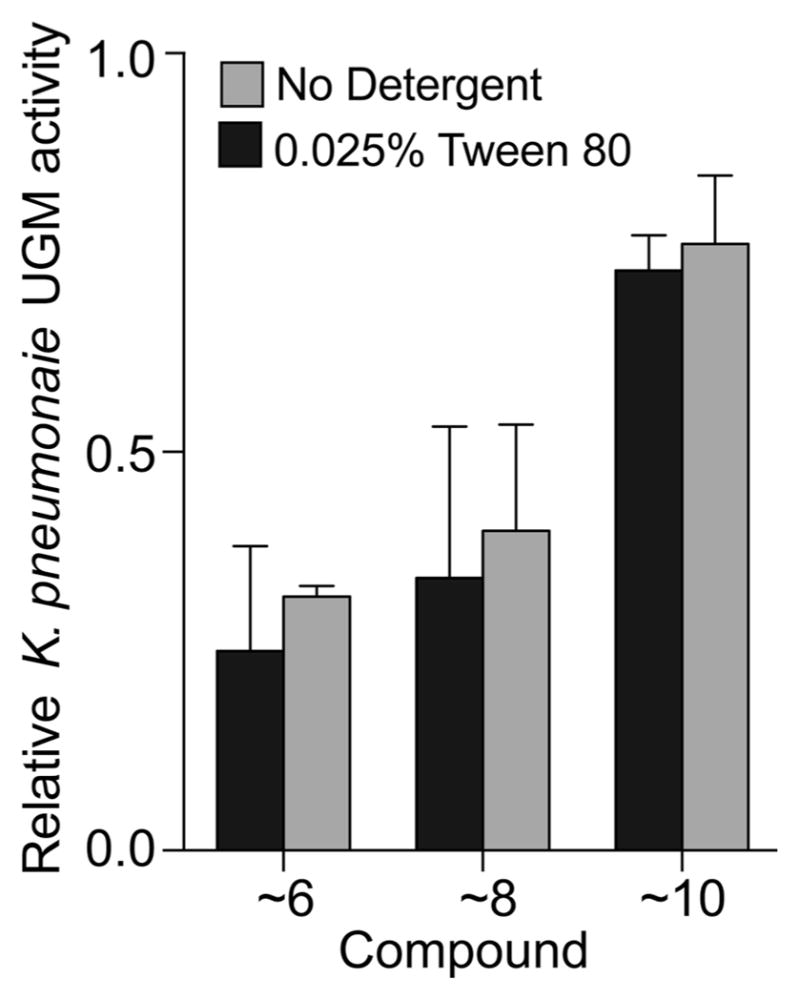
Inhibition of K. pneumoniae UGM by closest anionic analogs to docking hits in the empirically screened MLSMR library. Compounds were analyzed at 100 μM in the presence and absence of detergent to rule out nonspecific small molecule aggregation-based inhibition. See Table 2 for chemical structures and similarity scoring of the tested MLSMR compounds. Error bars represent standard deviation (n = 2).
Given our results, it is interesting to compare our HTS campaigns to the docking screen from a technical perspective. In total, almost 400 000 molecules have been tested empirically by HTS against the UGMs from K. pneumoniae and M. tuberculosis. Three active inhibitor chemotypes have been obtained with affinities ranging from 10 to 150 μM. Conversely, structure-based docking of a 10-fold larger library revealed three new inhibitor classes, all of which are dissimilar to those discovered by HTS and to each other (Figure 2, Figure S1; Tc < 0.32 for docking hits to any previously known inhibitor). These findings suggest that the identified compounds could not have been discovered by ligand-based screening methods, such as pharmacophore modeling.64 Together, our data are consistent with the growing body of literature that suggests virtual screening and HTS are complementary.37,58,65,66
Conclusions
A virtual screen afforded triazolothiadiazines that inhibit UGM and are more potent, specific, and active against mycobacteria than other inhibitors reported to date. Our X-ray structure of the UGM–compound 22 complex can guide the generation of new antimycobacterial agents and potent probes of UGM function. Moreover, the activity of our triazolothiadiazine inhibitors against C. elegans UGM should yield reagents for constructing novel probes and effective experimental screens for a broader scope of UGMs. Perhaps most importantly, our study suggests that the success of virtual screening with UGM compared to HTS was not due to exploring a larger chemical space—rather it provided the opportunity to reclaim HTS false negatives. These results highlight the complementarity of HTS and docking66 and provide a way forward for inhibitor discovery for nucleotide-sugar-dependent enzymes, an important and recalcitrant class of proteins.
