

Abstract
Advances in chlorin synthetic chemistry now enable the de novo preparation of diverse chlorin-containing molecular architectures. Five distinct molecular designs have been explored here, including hydrophobic bioconjugatable (oxo)chlorins; a hydrophilic bioconjugatable chlorin; a trans-ethynyl/iodochlorin building block; a set of chlorins bearing electron-rich (methoxy, dimethylamino, methylthio) groups at the 3-position; and a set of ten 3,13-disubstituted chlorins chiefly bearing groups with extended π-moieties. Altogether 23 new chlorins (17 targets, 6 intermediates) have been prepared. The challenge associated with molecular designs that encompass the combination of “hydrophilic, bioconjugatable and wavelength-tunable” chiefly resides in the nature of the hydrophilic unit.
Keywords: auxochrome, artificial photosynthesis, chlorophyll, hydroporphyrin, hydrophilic, photomedicine
INTRODUCTION
The central role of chlorophylls in plant photosynthesis has motivated studies of vast scope concerning the interplay of molecular structure, supramolecular chemistry, optical spectra, photochemical properties, and electronic structure in this family of macrocycles. Synthetic chemistry has played a very strong supporting role in such studies by making available diverse chlorins that are not accessible via naturally occurring biosynthetic pathways. Studies in artificial photosynthesis provide both a direct biomimetic mirror for natural photosynthesis and also enable fundamental studies of excited-state energy- and electron-transfer processes that are not possible with the natural systems.
Two distinct methods for gaining access to chlorin macrocycles entail semisynthesis, where naturally occurring tetrapyrroles (e.g., heme, chlorophylls, bacteriochlorophylls) provide the initial starting material [1–4], and de novo synthesis, where the macrocycle is created from relatively simple, acyclic starting materials [5–7]. Semisynthesis has the advantage of abundant feedstocks but the disadvantage of a full complement of β-pyrrolic substituents, which can limit synthetic maneuverability. De novo syntheses have the advantage of a tabula rasa thereby enabling – at least in principle – full synthetic control over all substituents, but the disadvantage of a considerable amount of synthetic work to construct and derivatize the chlorin macrocycle.
An offshoot of synthetic studies in chlorin chemistry has entailed the preparation of chlorins for use in photomedicine, which encompasses diagnostics, imaging and therapeutics. Our research groups have been engaged in the de novo synthesis of chlorins. In Raleigh, we have been working to develop methodology for the preparation of chlorins for diverse photochemical studies [8–22]. In Baltimore, we have been working to design and synthesize chlorins for use in photomedicine, particularly laser-guided surgery [23–28]. A common challenge in all such studies is the ability to (1) control overall molecular polarity, (2) dial-in the position of the long-wavelength absorption band (which determines the maximum energy for photochemical processes emanating from the excited singlet state), and (3) install one or more reactive functional groups for attachment to other moieties such as proteins, surfaces, or other molecular entities.
In this paper, we describe the synthesis of a collection of chlorins for use in diverse studies. The paper is divided into three sections. The sections encompass (I) the synthesis of lipophilic or hydrophilic chlorins that bear a single (bio)conjugatable group; (II) the synthesis of a chlorin bearing two reactive aryl functional groups attached to opposing β-pyrrole rings; and (III) the synthesis and spectral properties of diverse 3-substituted or 3,13-disubstituted chlorins. Studies of functional properties of the chlorins are beyond the scope of the present article. Taken together, this work provides an overview of the rich capabilities of chlorin synthetic chemistry, and provides milestones concerning versatility of molecular design of chromophores with red-region spectral features.
RESULTS AND DISCUSSION
The methodology for preparing chlorins is outlined in Scheme 1. Chlorin formation entails the convergent joining of an Eastern half and a Western half under acid-catalyzed condensation followed by metal-mediated oxidative cyclization [8,10]. The Eastern half is a dipyrromethane bearing an α-bromo atom and an α-carbinol or carboxaldehyde; the linear tetrapyrrole derived from the former is a tetrahydrobilene-a whereas the latter is a tetrahydrobiladiene-ab. The Western half is a tetrahydrodipyrrin that contains the geminal-dimethyl-substituted pyrroline ring [29]. The resulting zinc chlorin can be modified in a number of ways, including conversion to the 17-oxochlorin [11], derivatization at the 15-position [12,14], alteration of the various pyrrolic substituents [13,15,17], de/remetalation, and various combinations of such approaches. By use of these and other approaches, access to each peripheral position of the chlorin macrocycle is now available [22]. Complementary routes to analogous chlorins have been developed by the groups of Battersby [30,31], Jacobi [32–36] and Montforts [37–44].
