

Abstract
Specific niches within the lymphoma tumor microenvironment (TME) provide sanctuary for subpopulations of tumor cells through stromal cell–tumor cell interactions. These interactions notably dictate growth, response to therapy and resistance of residual malignant B cells to therapeutic agents. This minimal residual disease (MRD) remains a major challenge in the treatment of B-cell malignancies and contributes to subsequent disease relapse. B-cell receptor (BCR) signaling has emerged as essential mediator of B-cell homing, survival and environment-mediated drug resistance (EMDR). Central to EMDR are chemokine- and integrin-mediated interactions between lymphoma and the TME. Further, stromal cell–B cell adhesion confers a sustained BCR signaling leading to chemokine and integrin activation. Recently, the inhibitors of BCR signaling have garnered a substantial clinical interest because of their effectiveness in B-cell disorders. The efficacy of these agents is, at least in part, attributed to attenuation of BCR-dependent lymphoma–TME interactions. In this review, we discuss the pivotal role of BCR signaling in the integration of intrinsic and extrinsic determinants of TME-mediated lymphoma survival and drug resistance.
Keywords: BCR, microenviroment, lymphoma, drug resistance
Introduction
Despite advances in clinical care, most mature B-cell malignancies remain incurable. This is, in part, because of the complex growth and survival signaling provided by the tumor microenvironment (TME). Within the context of genetic diseases, specific mature B-cell malignancies depend on the microenvironment for survival, proliferation and resistance of residual malignant cells to therapy. This minimal residual disease (MRD) remains the major challenge for long-term in the treatment of B-cell lymphomas and related disorders. In a number of mature B-cell malignancies early transforming events are not ‘sufficient’ to foster environment-independent growth. This is likely the case in chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), diffuse large B-cell lymphoma (DLBCL), mucosa-associated lymphoid tissue (MALT), mantle cell lymphoma (MCL) and multiple myeloma (MM). In these tumor subtypes, interdependence exists between malignant cells and the specific TME. The microenvironment provides support via extrinsic soluble and physical factors including cytokines, chemokines and adhesion to ECM (extracellular matrix) components as well as to heterogeneous populations of fibroblast-like stromal cells (Figure 1).1–5 These factors facilitate B-cell migration and adhesion (homing) in specific niches.6,7 Extrinsic stimuli, in turn, are translated into intrinsic biochemical signaling cascades. Of critical importance, the B-cell receptor (BCR) and its downstream effectors are emerging as central modulators of B-cell homing, survival and drug resistance within the context of B-cell microenvironment.8–12 As such, the BCR may serve as a central hub at the crossroads between extrinsic and intrinsic events.
Figure 1.
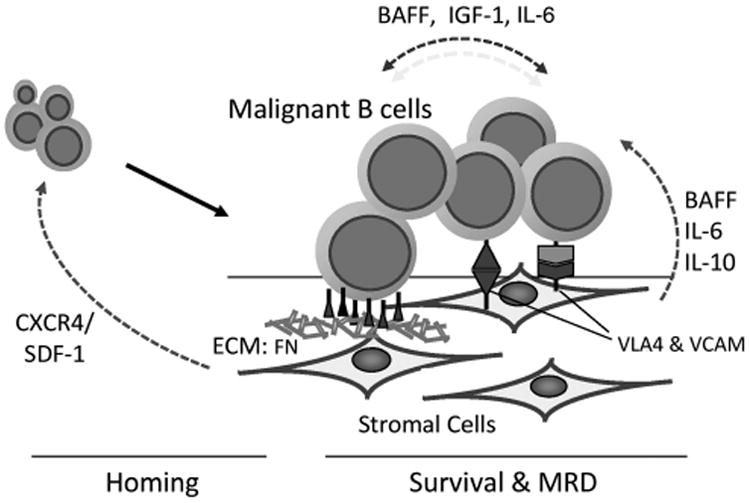
The malignant B cell and the TMEs. Malignant B cells have a dependent relationship with the TME. The TME consists of a host of extracellular matrix (ECM) components and supportive fibroblast-like stromal cells including mesenchymal stromal cells, dendritic cells, tumor-associated macrophages (nurse-like cells), osteoclasts, osteoblasts and endothelial cells among others. These cells contribute to both the soluble and physical dynamics of the TME. The soluble factors CXCR4, BAFF (B-cell activating factor), interleukin (IL)-6 and insulin-like growth factor-1 are produced by the tumor cells and the cellular components of the TME. These can function to recruit tumor cells to the TME where malignant B cells are afforded a survival advantage. Malignant B-cell survival, therapy resistance and MRD is controlled by the dynamic interplay between these chemokine networks and cellular adhesion molecules. Extrinsic stimuli, in turn, are translated to intrinsic biochemical signaling cascades modulating B-cell homing, survival and drug resistance within the context of B-cell microenvironment, with the BCR emerging as a critical component of these signaling networks.