EXPERIMENTAL SECTION
Methods are summarized briefly below. See the Supporting Information for experimental detail. DLS, aggregation screens, X-ray crystallography, and mammalian cytotoxicity assays are described exclusively in the Supporting Information.
Molecular Docking and Ligand Selection
Docking was performed with DOCK3.6.41,42 Sampling parameters were varied and assessed by enrichment of known ligands over decoys. The docking-property-matched decoys were calculated by the DUD-E server.49 For the final virtual screen, ZINC’s35 pregenerated lead-like subset (subset 1), containing 4.6 million molecules, was docked. The top 500 molecules from the docking screen were filtered using Marvin (ChemAxon), a SMARTS filter, and chemical logic. Compound similarity was calculated using ECFP4-based Tanimoto coefficients67 as implemented in Pipeline-Pilot.68 See the Supporting Information for compound sources.
Recombinant Protein Expression and Purification
UGM homologues from K. pneumoniae and M. tuberculosis were produced in E. coli and purified as previously described20,21 with slight modifications (see the Supporting Information). C. diphtheriae UGM was cloned into the pMALc5x vector, expressed in E. coli ER2523, and purified using a previously described procedure for the K. pneumoniae UGM.45
UGM in Vitro Inhibition
The enzymatic activity of each recombinant UGM was monitored using an HPLC-based assay.10,52 Small molecules were added from DMSO stocks to a final concentration of 1% DMSO. Reactions were analyzed using a CarboPac PA-100 column (Thermo Scientific) on an Agilent 1260 Infinity HPLC using an isocratic elution.
Small Molecule Binding to UGM
A fluorescence polarization assay was used to measure ligand binding to K. pneumoniae UGM,19,20 and data were fit to a one-site binding nonlinear regression in GraphPad Prism 6.
Microbroth Dilution Assay
A starter culture of M. smegmatis mc2155 (ATCC 700084) was grown to saturation at 30 °C in Middlebrook 7H9 broth with albumin dextrose catalase enrichment and 0.05% Tween 80. Small molecules or DMSO (vehicle control) were added to wells of a sterile 96-well plate. Cells were diluted into fresh media and added to the plate to a final 1% DMSO concentration. After growth (46 h) in a 30 °C shaking incubator, cell viability was measured using an alamarBlue assay (Invitrogen).
Disk Difiusion Assay
Saturated starter cultures were diluted to an OD600 of 0.02 in fresh media and plated. Sterile disks (6 mm diameter, Whatman) infused with compound (400 nmol) were placed on the surface of the inoculated solid media. E. coli plates were incubated at 37 °C overnight. Plates with M. smegmatis were incubated 3 additional days at room temperature, and M. tuberculosis plates were incubated 2 weeks at 37 °C. Zones of inhibition were measured from the outer edge of the cloning disk to the border of cell growth.
Supplementary Material
Acknowledgments
This research was supported by R01 AI063596 (L.L.K.), R01 GM59957 (B.K.S.), and R01 GM71630) (B.K.S.). V.A.K. and D.A.W. thank the NSF Graduate Research Fellowship Program for funding (DGE-1256259). N.L. was supported by an EMBO long-term fellowship (ALTF-1121-2011). K.W. was supported by a fellowship from the Development and Promotion of Science and Technology Talents Project of Thailand. S.A.M. was supported by NIH grant (R21AI090308) to A.M.T. We would like to thank M.A. Martinez-Farias and M.R. Levengood for their work on the MLPCN screen. We also thank Valerie J. Winton for her assessment of compound purity. X-ray data were measured at beamline X25 of the National Synchrotron Light Source, which is supported principally by the Offices of Biological and Environmental Research and Basic Energy Sciences of the U.S. Department of Energy, and from the National Center for Research Resources (P41RR012408) and the National Institute of General Medical Sciences (P41GM103473) of the NIH.
Footnotes
Notes
The authors declare the following competing financial interest: L.L.K., V.A.K., N.L. and B.S.S. are authors on patent that focuses on UGM inhibitors as anti-microbial agents.
Accession Codes
The PDB ID of C. diphtheriae UGM bound to inhibitor 22 is 4XGK.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.5b00370.