Scheme 1.
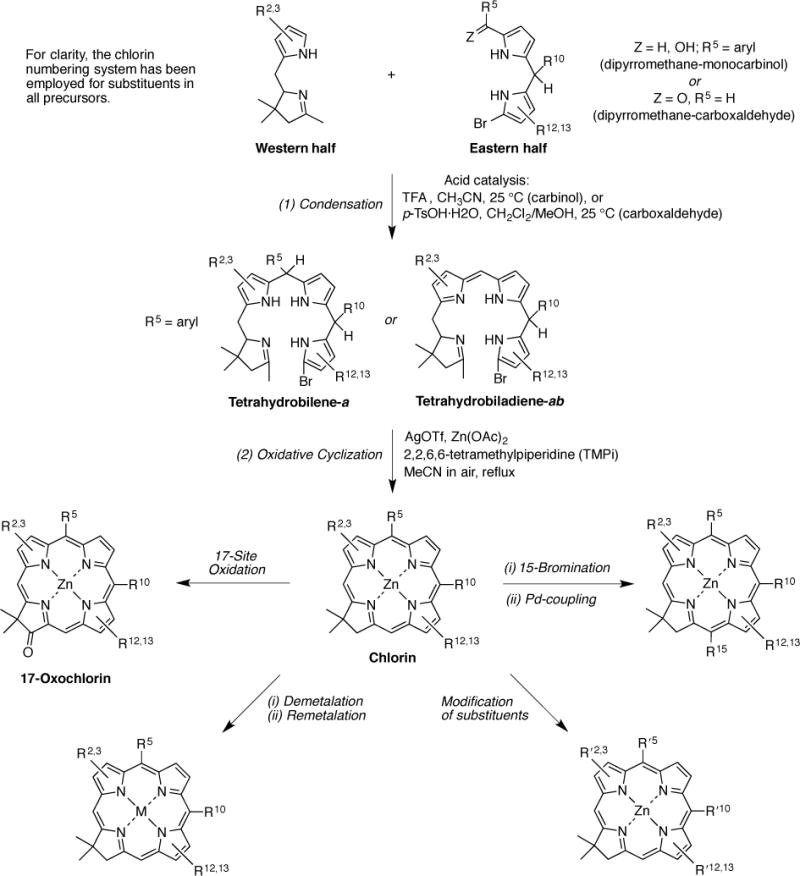
Synthetic methods employed herein.
I. Bioconjugatable chlorins
A. Lipophilic (bio)conjugatable chlorins
The synthesis of a lipophilic chlorin bearing a single bioconjugatable group can be achieved by incorporation of the latter in protected form as a substituent on one of the meso-aryl units. A key design issue is that the bioconjugatable motif must be able to survive the conditions for formation of the Eastern half and conversion to the chlorin. Two examples are shown here, which include a phenol and an arylcarboxylic acid as bioconjugatable groups.
The synthesis of an oxochlorin–phenol is shown in Scheme 2. The synthesis of the Eastern half begins with dipyrromethane 1 [45–47], which contains the protected phenol. Treatment of 1 with EtMgBr at room temperature for 10 min followed by Mukaiyama reagent 2 [48] at −78 °C for 20 min gave the 1-acyldipyrromethane 3 in crude form. The latter was complexed with 9-BBN-OTf in the presence of triethylamine (TEA) to afford the complex 4 in 73% yield (two steps from 1). Such 1-acyldipyrromethane–boron complexes are more readily handled and derivatized given the masking of the α-acylpyrrole motif [48,49]. Bromination of 4 using NBS following a standard procedure [48] afforded the compound 5 in 77% yield. Compound 5 was used directly for the chlorin-forming reaction without decomplexation of the boron moiety. Thus, treatment of 5 with NaBH4 gave the 1-bromo-dipyrromethane-monocarbinol (5–OH, not shown), the Eastern half. The crude Eastern half was combined with Western half 6 [10] and subjected to two-step one-flask chlorin-forming conditions [10] to afford the zinc chlorin ZnC-1 in 36% yield. The ZnC-1 was converted to zinc oxochlorin ZnC-2 following a standard procedure [11]. Reaction of ZnC-2 with BBr3 in CH2Cl2 at room temperature [50] followed by treatment with Zn(OAc)2·2H2O afforded the zinc oxochlorin–phenol ZnC-3.