Each B-cell malignancy is the consequence of genetic and epigenetic anomalies occurring at different stages of B-cell lymphopoiesis. Therefore, each malignancy is associated with a distinct, yet inter-related TME. B-cell homing to the bone marrow and/or secondary lymphoid organs as well as niches within these tissues is controlled by the dynamic interplay between chemokine networks and cellular adhesion molecules. In B-cell cancers an expanding body of evidence has revealed a central role BCR signaling events have in coordinating tumor cell–TME interactions.5,8,13 As such, BCR and downstream signaling effectors are emerging as a novel therapeutic targets in B-cell lymphomas and MM. Within this review, we discuss the evidence that the BCR is a central modulator of the extrinsic B-cell microenvironment and intrinsic survival signaling pathways. Specifically, we will discuss the role of BCR in regulating the interplay between ‘outside-in’ and ‘inside-out’ signaling by CXCR4 and very late antigen-4 (VLA-4; α4/β1 integrin heterodimers); thus, the contribution of BCR to TME homing, EMDR (environment-mediated drug resistance), MRD, eventual therapy resistance and relapse.5,6,14
B-Cell Receptor Signaling
The defining characteristic and functional component of B cells is the BCR. The BCR remains essential in normal B-cell development, B-cell survival and the evolution of B-cell malignancies.5,8 The BCR is a transmembrane protein located on the outer surface of B cells. It is a heterodimer composed of heavy-chain and light-chain immunoglobulins (Igs), CD79A/Igα and CD79B/Igβ. Upon ligation by antigen, the immune receptor tyrosine activation motif domains of CD79A and CD79B are phosphorylated by the src-family tyrosine kinase, LYN, and spleen tyrosine kinase, SYK.5,15 BCR phosphorylation facilitates recruitment of additional kinases and adapter proteins including Bruton's tyrosine kinase (BTK), the guanine exchange factor VAV protein, and the adapter proteins GRB2 and BLNK (B-cell linker) forming a large protein multimer or signalome.15 Signalome formation induces BCR-specific signaling via phospholipase C (PLC)-γ2, phosphatidylinositol (PI)-3 kinases and BTK.5,16 In an antigen-dependent or -independent manner, intrinsic signaling from the BCR is mediated by SYK, PLC-γ2, BTK and PI3Ks. PLC-γ2 is phosphorylated by SYK and BTK leading to the production of diacyglyerol and inositol 1,4,5 triphosphate second messengers.15,17 Diacyglyerol facilitates activation of protein kinase C, a key downstream effector of BCR signaling and positive regulator of nuclear factor (NF)-κB signaling.15 Inositol 1,4,5 triphosphate also facilitates specific downstream BCR events including calcium release from the endoplasmic reticulum.5,15 The BCR also signals through PI3K. Upon BCR activation, the p85 subunit is recruited to the plasma membrane and BCR signalome leading to the activation of the p110 delta subunit, phosphorylation of phosphatidylinositol 4,5 bisphosphate, leading to the recruitment of the kinases AKT and BTK. BTK is a member of Tec tyrosine kinase family initially linked to BCR signaling and B-cell development in X-linked agammaglobulinemia.5,15 BTK is activated following phosphorylation by SYK and LYN.17,18 BTK activation mediates downstream signaling via both PI3K/AKT as well as NF-κB signaling via activation of the IκB kinase.15,19 CD79A/CD79B, LYN, SYK, PLCγ2, PI3Ks and BTK comprise the core intrinsic signaling determinants of BCR.
Sustained activation of BCR signaling and its effectors has been demonstrated to be critical in a number of mature B-cell malignancies.15,20 In activated B cell, diffuse large B-cell lymphoma activating CD79A and CD79B immune receptor tyrosine activation motif mutations have been shown to correlate with sensitivity to BTK inhibition.21 However, even these molecular events require sustained stimulation by foreign (or self-) antigens and/or the TME for BCR activation emphasizing the role of the TME in the survival of malignant cells.21 Continuous BCR signaling within the context of the TME also modulates key extrinsic and intrinsic pathways. BCR signaling mediates the inside-out activation of α4/β1 integrins and CXCR4 facilitating B-cell adhesion.9,11 BCR signaling components BTK and PLCγ2 are targets of outside-in activation by the CXCL12/stromal cell-derived factor (SDF)-1 and integrins promoting B-cell migration and adhesion.5,22 These observations suggest a role for the BCR at the crossroads of key components of malignant B-cell homing within the TME. This hypothesis is further supported by the effectiveness in B-cell disorders treated with BCR signaling inhibitors specifically targeting BTK, SYK and PI3Kδ alone and in combination with cytotoxic agents.10,23–28 These anti-BCR signaling agents facilitate transient lymphocytosis and decreased lymph node size attributable to the attenuation of BCR-dependent lymphoma–TME interactions (decreased homing). It is also important to note that the BCR signaling determinants BTK, PI3K, SYK and PLCγ are also targets of outside-in signaling following stimulation of B cells by effectors of the TME (for example, chemokines, cytokines and integrins).5,15,22
TME, Homing (Migration and Adhesion) and BCR Signaling in B-Cell Malignancies
The TME consists of a host of ECM components and supportive fibroblast-like stromal cells including mesenchymal stromal cells, dendritic cells, tumor-associated macrophages (nurse-like cells), osteoclasts, osteoblasts and endothelial cells among others. Niche cells contribute to trafficking and adhesion of malignant cells to/within the TME via the dynamic interplay between chemokine networks and cellular adhesion molecules, with the BCR emerging as a critical component of these signaling networks.
Migration and adhesion of malignant and normal B cells to the bone marrow and secondary lymphoid tissues are modulated by the homeostatic chemokine CXCL12/stromal cell-derived factor-1(SDF-1) and its cognate receptor CXCR4 and subsequent inside-out regulation of integrin expression and function.29,30 Ligation of the membrane-spanning G-protein-associated receptor CXCR4 with CXCL12/SDF-1 modulates intracellular pathways integral to chemotaxis, cell proliferation and cell survival.7,29,30 CXCR4 signals through heterotrimeric G proteins (Gα, Gβ and Gγ) facilitating the activation of (1) PI3K-Akt, (2) Raf/Ras/MEK1/2/ERK1/2, (3) the small GTPases Rac and Rho, and (4) PLC and production of second messengers inositol 1,4,5 triphosphate and diacyglyerol signaling cascades.30,31 One of the key functions of CXCR4 is to modulate homing of B cells to CXCL12/SDF-1-rich regions of TME via two mechanisms: (1) increased expression of integrin subunits and (2) inside-out activation of integrins.7,11,25,31–35 Ngo et al.32 demonstrated that the bone marrow localization of lymphoplasmacytic cells correlated with the increased expression of CXCR4 and VLA-4 relative to peripheral circulating tumor cells, indicating that increased expression of these determinants correlated with homing to the bone marrow. Further, loss of CXCR4 expression correlates with B-cell migration from the bone marrow niche.36 In addition to expression, CXCR4 modulates integrin ligand affinity through inside-out activation of VLA-4 in a manner dependent upon the GTPases RhoA and Rac1.31 Integrins are expressed on lymphocytes as monomers constrained in a ‘bent’ (inactive) confirmation. In response to stimuli, integrins undergo conformational changes to an ‘open’ (active) confirmation and undergo membrane clustering.11,34,37,38 In the active confirmation, integrins are competent for high-affinity adhesion. The converse can also occur, integrins trigger signals for both increased CXCR4 expression and function.39 To this end, the relationship between CXCR4 and VLA-4 is complex constituting bidirectional outside-in and inside-out signaling systems. Accumulating evidence indicates that the BCR is integral in this bidirectional signaling between chemokine receptors and integrins contributing to lymphoma migration and cell adhesion in B-cell malignancies, such as CLL, MCL and MM.5,6,32
The BCR represents a central modulator of the interplay between the intrinsic and extrinsic CXCR4/CXCL12-mediated migration and VLA-4-mediated adhesion (Figure 2).30,34 Quiroga et al.40 demonstrated that BCR activation enhances CLL cell homing in tissue microenvironments. Further, in CLL cells the tyrosine kinase inhibitor dasatinib has been shown to decrease CXCL12-induced migration in a manner similar to direct BCR inhibitors.41 Specific targeting of SYK with the inhibitor R406 attenuated CXCL12/SDF-1-mediated malignant B-cell homing signals and AKT phosphorylation.17 Inhibition of PI3Kδ with idelalisib/GS1101 inhibited the expression of specific chemokines and cytokines amplified in tumor nurse-like cell coculture models.29 The BCR has also been shown to induce VLA-4 high-affinity binding via inside-out signaling.5,11,40 Spaargaren et al.11 demonstrated that BCR signaling-induced high-affinity binding state of VLA-4 was dependent on the sequential activation of the components of the BCR effectors LYN, SYK, PI3Kδ, BTK and PLCγ2-mediated release of calcium. These data underscore the importance of BCR signaling in the upstream modulation of interactions between lymphocytes and the microenvironment.