Protocols for the structure-based virtual screen, protein production, and crystallization, as well as supporting figures (PDF)
References
- 1.Richards MR, Lowary TL. Chemistry and Biology of Galactofuranose-Containing Polysaccharides. ChemBioChem. 2009;10:1920–1938. doi: 10.1002/cbic.200900208. [DOI] [PubMed] [Google Scholar]
- 2.Shankar-Sinha S, Valencia GA, Janes BK, Rosenberg JK, Whitfield C, Bender RA, Standiford TJ, Younger JG. The Klebsiella pneumoniae O antigen contributes to bacteremia and lethality during murine pneumonia. Infect Immun. 2004;72:1423–1430. doi: 10.1128/IAI.72.3.1423-1430.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density muta-genesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 4.Soltero-Higgin M, Carlson EE, Gruber TD, Kiessling LL. A unique catalytic mechanism for UDP-galactopyranose mutase. Nat Struct Mol Biol. 2004;11:539–543. doi: 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]
- 5.Nassau PM, Martin SL, Brown RE, Weston A, Monsey D, McNeil MR, Duncan K. Galactofuranose biosynthesis in Escherichia coli K-12: identification and cloning of UDP-galactopyranose mutase. J Bacteriol. 1996;178:1047–1052. doi: 10.1128/jb.178.4.1047-1052.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders DAR, Staines AG, McMahon SA, McNeil MR, Whitfield C, Naismith JH. UDP-galactopyranose mutase has a novel structure and mechanism. Nat Struct Biol. 2001;8:858–863. doi: 10.1038/nsb1001-858. [DOI] [PubMed] [Google Scholar]
- 7.Pan F, Jackson M, Ma Y, McNeil M. Cell Wall Core Galactofuran Synthesis Is Essential for Growth of Mycobacteria. J Bacteriol. 2001;183:3991–3998. doi: 10.1128/JB.183.13.3991-3998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh PF, Lin TL, Yang FL, Wu MC, Pan YJ, Wu SH, Wang JT. Lipopolysaccharide O1 Antigen Contributes to the Virulence in Klebsiella pneumoniae Causing Pyogenic Liver Abscess. PLoS One. 2012;7:e33155. doi: 10.1371/journal.pone.0033155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tefsen B, Ram AF, van Die I, Routier FH. Galactofuranose in eukaryotes: aspects of biosynthesis and functional impact. Glycobiology. 2012;22:456–469. doi: 10.1093/glycob/cwr144. [DOI] [PubMed] [Google Scholar]
- 10.Wesener DA, May JF, Huffman EM, Kiessling LL. UDP-galactopyranose mutase in nematodes. Biochemistry. 2013;52:4391–4398. doi: 10.1021/bi400264d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beverley SM, Owens KL, Showalter M, Griffith CL, Doering TL, Jones VC, McNeil MR. Eukaryotic UDP-galactopyranose mutase (GLF gene) in microbial and metazoal pathogens. Eukaryotic Cell. 2005;4:1147–1154. doi: 10.1128/EC.4.6.1147-1154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.World Health Organization Annual TB Report 2014 [Google Scholar]
- 13.Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, Holmes EC, Birren B, Galagan J, Feldman MW. The Role of Selection in Shaping Diversity of Natural M. tuberculosis Populations. PLoS Pathog. 2013;9:14. doi: 10.1371/journal.ppat.1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crick DC, Mahapatra S, Brennan PJ. Biosynthesis of the arabinogalactan-peptidoglycan complex of Mycobacterium tuberculosis. Glycobiology. 2001;11:107R–118R. doi: 10.1093/glycob/11.9.107r. [DOI] [PubMed] [Google Scholar]
- 15.Angala SK, Belardinelli JM, Huc-Claustre E, Wheat WH, Jackson M. The cell envelope glycoconjugates of Mycobacterium tuberculosis. Crit Rev Biochem Mol Biol. 2014;49:361–399. doi: 10.3109/10409238.2014.925420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borrelli S, Zandberg WF, Mohan S, Ko M, Martinez-Gutierrez F, Partha SK, Sanders DA, Av-Gay Y, Pinto BM. Antimycobacterial activity of UDP-galactopyranose mutase inhibitors. Int J Antimicrob Agents. 2010;36:364–368. doi: 10.1016/j.ijantimicag.2010.06.030. [DOI] [PubMed] [Google Scholar]