Scheme 2.
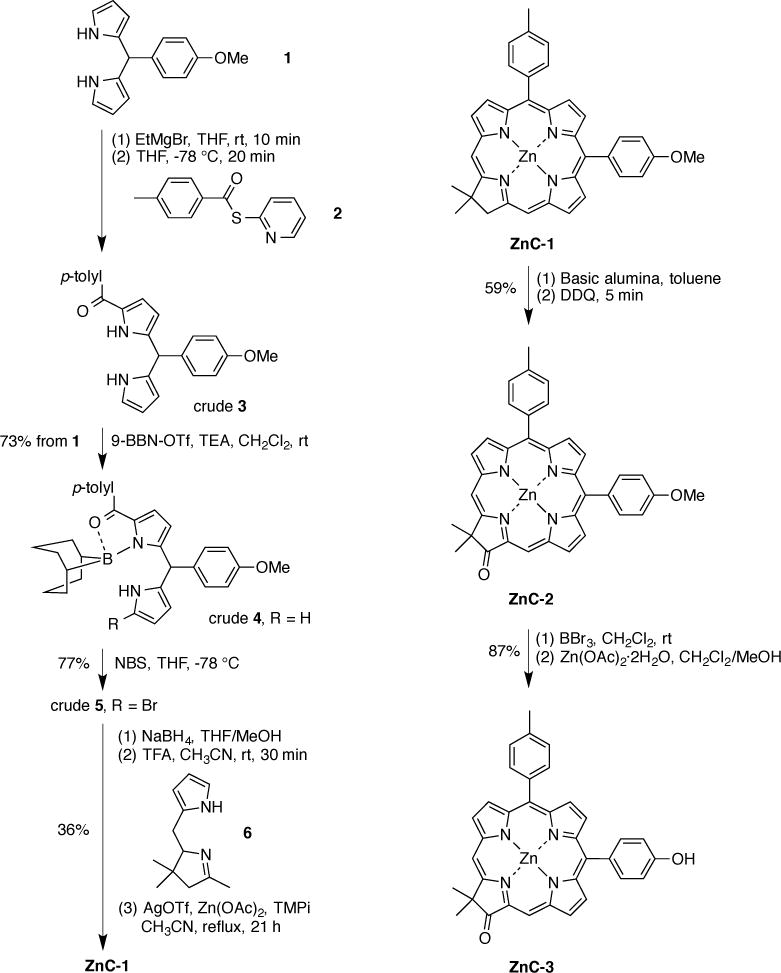
Synthesis of an oxochlorin–phenol.
The synthesis of a chlorin–carboxylic acid followed the same synthetic approach. The resulting chlorin–ester (ZnC-4) [10] was treated with KF to cleave the trimethylsilylethyl unit under relatively mild conditions, thereby affording the chlorin–carboxylic acid ZnC-5 (Scheme 3). It warrants mention that both the oxochlorin–phenol and the chlorin–carboxylic acid are amphiphilic given the presence of the polar bioconjugatable group attached to the lipophilic chlorin. As such, both compounds if not biconjugated are expected to assemble at an aqueous–surfactant interface (e.g., micelle, lipid bilayer membrane, liposome) thereby representing an additional application for studies with such chlorins. In this regard, Jacobi and Ehrenberg have studied the membrane depth penetration of members of a set of amphiphilic chlorins that bear alkanoic chains of a range of lengths [36].
Scheme 3.
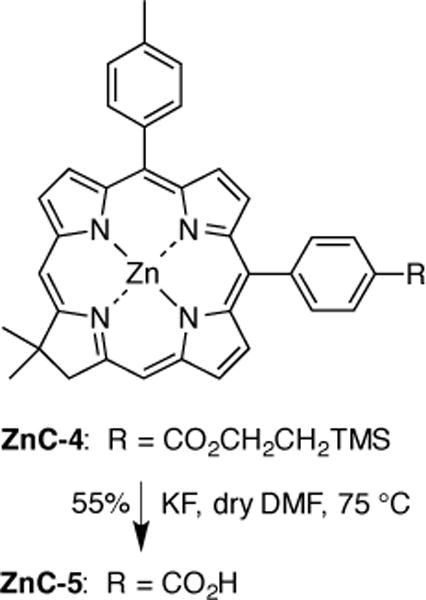
Synthesis of a chlorin–carboxylic acid.