Figure 2.
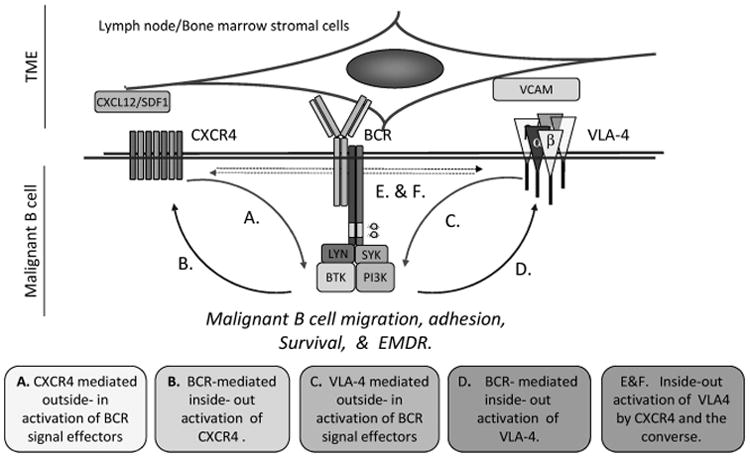
The B-cell receptor (BCR) is a central mediator of malignant B-cell homing, survival and EMDR. B-cell homing to the bone marrow and/or secondary lymphoid organs as well as niches within these tissues is controlled by the dynamic interplay between chemokine networks and cellular adhesion molecule. The BCR regulates the interplay between ‘outside-in’ and ‘inside-out’ signaling by CXCR4 and VLA-4 (α4/β1 integrin heterodimers). As such, the BCR is positioned as a central modulator of homing, environment-mediated drug resistance (EMDR), MRD, eventual therapy resistance and relapse within the context of the TME. Continuous BCR signaling within the context of the TME modulates key extrinsic and intrinsic pathways. BCR signaling mediates the inside-out activation of α4/β1 integrins facilitating B-cell adhesion. BCR signaling components Bruton's tyrosine kinase (BTK) and phospholipase C (PLC)-γ2 are targets of outside-in activation by the CXCL12/SDF-1 and integrins promoting B-cell migration and adhesion. These observations suggest a role for the BCR at the crossroads of key components of malignant B-cell homing within the TME. This hypothesis is further supported by the striking responses in B-cell disorders treated with BCR signaling inhibitors specifically targeting BTK, SYK and PI3Kδ alone and in combination with cytotoxic agents. It is apparent that the TME also influences the BCR signaling.
Importantly, it is becoming apparent that the TME also influences the BCR signaling. Carrasco and Batista42 showed B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with vascular cell adhesion molecule-1. Vascular cell adhesion molecule-1, the major ligand for VLA-4, is highly expressed on the surface of follicular dendritic cells; as such, VLA-4 acts not only as an initial tether for the B cells allowing antigen recognition by the BCR, but also in signaling synergistically with the BCR to promote tight adhesion and to enhance B-cell activation and survival. In addition, the chemokine CXCL12/SDF-1 also induces outside-in activation of BCR signaling effectors BTK, PLC-γ, SYK and PI3K.22,43,44 Taken together, these results suggest that the BCR is also a downstream target of the coordinated action of chemokines and integrins. These data further support the pivotal role of BCR signaling in the integration of intrinsic and extrinsic determinants of TME.
TME-Mediated Survival, EMDR and BCR Signaling in B-Cell Malignancies
Microenvironment-associated survival is modulated by a host of both soluble factors (cytokines, chemokines and growth factors) and physical factors (ECM components and juxtaposed cellular components). Given the central role of the BCR in B-cell homeostasis and TME interactions, it is not surprising that BCR signaling is also critical for EMDR.
Soluble factor(s)-mediated drug resistance
There are a number of soluble factors associated with malignant B-cell survival, including interleukin (IL)-6, B-cell activating factor (BAFF), IL-10 and CXCL12/SDF-1 to list a few. Within this section, we will discuss evidence that BAFF and IL-6 are critical examples of soluble TME determinants. BAFF targets B lymphocytes, promoting proliferation, activation, differentiation, survival and stimulation of Ig production both in vitro and in vivo.45 The role of BAFF in B-cell homeostasis became evident from studies in BAFF-null and BAFF-transgenic mice.46–49 Mice lacking BAFF have reduced circulating mature B cells; in contrast, BAFF-overexpressing mice have increased numbers of mature splenic and lymph node B cells, elevated Ig levels and manifestations of autoimmune disease.47,50 BAFF also affords malignant B cells a resistance to therapeutics, as demonstrated by reports in CLL and MM. Kern et al.51 demonstrated that addition of soluble BAFF protected B-CLL cells against spontaneous and drug-induced apoptosis; conversely, addition of soluble anti-BAFF antibodies enhanced CLL cell apoptosis. In MM, Moreaux et al.52 demonstrated that BAFF modulated the survival of primary MM cells and attenuated dexamethasone-induced apoptosis. Our group also demonstrated that B-cell lymphoma adhesion-induced BAFF expression by bone marrow stromal cells (BMSCs) stimulated a NF-κB survival pathway and subsequently protected various lymphoma cells from apoptosis.1 This observation was further supported by a recent report revealing that mesenchymal stromal cells protect MCL cells from spontaneous and drug-induced apoptosis and long-term expansion through secretion of BAFF and activation of the canonical and non-canonical NF-κB pathways.53 Taken together, B cell and stromal cell interactions create sanctuary niches for lymphomas with high BAFF availability. Importantly, the BCR was recently demonstrated to be critical for BAFF-dependent B-cell survival.54 BAFF-induced SYK and PI3K-mediated B-cell survival required intact BCR and CD79A indicating that sustained BCR signaling was required for soluble factor-dependent B-cell survival.