- 17.Dykhuizen EC, Kiessling LL. Potent ligands for prokaryotic UDP-galactopyranose mutase that exploit an enzyme subsite. Org Lett. 2009;11:193–196. doi: 10.1021/ol802094p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dykhuizen EC, May JF, Tongpenyai A, Kiessling LL. Inhibitors of UDP-galactopyranose mutase thwart mycobacterial growth. J Am Chem Soc. 2008;130:6706–6707. doi: 10.1021/ja8018687. [DOI] [PubMed] [Google Scholar]
- 19.Soltero-Higgin M, Carlson EE, Phillips JH, Kiessling LL. Identification of inhibitors for UDP-galactopyranose mutase. J Am Chem Soc. 2004;126:10532–10533. doi: 10.1021/ja048017v. [DOI] [PubMed] [Google Scholar]
- 20.Carlson EE, May JF, Kiessling LL. Chemical probes of UDP-galactopyranose mutase. Chem Biol. 2006;13:825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Gruber TD, Borrok MJ, Westler WM, Forest KT, Kiessling LL. Ligand Binding and Substrate Discrimination by UDP-Galactopyranose Mutase. J Mol Biol. 2009;391:327–340. doi: 10.1016/j.jmb.2009.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Partha SK, Sadeghi-Khomami A, Slowski K, Kotake T, Thomas NR, Jakeman DL, Sanders DAR. Chemoenzymatic Synthesis, Inhibition Studies, and X-ray Crystallographic Analysis of the Phosphono Analog of UDP-Galp as an Inhibitor and Mechanistic Probe for UDP-Galactopyranose Mutase. J Mol Biol. 2010;403:578–590. doi: 10.1016/j.jmb.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 23.Scherman MS, Winans KA, Stern RJ, Jones V, Bertozzi CR, McNeil MR. Drug targeting Mycobacterium tuberculosis cell wall synthesis: development of a microtiter plate-based screen for UDP-galactopyranose mutase and identification of an inhibitor from a uridine-based library. Antimicrob Agents Chemother. 2003;47:378–382. doi: 10.1128/AAC.47.1.378-382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caravano A, Dohi H, Sinay P, Vincent SP. A new methodology for the synthesis of fluorinated exo-glycals and their time-dependent inhibition of UDP-galactopyranose mutase. Chem -Eur J. 2006;12:3114–3123. doi: 10.1002/chem.200500991. [DOI] [PubMed] [Google Scholar]
- 25.Itoh K, Huang ZS, Liu HW. Synthesis and analysis of substrate analogues for UDP-Galactopyranose mutase: Implication for an oxocarbenium ion intermediate in the catalytic mechanism. Org Lett. 2007;9:879–882. doi: 10.1021/ol0631408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liautard V, Desvergnes V, Itoh K, Liu HW, Martin OR. Convergent and stereoselective synthesis of iminosugar-containing Galf and UDP-Galf mimicks: Evaluation as inhibitors of UDP-Gal mutase. J Org Chem. 2008;73:3103–3115. doi: 10.1021/jo8001134. [DOI] [PubMed] [Google Scholar]
- 27.Ansiaux C, N’Go I, Vincent SP. Reversible and Efficient Inhibition of UDP-Galactopyranose Mutase by Electrophilic, Constrained and Unsaturated UDP-Galactitol Analogues. Chem - Eur J. 2012;18:14860–14866. doi: 10.1002/chem.201202302. [DOI] [PubMed] [Google Scholar]
- 28.El Bkassiny S, N’Go I, Sevrain CM, Tikad A, Vincent SP. Synthesis of a Novel UDP-carbasugar as UDP-galactopyranose Mutase Inhibitor. Org Lett. 2014;16:2462–2465. doi: 10.1021/ol500848q. [DOI] [PubMed] [Google Scholar]
- 29.Van Straaten KE, Kuttiyatveeti JRA, Sevrain CM, Villaume SA, Jimenez-Barbero J, Linclau B, Vincent SP, Sanders DAR. Structural Basis of Ligand Binding to UDP-Galactopyranose Mutase from Mycobacterium tuberculosis Using Substrate and Tetrafluorinated Substrate Analogues. J Am Chem Soc. 2015;137:1230–1244. doi: 10.1021/ja511204p. [DOI] [PubMed] [Google Scholar]
- 30.Kuppala R, Borrelli S, Slowski K, Sanders DAR, Kartha KPR, Pinto BM. Synthesis and biological evaluation of nonionic substrate mimics of UDP-Galp as candidate inhibitors of UDP galactopyranose mutase (UGM) Bioorg Med Chem Lett. 2015;25:1995–1997. doi: 10.1016/j.bmcl.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 31.Tangallapally RP, Yendapally R, Lee RE, Hevener K, Jones VC, Lenaerts AJM, McNeil MR, Wang YH, Franzblau S, Lee RE. Synthesis and evaluation of nitro-furanylamides as novel antituberculosis agents. J Med Chem. 2004;47:5276–5283. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 32.Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481–483. doi: 10.1038/513481a. [DOI] [PubMed] [Google Scholar]