B. Hydrophilic bioconjugatable chlorins
A general challenge in tetrapyrrole chemistry concerns appropriate molecular designs to impart high solubility in either organic or aqueous solution. The core unsaturated, unsubstituted tetrapyrrole skeleton, porphine, is hideously insoluble. The solubility increases upon ‘bulking up” the tetrapyrrole framework with substituents that suppress aggregation phenomena. Examples include β-alkyl (e.g., octaethylporphyrin), meso-aryl (e.g., meso-tetraphenylporphyrin), and meso-swallowtail (i.e., symmetrically branched alkyl) [51] substituents for solubilization in organic media. The use of meso-aryl units that bear ortho-substituents (e.g., methyl, ethyl, benzyloxy) provide increased solubility in organic media versus that of the unsubstituted phenyl unit [52]. The ortho-substituents project above and below the plane of the macrocycle (Figure 1). An extension of this approach of “facial encumbrance” has afforded solubilization of porphyrins and chlorins [19] in aqueous media. Thus, 2,6-disubstituted aryl groups or swallowtail substituents with terminal polar groups (e.g., phosphate, phosphonate) have been found to impart water solubility of synthetic chlorins, as shown for the chlorins C-6 and C-7 shown in Figure 1 [19].
Figure 1.
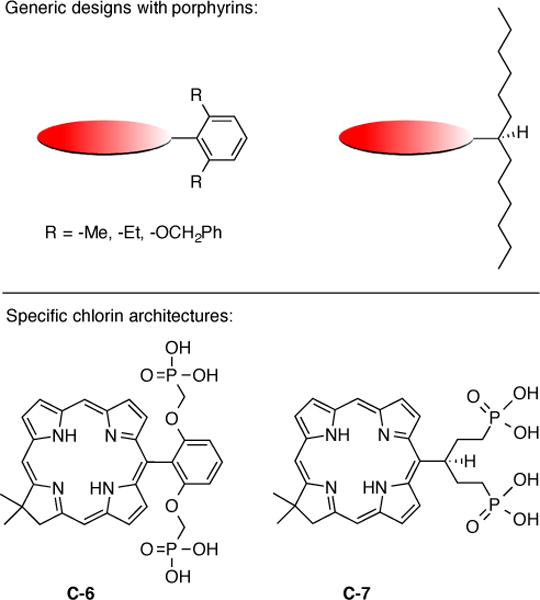
Facial encumbrance to achieve increased solubility of porphyrins in organic media (top) [51,52]. Extension of the approach for solubilization of chlorins in aqueous media (bottom) [19].
Our first approach to a hydrophilic bioconjugatable chlorin focused on a molecular architecture that (1) represented a hybrid of the meso-aryl and swallowtail motifs, (2) incorporated a carboxylic acid for bioconjugation, and (3) could be incorporated to the chlorin macrocycle via Suzuki coupling. Precedence in considering this route include phenol alkylation with diethyl chloromethylphosphonate [53] as well as Suzuki coupling with hindered aryl units [54] (including 2,6-dialkoxyaryl halides with aryl boronic acids [55]), albeit under forcing conditions. Thus, the commercially available methyl 4-bromo-3,5-dihydroxybenzoate (I) was alkylated [19,56] with TfOCH2PO(OEt)2 (II) [57] to give methyl 4-bromo-3,5-bis(diethylphosphonomethoxy)benzoate (7) in 92% yield (Scheme 4). Compound 7 is a potential snap-on motif that bears the requisite facially encumbering phosphonate groups and the bioconjugatable carboxylate unit. A homologue of 7 containing a longer alkyl spacer between the alkoxyarene and the phosphonate moiety also was prepared by alkylation of I with diethyl 3-bromopropylphosphonate (III) to give 8. Attempts to convert synthon 7 or 8 to the boronate derivative, however, were unsuccessful.
Scheme 4.
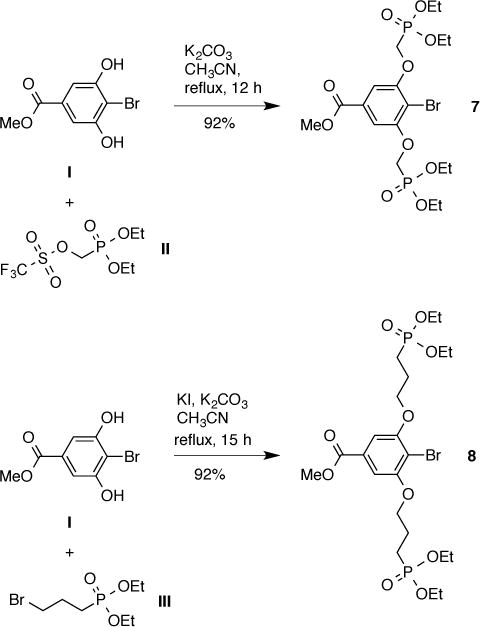
Synthesis of candidate snap-on units for water solubilization and bioconjugatability.