IL-6 remains the prototypical growth and survival factor in myeloma as well as B-cell lymphomas.55,56 IL-6 binds its receptors on lymphoma cells to induce drug resistance.57,58 IL-6 binding and receptor multimerization facilitates the phosphorylation of gp130 and initiation of three major signaling pathways in MM cells: (1) the Ras/Raf–mitogen-activated protein kinase kinase–ERK (extracellular signal-related kinase) 1/2 pathway; (2) the PI3K pathway; and (3) the JAK (Janus kinase)/STAT3 (signal transducer and activator of transcription) pathway. Each of these signal transduction pathways has been implicated in IL-6-mediated resistance to both physiological and chemotherapy-mediated apoptosis.59,60 IL-6 was found to contribute to this effect by increasing the expression of the anti-apoptotic protein FLIP. Earlier work by Catlett-Falcone et al.60 demonstrated that IL-6-induced STAT3 signaling protects myeloma cells from FAS-mediated apoptosis by upregulating transcription of the anti-apoptotic molecule BCL-2-like protein 1 (BCL-XL). Zhang et al.19 demonstrated that autocrine and/or paracrine IL-6 is involved in MCL growth or survival, and clearly showed that IL-6 is an important cytokine involved in MCL growth, survival and EMDR. Importantly, Baran-Marszak et al.61 demonstrated that the BCR activity was critical for IL-6-specific STAT3-dependent MCL survival. Taken together, these studies indicate that ‘optimal’ BAFF and IL-6-induced B-cell survival may be dependent on intact and sustained BCR activity.
Cell adhesion-mediated drug resistance
Cell adhesion-mediated drug resistance is defined by the drug resistance modulated by adhesion of tumor cells to the extracellular matrix protein fibronectin via β1 integrins or adjacent cells.62 Studies have suggested that the binding of the b1 integrins, VLA-4 and VLA-5 may be involved in protecting malignant B cells from apoptosis.63 The binding of CLL patient isolates to fibronectin resulted in an increase of the anti-apoptotic protein Bcl-2 and an increase in the cellular ratio of Bcl-2 to Bax, compared with cells grown in suspension.64 Pedersen et al.65 showed that stromal cell-induced survival of CLL cells was, in part, through the upregulation of Mcl-1. Lwin et al.66 demonstrated that direct contact with the human BMSC line 5 (HS-5) afforded protection from spontaneous and drug-induced apoptosis in various lymphomas including large cell lymphoma and MCL. In MM, cell adhesion-mediated drug resistance-DR was linked to activation of NF-κB signaling, specifically RELB/p50.67 Stroma-induced activation of NF-κB and the upregulation of NF-κB-regulated anti-apoptotic proteins XIAP and cIAP contribute to EMDR. Interactions with the bone marrow microenvironment have also been shown to affect physiological mediators of cell death such as CD95 (FAS).68,69 These effects were mediated by the regulation of the anti-apoptotic protein c-FLIPL.68,69 In addition, G1 cell cycle arrest induced by β1 integrin adhesion of myeloma cells is associated with drug resistance via the post-translational upregulation of p27 and inactivation of cyclin-associated kinase activity.70 Subsequently, Lwin et al.71 showed that adhesion-mediated upregulation of p27 was mediated by proteasomal degradation of S-phase kinase-associated protein 2, a negative regulator of p27 protein stability.
Soluble and physical determinants of the bone marrow microenvironment both confer the EMDR phenotype in vitro. However, logic dictates that these soluble and physical environmental effectors function in concert in vivo. This network involves autocrine/paracrine signaling as well as regulatory events mediated by adhesion of lymphoma cells to stromal cells. Tumor-stimulated production of soluble factors by the stroma increases EMDR not only by directly upregulating anti-apoptotic molecules but also by inducing increased integrin expression and/or affinity for their ligands on tumor cells. Soluble factors not only increase integrin affinity by inside-out signaling, but integrin-mediated adhesion also increases the activation of cytokine signaling pathways. Shain et al.2 demonstrated that adhesion of myeloma cell lines to immobilized fibronectin through β1 integrin amplifies IL-6-induced STAT3 signaling, demonstrating that soluble and physical factors cooperate in conferring EMDR. Together, these results suggest that examination of crosstalk between intra-cellular signaling networks may identify specific components and contribute more (or less) significantly to malignant B-cell therapy resistance and proliferation. To this end, identification of prominent signaling molecules under co-stimulatory conditions may direct us toward more appropriate drug targets. Emerging evidence indicates that the BCR represents a critical point of integration between the extrinsic effectors of the TME and intrinsic survival signaling pathways. More specifically, the BCR contributes to the interplay between outside-in and inside-out signaling regulation of CXCR4, integrins and subsequent EMDR.5,6
BCR signaling and EMDR
Several lines of evidence demonstrated that lymphoma TME activates BCR signaling and confers a sustained BCR signal in lymphoma microenvironment. Although BCR signaling is generally thought to depend on ligand-induced aggregation, additional studies highlight the important role of continuous BCR maintenance or survival signals in the absence of receptor engagement. Lam et al.20 first demonstrated that the inducible loss of murine BCR resulted in the death of peripheral B cells, highlighting the requirement for continued BCR expression for B-cell survival.72 Further, the selective excision of the Igα immune receptor tyrosine activation motif and ablation of Igα signaling led to the loss of mature B cells, further emphasizing the role of chronic BCR signaling in B-cell survival and maturation.15 Gene expression profiles from CLL cells comparing peripheral blood versus lymph node-resident tumors cells demonstrated a significant upregulation of BCR pathway genes in the lymph node microenvironment relative to circulating cells involving SYK phosphorylation and NF-κB and NFAT signaling.73 Coculture experiments with stromal cells have also shown activation of SYK and AKT in lymphoma cells.54 These data indicate that sustained BCR signaling confers survival and proliferation signals in