- 33.Helm JS, Hu Y, Chen L, Gross B, Walker S. Identification of active-site inhibitors of MurG using a generalizable, high-throughput glycosyltransferase screen. J Am Chem Soc. 2003;125:11168–11169. doi: 10.1021/ja036494s. [DOI] [PubMed] [Google Scholar]
- 34.Abagyan R, Totrov M. High-throughput docking for lead generation. Curr Opin Chem Biol. 2001;5:375–382. doi: 10.1016/s1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- 35.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferreira RS, Simeonov A, Jadhav A, Eidam O, Mott BT, Keiser MJ, McKerrow JH, Maloney DJ, Irwin JJ, Shoichet BK. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J Med Chem. 2010;53:4891–4905. doi: 10.1021/jm100488w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 39.Muddana HS, Fenley AT, Mobley DL, Gilson MK. The SAMPL4 host-guest blind prediction challenge: an overview. J Comput-Aided Mol Des. 2014;28:305–317. doi: 10.1007/s10822-014-9735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karunan Partha S, Sadeghi-Khomami A, Cren S, Robinson RI, Woodward S, Slowski K, Berast L, Zheng B, Thomas NR, Sanders DAR. Identification of Novel Inhibitors of UDP-Galactopyranose Mutase by Structure-Based Virtual Screening. Mol Inf. 2011;30:873–883. doi: 10.1002/minf.201100085. [DOI] [PubMed] [Google Scholar]
- 41.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem. 2005;5:739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mysinger MM, Shoichet BK. Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 43.Dhatwalia R, Singh H, Oppenheimer M, Karr DB, Nix JC, Sobrado P, Tanner JJ. Crystal structures and small-angle x-ray scattering analysis of UDP-galactopyranose mutase from the pathogenic fungus Aspergillus fumigatus. J Biol Chem. 2012;287:9041–9051. doi: 10.1074/jbc.M111.327536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dhatwalia R, Singh H, Oppenheimer M, Sobrado P, Tanner JJ. Crystal structures of Trypanosoma cruzi UDP-galactopyranose mutase implicate flexibility of the histidine loop in enzyme activation. Biochemistry. 2012;51:4968–4979. doi: 10.1021/bi300498c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beis K, Srikannathasan V, Liu H, Fullerton SWB, Bamford VA, Sanders DAR, Whitfield C, McNeil MR, Naismith JH. Crystal Structures of Mycobacteria tuberculosis and Klebsiella pneumoniae UDP-Galactopyranose Mutase in the Oxidised State and Klebsiella pneumoniae UDP-Galactopyranose Mutase in the (Active) Reduced State. J Mol Biol. 2005;348:971–982. doi: 10.1016/j.jmb.2005.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gruber TD, Westler WM, Kiessling LL, Forest KT. X-ray crystallography reveals a reduced substrate complex of UDP-galactopyranose mutase poised for covalent catalysis by flavin. Biochemistry. 2009;48:9171–9173. doi: 10.1021/bi901437v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karunan Partha S, van Straaten KE, Sanders DA. Structural basis of substrate binding to UDP-galactopyranose mutase: crystal structures in the reduced and oxidized state complexed with UDP-galactopyranose and UDP. J Mol Biol. 2009;394:864–877. doi: 10.1016/j.jmb.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 48.Poulin MB, Shi Y, Protsko C, Dalrymple SA, Sanders DA, Pinto BM, Lowary TL. Specificity of a UDP-GalNAc pyranose-furanose mutase: a potential therapeutic target for Campylobacter jejuni infections. ChemBioChem. 2014;15:47–56. doi: 10.1002/cbic.201300653. [DOI] [PubMed] [Google Scholar]
- 49.Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012;55:6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teague SJ, Davis AM, Leeson PD, Oprea T. The Design of Leadlike Combinatorial Libraries. Angew Chem, Int Ed. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 51.Chad JM, Sarathy KP, Gruber TD, Addala E, Kiessling LL, Sanders DAR. Site-directed mutagenesis of UDP-Galactopyranose mutase reveals a critical role for the active-site, conserved arginine residues. Biochemistry. 2007;46:6723–6732. doi: 10.1021/bi7002795. [DOI] [PubMed] [Google Scholar]