The chlorin examined for pilot studies, C-8 [17], smoothly underwent bromination at the 15-position to give C-9 [17] followed by Pd-mediated borylation [58] to give chlorin–boronate C-10 (Scheme 5). On the other hand, Pd-mediated coupling of C-10 with the 2,6-dialkoxyaryl bromide (7 or 8) proceeded quite poorly. The product in each case (C11 or C12) was suggested by a molecular ion peak upon ESI-MS analysis, but the trace quantity thwarted isolation. Alternatively, attachment of 1,3-dimethoxy-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene [59,60] (derived here from 2,6-dimethoxyphenyl boronic acid and pinacol) to the 15-position of the chlorin was investigated but found non-advantageous versus incorporation of the 2,6-dimethoxyaryl unit via the dipyrromethane in the Eastern half synthesis. The failure to incorporate the snap-on motif prompted return to an established route wherein the facially encumbering diphosphonate group is installed via the use of the Eastern half.
Scheme 5.
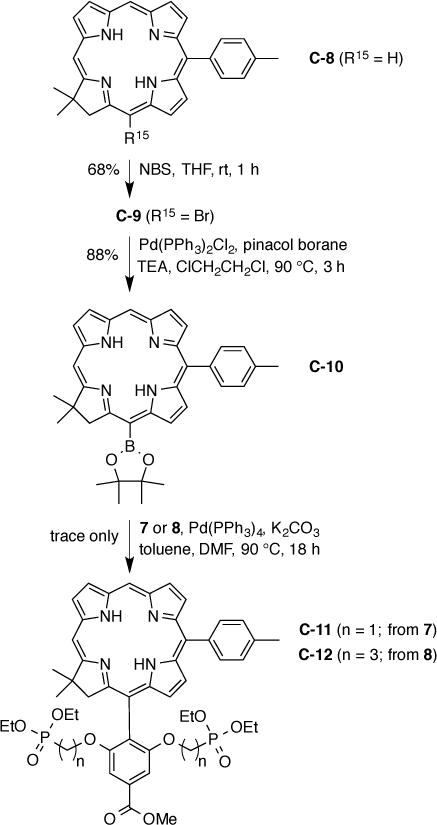
Attempted snap-on of tri-substituted aryl motif for solubilization and conjugation.
Chlorin C-13 is readily prepared and contains a 2,6-dimethoxyphenyl unit at the 10-position; cleavage of the methyl ethers reveals the diphenol, which is alkylated with II [19]. In the present work, the resulting bis(diethylphosphonomethoxy)phenylchlorin C-14 was further manipulated to install a bioconjugatable handle. Bromination [14,17] of C-14 led to the 15-bromo-10-arylchlorin C-15 in 96% yield. Suzuki coupling of C-15 with boronic ester IV under standard conditions employed for porphyrins [61,62], chlorins [12] and bacteriochlorins [63] led to the formation of chlorin C-16 in 50% yield. Saponification followed by deprotection of the ethyl phosphonate groups led to the water-soluble bioconjugatable chlorin C-17 as a green solid in excellent yield (Scheme 6). The 1H NMR spectrum in CD3OD indicated exchange of 7 protons (two NH, one CO2H, four P–OH), as expected, but upon integration only 26 of the remaining 29 protons were identified. The characteristic chlorin absorption spectrum and accurate (negative ion) mass spectrum support a provisional assignment of the target compound. In summary, this approach offers a rational route to a chlorin bearing a facially encumbering bis(phosphonate) group and a single bioconjugatable group.
Scheme 6.