the bone marrow and lymph node microenvironment niches. Further evidence that supports the importance of BCR signaling pathway in microenvironment-induced survival signals comes from the in vitro effects of BCR kinase inhibitors. Buchner et al.17 demonstrated that the SYK inhibitor R406 blocked stroma-mediated lymphoma survival, reduced homing and CXCL12-induced AKT activation. Similarly, Hoellenriegel et al.27 revealed that the PI3Kδ inhibitor idelalisib/GS1101 inhibits CLL cell viability in coculture with nurse-like cells and downregulates chemokines and cytokines amplified in this coculture model. Similarly, idelalisib/GS1101 attenuated myeloma cell growth and EMDR conferred by IL-6, insulin-like growth factor-1 and BMSC coculture.74 Further, idelalisib/GS1101 demonstrated synergistic activity in combination with bortezomib in myeloma cells. The BTK inhibitor ibrutinib/PCI32765 also antagonizes microenvironment signals.10,15,23,44,75 Herman et al.23 demonstrated that ibrutinib/PCI32765 blocked BAFF, IL-6 and IL-10 expression as well as CLL cell adhesion to FN (fibronectin) and stromal cells. In myeloma, ibrutinib/PCI32765 was most potent against stromal-dependent myeloma cells in coculture with patient-derived BMSCs or osteoclasts, suggesting that ibrutinib-mediated cytotoxicity was the result of targeting the bone marrow microenvironment and potentially BCR signaling.10 Ibrutinib strongly reduced secretion of multiple cytokines and chemokines in myeloma cocultures with BMSCs.10 The authors further demonstrated that attenuation of BTK signaling decreased myeloma cell growth in in vivo models of the TME. Rushworth et al.75 demonstrated that ibrutinib/PCI32765 and the BTK inhibitor LFM-A13 enhanced bortezomib and lenalidomide cytotoxicity in myeloma. Collectively, these data indicated that the BCR and its signaling effectors are critical to orchestrating malignant B-cell survival and EMDR.
Concluding Remarks: Implication to target BCR Signaling for Lymphoma Treatment
By elucidating the role of the TME in the pathogenesis of B-cell malignancies, recent studies have provided the framework for identifying and validating novel therapies that target both lymphoma cells and the TME. The studies reviewed here support that both the extrinsic and intrinsic determinants have a central role in the survival, drug resistance and progression of B-cell disorders. The extrinsic signals are generated by the lymphoma microenvironment and include chemokine receptors (CXCR4) and adhesion molecules (VLA-4). The intrinsic factors encompass biochemical signaling determinants of cell cycle and prosurvival pathways. To this end, targeting the malignant TME and overcoming MRD and EMDR can be implemented through several strategies. Optimally, this strategy would target a critical regulatory component of the dynamic relationship between malignancy and TME. The data discussed in this review indicate that the BCR is a central hub for the integration between the extrinsic B-cell microenvironment and the intrinsic signaling pathways. More specifically, the BCR orchestrates the interplay between outside-in and inside-out by CXCR4, integrins and other key effectors of the TME, thereby having a critical role in malignant B-cell homing, survival and EMDR. Therefore, targeting the BCR pathway molecules will attenuate growth and survival signals emanating from both B-cell intrinsic abnormalities and from the TME, serving as a novel ‘double-hit’ strategy: targeting both BCR-regulated survival signaling and BCR-regulated lymphoma–TME interactions releasing lymphoma cells from their microenvironment, resulting in sensitization and enhanced cytotoxic killing. This hypothesis has been substantiated by recent clinical trials of BCR inhibitors in B-cell lymphoma patients with encouraging results (Figure 3). Recently, early-stage clinical trials with the SYK inhibitor FosD,76 the BTK inhibitor ibrutinib77 and the PI3Kδ inhibitor GS1101/idelalisib78 revealed that patients with CLL and some B-cell lymphomas are particularly sensitive to inhibitors of BCR-associated kinases. Clinical responses are characterized by an early redistribution of tissue-resident CLL cells into the blood, resulting in rapid resolution of lymphadenopathy and organomegaly, along with a transient surge in lymphocytosis during the first weeks of therapy consistent with the attenuated B-cell migration and adhesion to the TME.77,79 Subsequently, the antigrowth and antisurvival activities of these agents become more apparent and resulted in the normalization of lymphocyte counts and remissions in a majority of patients consistent with attenuated EMDR. The encouraging clinical and preclinical results obtained with FosD, PCI32765 and CAL-101/GS1101 support the idea that therapeutic targeting of BCR signaling pathways is an effective strategy for treatment of CLL and other B-cell malignancies.
Figure 3.
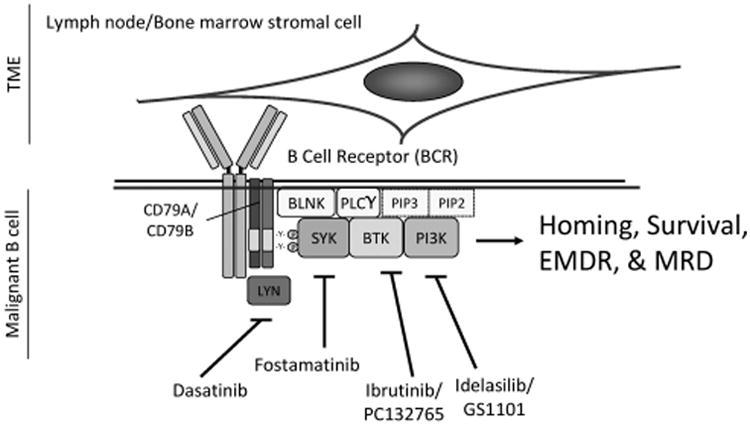
Targeting BCR signaling attenuates homing, survival, EMDR and MRD of malignant B cells. The BCR is a transmembrane protein located on the outer surface of B cells. It is a heterodimer composed of heavy-chain and light-chain Igs, CD79A and CD79B. Upon ligation by antigen, the immune receptor tyrosine activation motif domains of CD79A and CD79B are phosphorylated by the src-family tyrosine kinase, LYN, and spleen tyrosine kinase, SYK. BCR phosphorylation facilitates recruitment of additional kinases and adapter proteins including Bruton's tyrosine kinase (BTK), B-cell linker (BLNK) and other adapter proteins forming a large protein multimer or signalome. CD79A/CD79B, LYN, SYK, PLCγ2, PI3Ks and BTK comprise the core intrinsic signaling determinants of BCR. The data discussed in this review indicate that the BCR is a central hub for the integration between the extrinsic B-cell microenvironment and the intrinsic signaling pathways, thereby playing a central role in malignant B-cell homing, survival and EMDR. To this end, targeting the BCR pathway will attenuate growth and survival signals emanating from both B-cell intrinsic abnormalities and from the TME. BCR-targeting strategies using SYK inhibitor fosamatinib, the BTK inhibitor ibrutinib and the PI3Kδ inhibitor GS1101/idelalisib revealed that patients with malignant B-cell disorders are particularly sensitive to inhibitors of BCR-associated kinases.