- 52.Lee R, Monsey D, Weston A, Duncan K, Rithner C, McNeil M. Enzymatic Synthesis of UDP-Galactofuranose and an Assay for UDP-Galactopyranose Mutase Based on High-Performance Liquid Chromatography. Anal Biochem. 1996;242:1–7. doi: 10.1006/abio.1996.0419. [DOI] [PubMed] [Google Scholar]
- 53.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 54.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 55.Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. A high-throughput screen for aggregation-based inhibition in a large compound library. J Med Chem. 2007;50:2385–2390. doi: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- 56.Ferreira RS, Bryant C, Ang KK, McKerrow JH, Shoichet BK, Renslo AR. Divergent modes of enzyme inhibition in a homologous structure-activity series. J Med Chem. 2009;52:5005–5008. doi: 10.1021/jm9009229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beverley SM, Owens KL, Showalter M, Griffith CL, Doering TL, Jones VC, McNeil MR. Eukaryotic UDP-Galactopyranose Mutase (GLF Gene) in Microbial and Metazoal Pathogens. Eukaryotic Cell. 2005;4:1147–1154. doi: 10.1128/EC.4.6.1147-1154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barelier S, Eidam O, Fish I, Hollander J, Figaroa F, Nachane R, Irwin JJ, Shoichet BK, Siegal GD. Increasing chemical space coverage by combining empirical and computational fragment screens. ACS Chem Biol. 2014;9:1528–1535. doi: 10.1021/cb5001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Da Fonseca I, Qureshi IA, Mehra-Chaudhary R, Kizjakina K, Tanner JJ, Sobrado P. Contributions of unique active site residues of eukaryotic UDP-galactopyranose mutases to substrate recognition and active site dynamics. Biochemistry. 2014;53:7794–7804. doi: 10.1021/bi501008z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dhatwalia R, Singh H, Oppenheimer M, Karr DB, Nix JC, Sobrado P, Tanner JJ. Crystal structures and small-angle x-ray scattering analysis of UDP-galactopyranose mutase from the pathogenic fungus Aspergillus fumigatus. J Biol Chem. 2012;287:9041–9051. doi: 10.1074/jbc.M111.327536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reynolds CH, Bembenek SD, Tounge BA. The role of molecular size in ligand efficiency. Bioorg Med Chem Lett. 2007;17:4258–4261. doi: 10.1016/j.bmcl.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 62.Koplin R, Brisson JR, Whitfield C. UDP-galactofuranose Precursor Required for Formation of the Lip-opolysaccharide O Antigen of Klebsiella pneumoniae Serotype O1 Is Synthesized by the Product of the rfbDKPO1 Gene. J Biol Chem. 1997;272:4121–4128. doi: 10.1074/jbc.272.7.4121. [DOI] [PubMed] [Google Scholar]
- 63.Muchmore SW, Debe DA, Metz JT, Brown SP, Martin YC, Hajduk PJ. Application of belief theory to similarity data fusion for use in analog searching and lead hopping. J Chem Inf Model. 2008;48:941–948. doi: 10.1021/ci7004498. [DOI] [PubMed] [Google Scholar]
- 64.Ripphausen P, Nisius B, Bajorath J. State-of-the-art in ligand-based virtual screening. Drug Discovery Today. 2011;16:372–376. doi: 10.1016/j.drudis.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 65.Babaoglu K, Simeonov A, Irwin JJ, Nelson ME, Feng B, Thomas CJ, Cancian L, Costi MP, Maltby DA, Jadhav A, Inglese J, Austin CP, Shoichet BK. Comprehensive mechanistic analysis of hits from high-throughput and docking screens against beta-lactamase. J Med Chem. 2008;51:2502–2511. doi: 10.1021/jm701500e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klebe G. Virtual screening: An alternative or complement to high throughput screening? Preface. Perspect Drug Discovery Des. 2000;20:7–11. [Google Scholar]
- 67.Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 68.Pipeline Pilot, version 6.1. SciTegic Inc; San Diego, CA: [Google Scholar]
- 69.Yung-Chi C, Prusoff W. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.