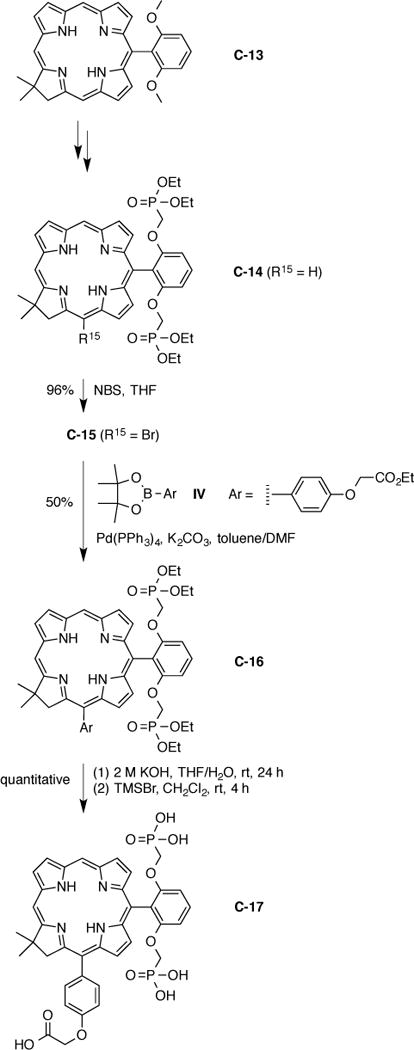
Hydrophilic bioconjugatable chlorin–carboxylic acid.
II. β-Disubstituted chlorin building block
Chlorins bearing two orthogonal functional groups enable successive derivatization. Two such functional groups at diametrically opposed positions are particularly attractive for the construction of linear multipigment arrays that function as energy-transfer cascades or hole-hopping relays. We have found that the extent of electronic communication between porphyrins of a given electronic composition depends on the site of attachment of a linker on the macrocycle [64]. The availability of chlorins with diverse substitution patterns would enable study of such electronic effects in chlorins, complementing our prior studies with porphyrins. For the construction of linear arrays, substitution at opposing β-pyrrole positions is quite attractive.
We previously prepared several β-substituted chlorins wherein each bears an ethyne group and a halogen atom in diametrically substituted (2,12-; 3,13-) positions; the chlorins include ZnC-18 [9], ZnC-19 [13], and ZnC-20 [65] (Chart 1). Here we present the synthesis of an analogue of ZnC-18 that contains a 3,5-di-tert-butylphenyl group (rather than a p-tolyl group) at the 5-position to impart increased solubility in organic solvents. While in principle either the 2,12- or 3,13-positions can be employed, given the bulk of the 3,5-di-tert-butylphenyl group at the 5-position, we chose the 2,12-positons for installation of the iodo and ethynyl groups. The synthesis of such trans-β-substituted chlorin building blocks requires the availability of β-substituted Eastern and Western halves. The synthesis also makes use of improved methods, including the intermediacy of a tetrahydrodipyrrin as the Western half (versus the dihydrodipyrrin employed previously).
Chart 1.
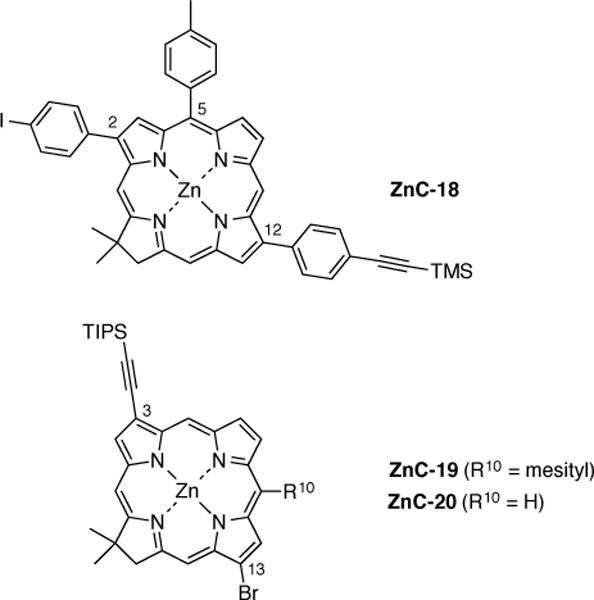
Prior trans-disubstituted chlorin building blocks.