However, these exciting results represent only an initial step to optimal clinical success with BCR-targeting agents. It will be important to consider the merit of targeting other molecules within the BCR signal pathways in the setting of therapy resistance. Targeting downstream BCR signaling pathways hold advantage of direct modulation of the cell survival and proliferation machinery; however, clinical evidence suggests that the effect of BCR inhibitors (as single agents) in aggressive lymphomas is modest in terms of duration of clinical efficacy, although encouraging response rates relapse remains inevitable.76,77,79 These clinical failures are the consequence of therapy resistance and the evolution/selection of compensatory survival pathways. As discussed, the complexity of the survival networks induced by the TME mitigates the prolonged success of single-agent therapy. Therefore, dual targeting of parallel survival pathways or sequentially activated effectors of a single pathway may be necessary. From this perspective, the continued success of BCR inhibitor therapy in lymphoma will require the rational design of combination of targeted agents, a deep understanding of the nature of signaling pathways and their interactions with TME.
Finally, the optimal therapeutic setting (or sequencing) of BCR inhibitors also needs to be assessed. The bane of cancer remains MRD. MRD represents important limitations in our technology to detect ‘small’ populations of disease as well as limitations in our therapeutic armamentarium. This residual population of cells represents cells that have evaded therapy. Subsequent expansion of these residual cells correlates with relapse frequently, with a MDR phenotype making salvage therapy less successful. Although the precise biological rationale for MRD is complex, it has been proposed that the TME and EMDR are important determinants contributing to MRD and eventual acquisition of multidrug resistance. Targeting BCR, a central orchestrator of the TME, will foster greater success in the eradication of B-cell tumors via a reduction in MRD. Currently, only rituximab is utilized as a maintenance agent in non-hodgkins lymphoma (NHL). With the development of well-tolerated oral agents such as the BCR inhibitors, their use in a maintenance setting is very attractive. Further rationale for the use of BCR inhibitors in B-cell malignancies is supported by the success of maintenance therapy in other myeloma with lenalidomide, with significant improvements in long-term disease control.80,81 As clinical use of BCR inhibitors continues to evolve, we must (1) define optimal combination strategies with current chemotherapeutics, (2) identify mechanisms of resistance (compensatory pathways) for the development of success design of multiagent-targeted therapies and (3) determine the optimal utilization of BCR inhibitors in therapy (primary therapy, maintenance and/or salvage). To determine these issues, continued bench-to-bedside (and bedside-to-bench) research will be needed to continue to define the role of BCR in B-cell malignancies and their interactions with the TME.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Lwin T, Crespo LA, Wu A, Dessureault S, Shu HB, Moscinski LC, et al. Lymphoma cell adhesion-induced expression of B cell-activating factor of the TNF family in bone marrow stromal cells protects non-Hodgkin's B lymphoma cells from apoptosis. Leukemia. 2009;23:170–177. doi: 10.1038/leu.2008.266. [DOI] [PubMed] [Google Scholar]
- 2.Shain KH, Yarde DN, Meads MB, Huang M, Jove R, Hazlehurst LA, et al. Beta1 integrin adhesion enhances IL-6-mediated STAT3 signaling in myeloma cells: implications for microenvironment influence on tumor survival and proliferation. Cancer Res. 2009;69:1009–1015. doi: 10.1158/0008-5472.CAN-08-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med. 2010;16:483–489. doi: 10.1038/nm.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall BM, Gibson LF. Regulation of lymphoid and myeloid leukemic cell survival: role of stromal cell adhesion molecules. Leukemia Lymphoma. 2004;45:35–48. doi: 10.1080/1042819031000139620. [DOI] [PubMed] [Google Scholar]
- 5.Woyach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120:1175–1184. doi: 10.1182/blood-2012-02-362624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger JA. Targeting the microenvironment in chronic lymphocytic leukemia is changing the therapeutic landscape. Curr Opin Oncol. 2012;24:643–649. doi: 10.1097/CCO.0b013e3283589950. [DOI] [PubMed] [Google Scholar]
- 7.Azab AK, Runnels JM, Pitsillides C, Moreau AS, Azab F, Leleu X, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113:4341–4351. doi: 10.1182/blood-2008-10-186668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. 2009;114:3367–3375. doi: 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byrd JC. Therapeutic targeting of B-cell receptor signaling pathways. Blood. 2012;120:21. doi: 10.1182/blood-2012-02-362624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tai YT, Chang BY, Kong SY, Fulciniti M, Yang G, Calle Y, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120:1877–1887. doi: 10.1182/blood-2011-12-396853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, et al. The B cell antigen receptor controls integrin activity through Btk and PLC gamma 2. J Exper Med. 2003;198:1539–1550. doi: 10.1084/jem.20011866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herreros B, Sanchez-Aguilera A, Piris MA. Lymphoma microenvironment: culprit or innocent? Leukemia. 2008;22:49–58. doi: 10.1038/sj.leu.2404970. [DOI] [PubMed] [Google Scholar]
- 13.Staudt LM. Chronic active B-cell receptor signaling in lymphoma. ASH Annual Meeting Abstracts. 2012;120(21):SCI-26. [Google Scholar]
- 14.Hall BM, Fortney JE, Taylor L, Wood H, Wang L, Adams S, et al. Stromal cells expressing elevated VCAM-1 enhance survival of B lineage tumor cells. Cancer Lett. 2004;207:229–239. doi: 10.1016/j.canlet.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 15.Young RM, Staudt LM. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov. 2013;12:229–243. doi: 10.1038/nrd3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng PC, Dykstra ML, Mitchell RN, Pierce SK. A role for lipid rafts in B cell antigen receptor signaling and antigen targeting. J Exper Med. 1999;190:1549–1560. doi: 10.1084/jem.190.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholome K, Burger M, et al. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69:5424–5432. doi: 10.1158/0008-5472.CAN-08-4252. [DOI] [PubMed] [Google Scholar]
- 18.Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, et al. BTK regulates PtdIns-4,5-P2 synthesis: importance for calcium signaling and PI3K activity. Immunity. 2003;19:669–678. doi: 10.1016/s1074-7613(03)00297-8. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Yang J, Qian J, Li H, Romaguera JE, Kwak LW, et al. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood. 2012;120:3783–3792. doi: 10.1182/blood-2012-04-424630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam KP, Rajewsky K. Rapid elimination of mature autoreactive B cells demonstrated by Cre-induced change in B cell antigen receptor specificity in vivo. Proc Natl Acad Sci USA. 1998;95:13171–13175. doi: 10.1073/pnas.95.22.13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–U97. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Gorter DJJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton's tyrosine kinase and phospholipase C gamma 2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26:93–104. doi: 10.1016/j.immuni.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 23.Herman SEM, Farooqui M, Bezabhie R, Aue G, Wiestner A. In vivo effects of ibrutinib on BCR signaling, tumor cell activation and proliferation in blood and tissue-resident cells of chronic lymphocytic leukemia patients. ASH Annual Meeting Abstracts. 2012;120:185. [Google Scholar]
- 24.Hoellenriegel J, Coffey GP, Sinha U, Pandey A, Sivina M, Ferrajoli A, et al. Selective, novel spleen tyrosine kinase (Syk) inhibitors suppress chronic lymphocytic leukemia B-cell activation and migration. Leukemia. 2012;26:1576–1583. doi: 10.1038/leu.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114:4441–4450. doi: 10.1182/blood-2009-07-233718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoellenriegel J, O'Brien S, Keating MJ, Wierda WG, Buggy JJ, Burger JA. In vivo inhibition of BCR activation in high-risk CLL patients on therapy with Bruton's tyrosine kinase inhibitor ibrutinib: correlative studies from an ongoing phase 2 clinical trial. ASH Annual Meeting Abstracts. 2012;120:186. [Google Scholar]
- 27.Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118:3603–3612. doi: 10.1182/blood-2011-05-352492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vij R, Chang BY, Berdeja JG, Huff CA, Lendvai N, Tai YT, et al. Early changes in cytokines, chemokines and indices of bone metabolism in a phase 2 study of the bruton tyrosine kinase (Btk) inhibitor, ibrutinib (PCI-32765) in patients with relapsed or relapsed/refractory multiple myeloma (MM) Blood. 2012;120:21. [Google Scholar]
- 29.Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]
- 30.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 31.Azab AK, Azab F, Blotta S, Pitsillides CM, Thompson B, Runnels JM, et al. Rho-A and Rac-1 GTPases play major and differential roles in SDF1-induced cell adhesion and chemotaxis in multiple myeloma. Blood. 2009;114:619–629. doi: 10.1182/blood-2009-01-199281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ngo HT, Leleu X, Lee J, Jia X, Melhem M, Runnels J, et al. SDF-1/CXCR4 and VLA-4 interaction regulates homing in Waldenstrom macroglobulinemia. Blood. 2008;112:150–158. doi: 10.1182/blood-2007-12-129395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanz-Rodriguez F, Hidalgo A, Teixido J. Chemokine stromal cell-derived factor-1alpha modulates VLA-4 integrin-mediated multiple myeloma cell adhesion to CS-1/fibronectin and VCAM-1. Blood. 2001;97:346–351. doi: 10.1182/blood.v97.2.346. [DOI] [PubMed] [Google Scholar]
- 34.Arana E, Harwood NE, Batista FD. Regulation of integrin activation through the B-cell receptor. J Cell Sci. 2008;121(Pt 14):2279–2286. doi: 10.1242/jcs.017905. [DOI] [PubMed] [Google Scholar]
- 35.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 36.Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of endothelial to mesenchymal transition-like features. Blood. 2012;119:5782–5794. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–11. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 38.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grzesiak JJ, Smith KC, Burton DW, Deftos LJ, Bouvet M. Integrin-mediated laminin-1 adhesion upregulates CXCR4 and IL-8 expression in pancreatic cancer cells. Surgery. 2007;141:804–814. doi: 10.1016/j.surg.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quiroga MP, Balakrishnan K, Kurtova AV, Sivina M, Keating MJ, Wierda WG, et al. B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood. 2009;114:1029–1037. doi: 10.1182/blood-2009-03-212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCaig AM, Cosimo E, Leach MT, Michie AM. Dasatinib inhibits CXCR4 signaling in chronic lymphocytic leukaemia cells and impairs migration towards CXCL12. PLoS One. 2012;7:e48929. doi: 10.1371/journal.pone.0048929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carrasco YR, Batista FD. B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with VCAM-1. EMBO J. 2006;25:889–899. doi: 10.1038/sj.emboj.7600944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niedermeier M, Hennessy BT, Knight ZA, Henneberg M, Hu J, Kurtova AV, et al. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009;113:5549–5557. doi: 10.1182/blood-2008-06-165068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Rooij MFM, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590–2594. doi: 10.1182/blood-2011-11-390989. [DOI] [PubMed] [Google Scholar]
- 45.Do RK, Hatada E, Lee H, Tourigny MR, Hilbert D, Chen-Kiang S. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exper Med. 2000;192:953–964. doi: 10.1084/jem.192.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bossen C, Cachero TG, Tardivel A, Ingold K, Willen L, Dobles M, et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood. 2008;111:1004–1012. doi: 10.1182/blood-2007-09-110874. [DOI] [PubMed] [Google Scholar]
- 47.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 48.Khare SD, Sarosi I, Xia XZ, McCabe S, Miner K, Solovyev I, et al. Severe B cell hyperplasia and autoimmune disease in TALL-1 transgenic mice. Proc Natl Acad Sci USA. 2000;97:3370–3375. doi: 10.1073/pnas.050580697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exper Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vora KA, Wang LC, Rao SP, Liu ZY, Majeau GR, Cutler AH, et al. Cutting edge: germinal centers formed in the absence of B cell-activating factor belonging to the TNF family exhibit impaired maturation and function. J Immunol. 2003;171:547–551. doi: 10.4049/jimmunol.171.2.547. [DOI] [PubMed] [Google Scholar]