A. Substituted Western halves
The β-arylpyrrole is conveniently prepared via a van Leusen reaction. Formylation of the resulting β-(p-iodophenyl)pyrrole [9] typically affords the 2-formyl-3-arylpyrrole and the 2-formyl-4-arylpyrrole (due to substitution at either the α- or α′-position), which must be separated and identified. Such distinction can be difficult by 1H NMR spectroscopy [9]. Accurate identification of the two products (or derivatives therefrom along the later steps of the pathway) is essential because the two patterns of substitution ultimately yield chlorin building blocks with substitution at different β-positions. Subsequent nitrovinyl condensation followed by reduction gave the 2-(2-nitroethyl)pyrrole 9 (Scheme 7). Michael reaction with mesityl oxide (V) gave the nitrohexanone (10), which upon treatment with zinc/acetic acid gave the tetrahydrodipyrrin-N-oxide (11) in 43% yield. The synthesis of the tetrahydrodipyrrin target(s) has been described previously in concise form [66], and 9 and V have been used previously to form the dihydrodipyrrin Western half [9]; the presentation here supplements the prior presentation with additional data and context.
Scheme 7.

Synthesis of β-substituted Western halves.
A single-crystal X-ray structure of 11 was obtained. The structure determination was not required to confirm the substitution pattern (Figure 2, Table 1), which was established previously by the X-ray structure of the 2-(2-nitroethyl)pyrrole 9 [9]. On the other hand, X-ray structure analysis of 11 adds to the very limited data available for hydrodipyrrins (considerable data are available for dipyrromethanes or dipyrrins, which lack the pyrroline ring). To our knowledge, structural data concerning hydrodipyrrins related to chlorin syntheses are available only for three tetrahydrodipyrrin–dibutylboron complexes [22], one dihydrodipyrrin [67], one dibutylboron complex of a dihydrodipyrrin–dibutylboron complex [22], and one nitrohexanone (a precursor for a dihydrodipyrrin or tetrahydrodipyrrin) [67].
Figure 2.
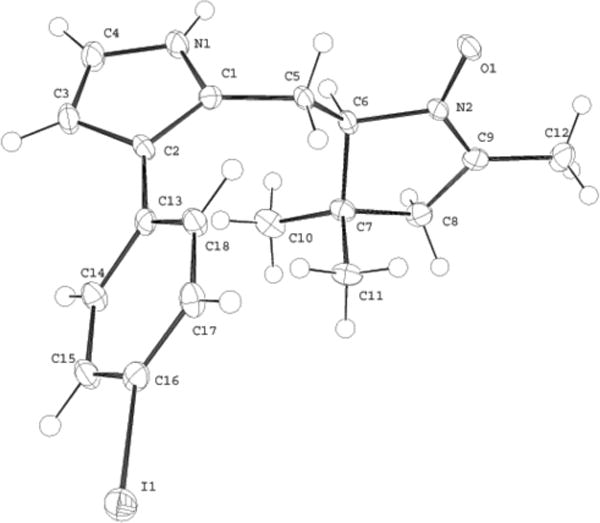
ORTEP drawing of 11, a Western half precursor. The ellipsoids are drawn at the 50% probability.
Table 1.
Summary of Crystal Data for 11
| Crystal data | |
|---|---|
| Formula | C18H21IN2O |
| Formula Weight (g/mol) | 408.28 |
| Crystal Dimensions (mm) | 0.58 × 0.24 × 0.06 |
| Crystal Color and Habit | colorless plate |
| Crystal System | monoclinic |
| Space Group | P 21/n |
| Temperature, K | 148 |
| a (Å) | 13.9428 (9) |
| b (Å) | 8.2765 (5) |
| c (Å) | 15.1619 (11) |
| α (°) | 90.0 |
| β (°) | 93.346 (8) |
| γ (°) | 90.0 |
| V (Å3) | 1746.66 (20) |
| Z | 4 |
| Dx (Mg m−3) | 1.553 |
| λ, Å, (MoKα) | 0.71073 |
| μ (cm−1) | 1.84 |
|
| |
| Data collection | |
|
| |
| No. of reflections measured | 3046 |
| No. of unique reflections | 3046 |
| No. of reflections with I > σ(I) | 2671 |
| θmax (°) | 25.00 |
| Index range | −16 ≤ h ≤ 16 |
|
| |
| Refinement | |
|
| |
| No. of reflections in least-squares | 2670 |
| No. of parameters in least-squares | 199 |
| R1a | 0.033 |
| wR2b | 0.043 |
| R1 (all data)a | 0.033 |
| wR2 (all data)b | 0.043 |
| GOFc | 1.54 |
| Maximum shift/error | 0.000 |
| Δρmax (e Å−3) | 1.17 |
| Δρmin (e Å−3) | −1.54 |
R1 = Σ(|Fo| − |Fc|)/Σ Fo
wR2 = [Σ(w(Fo − Fc)2)/Σ(wFo2)]½
GOF = [Σ(w(Fo − Fc)2)/(No. of reflections − No. of parameters)]½
Completion of the synthesis required deoxygenation of the N-oxide 11. The prior route to a β-substituted dihydrodipyrrin Western half [9] was used herein with modification to obtain the β-substituted tetrahydrodipyrrin counterpart. Thus, deoxygenation of N-oxide 11 yielded the β-substituted tetrahydrodipyrrin Western half 12 in 38% yield (Scheme 7). Sonogashira coupling of 12 with (trimethylsilyl)acetylene afforded the trimethylsilylethynyl substituted Western half 13 in 76% yield.