- 51.Kern C, Cornuel JF, Billard C, Tang R, Rouillard D, Stenou V, et al. Involvement of BAFF and APRIL in the resistance to apoptosis of B-CLL through an autocrine pathway. Blood. 2004;103:679–688. doi: 10.1182/blood-2003-02-0540. [DOI] [PubMed] [Google Scholar]
- 52.Moreaux J, Legouffe E, Jourdan E, Quittet P, Reme T, Lugagne C, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103:3148–3157. doi: 10.1182/blood-2003-06-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medina DJ, Goodell L, Glod J, Gelinas C, Rabson AB, Strair RK. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor kappaB pathways. Haematologica. 2012;97:1255–1263. doi: 10.3324/haematol.2011.040659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, et al. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity. 2013;38:475–488. doi: 10.1016/j.immuni.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lichtenstein A, Tu Y, Fady C, Vescio R, Berenson J. Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell Immunol. 1995;162:248–255. doi: 10.1006/cimm.1995.1076. [DOI] [PubMed] [Google Scholar]
- 56.Lu Y, Zhang J, Dai J, Dehne LA, Mizokami A, Yao Z, et al. Osteoblasts induce prostate cancer proliferation and PSA expression through interleukin-6-mediated activation of the androgen receptor. Clin Exper Metastasis. 2004;21:399–408. doi: 10.1007/s10585-005-0056-6. [DOI] [PubMed] [Google Scholar]
- 57.Gilbert LA, Hemann MT. Context-specific roles for paracrine IL-6 in lymphomagenesis. Genes Dev. 2012;26:1758–1768. doi: 10.1101/gad.197590.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–366. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thiel S, Sommer U, Kortylewski M, Haan C, Behrmann I, Heinrich PC, et al. Termination of IL-6-induced STAT activation is independent of receptor internalization but requires de novo protein synthesis. FEBS Lett. 2000;470:15–19. doi: 10.1016/s0014-5793(00)01276-x. [DOI] [PubMed] [Google Scholar]
- 60.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 61.Baran-Marszak F, Boukhiar M, Harel S, Laguillier C, Roger C, Gressin R, et al. Constitutive and B-cell receptor-induced activation of STAT3 are important signaling pathways targeted by bortezomib in leukemic mantle cell lymphoma. Haematologica. 2010;95:1865–1872. doi: 10.3324/haematol.2009.019745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 63.Shain KH, Dalton WS. Environmental-mediated drug resistance: a target for multiple myeloma therapy. Expert Rev Hematol. 2009;2:649–662. doi: 10.1586/ehm.09.55. [DOI] [PubMed] [Google Scholar]
- 64.de la Fuente MT, Casanova B, Garcia-Gila M, Silva A, Garcia-Pardo A. Fibronectin interaction with alpha4beta1 integrin prevents apoptosis in B cell chronic lymphocytic leukemia: correlation with Bcl-2 and Bax. Leukemia. 1999;13:266–274. doi: 10.1038/sj.leu.2401275. [DOI] [PubMed] [Google Scholar]
- 65.Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100:1795–1801. [PubMed] [Google Scholar]
- 66.Lwin T, Hazlehurst LA, Li Z, Dessureault S, Sotomayor E, Moscinski LC, et al. Bone marrow stromal cells prevent apoptosis of lymphoma cells by upregulation of anti-apoptotic proteins associated with activation of NF-kappaB (RelB/p52) in non-Hodgkin's lymphoma cells. Leukemia. 2007;21:1521–1531. doi: 10.1038/sj.leu.2404723. [DOI] [PubMed] [Google Scholar]
- 67.Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–2421. doi: 10.1038/sj.onc.1206315. [DOI] [PubMed] [Google Scholar]
- 68.Shain KH, Dalton WS. Cell adhesion is a key determinant in de novo multidrug resistance (MDR): new targets for the prevention of acquired MDR. Mol Cancer Ther. 2001;1:69–78. [PubMed] [Google Scholar]
- 69.Perez LE, Parquet N, Shain K, Nimmanapalli R, Alsina M, Anasetti C, et al. Bone marrow stroma confers resistance to Apo2 ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J Immunol. 2008;180:1545–1555. doi: 10.4049/jimmunol.180.3.1545. [DOI] [PubMed] [Google Scholar]
- 70.Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR) Oncogene. 2000;19:4319–4327. doi: 10.1038/sj.onc.1203782. [DOI] [PubMed] [Google Scholar]
- 71.Lwin T, Hazlehurst LA, Dessureault S, Lai R, Bai W, Sotomayor E, et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood. 2007;110:1631–1638. doi: 10.1182/blood-2006-11-060350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hallek M, Neumann C, Schaffer M, Danhauser-Riedl S, von Bubnoff N, de Vos G, et al. Signal transduction of interleukin-6 involves tyrosine phosphorylation of multiple cytosolic proteins and activation of Src-family kinases Fyn, Hck, and Lyn in multiple myeloma cell lines. Exper Hematol. 1997;25:1367–1377. [PubMed] [Google Scholar]
- 73.de Gorter DJJ, Vos JCM, Pals ST, Spaargaren M. The B cell antigen receptor controls AP-1 and NFAT activity through Ras-mediated activation of Ral. J Immunol. 2007;178:1405–1414. doi: 10.4049/jimmunol.178.3.1405. [DOI] [PubMed] [Google Scholar]
- 74.Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood. 2010;116:1460–1468. doi: 10.1182/blood-2009-06-222943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rushworth SA, Bowles KM, Barrera LN, Murray MY, Zaitseva L, MacEwan DJ. BTK inhibitor ibrutinib is cytotoxic to myeloma and potently enhances bortezomib and lenalidomide activities through NF-kappaB. Cell Signal. 2013;25:106–112. doi: 10.1016/j.cellsig.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 76.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115:2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stein R, Gupta P, Chen X, Cardillo TM, Furman RR, Chen S, et al. Therapy of B-cell malignancies by anti-HLA-DR humanized monoclonal antibody, IMMU-114, is mediated through hyperactivation of ERK and JNK MAP kinase signaling pathways. Blood. 2010;115:5180–5190. doi: 10.1182/blood-2009-06-228288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCarthy PL, Owzar K, Hofmeister CC, Hurd DD, Hassoun H, Richardson PG, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Eng J Med. 2012;366:1770–1781. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Eng J Med. 2012;366:1782–1791. doi: 10.1056/NEJMoa1114138. [DOI] [PubMed] [Google Scholar]