B. Substituted Eastern half
The synthesis of the β-substituted Eastern half began in the same manner as our prior synthesis of β-substituted dipyrromethanes [9,68]. A BOC-protected dipyrromethane (14) was obtained from 2-formyl-3-(4-iodophenyl)pyrrole through the protection of the pyrrolic nitrogen, reduction of the aldehyde and condensation with pyrrole (not shown) [9]. Treatment of the BOC-protected dipyrromethane 14 with 3.0 equiv of EtMgBr in THF followed by 3,5-di-tert-butylbenzoyl chloride afforded the monoacyldipyrromethane 15 in 45% yield (Scheme 8). Removal of the BOC group under standard conditions [69] gave 16. Sonogashira coupling [70] of 16 with (trimethylsilyl)acetylene afforded trimethylsilylethynyl–dipyrromethane 17 in 82% yield. Reaction of 17 with NBS at −78 °C gave the corresponding monobromo-monoacyl dipyrromethane 18 in 72% yield. Compound 18 serves as the precursor to the Eastern half dipyrromethane-monocarbinol 18-OH.
Scheme 8.
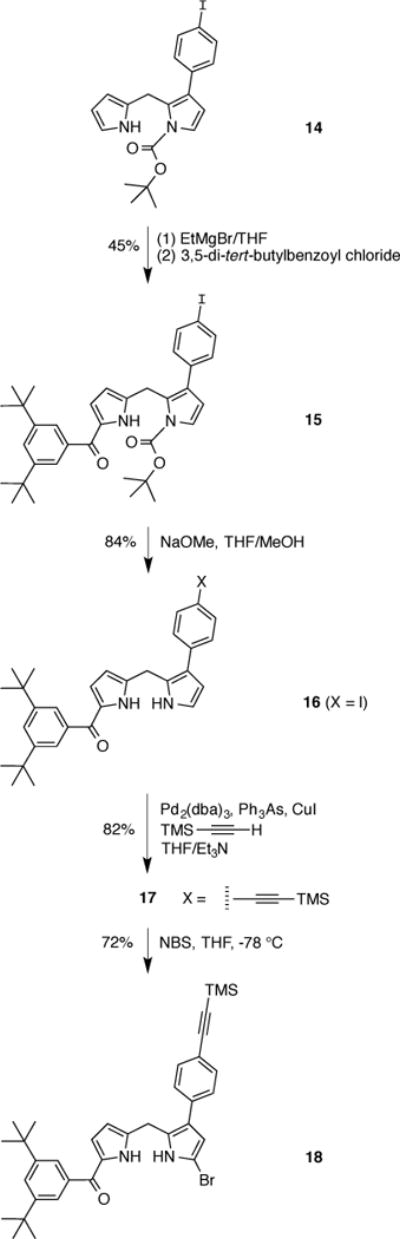
Synthesis of a β-substituted Eastern half.
C. Chlorin formation
The chlorin-forming reaction was carried out using a two-step two-flask procedure of acid-catalyzed condensation followed by metal-mediated oxidative cyclization. Thus, treatment of 18 with NaBH4 in methanol gave the corresponding dipyrromethane-monocarbinol (18-OH), which was used without extensive purification in the next step. The reaction of 12 and 18-OH employed early conditions for metal-mediated oxidative cyclization (using AgIO3, subsequently replaced with AgOTf [10]) afforded chlorin ZnC-21 in 8% yield (Scheme 9). The absorption spectrum was characteristic of a chlorin, with a molar absorption coefficient (log ε = 4.86 at 629 nm) in close accord with that of the p-tolylchlorin counterpart ZnC-18 (log ε = 4.90 at 629 nm) [13]. 1H NMR spectroscopy confirmed the expected structure: (i) the geminal dimethyl groups of ZnC-21 resonate as a singlet at δ 1.97 ppm, (ii) the CH2 in the reduced pyrrole ring gives rise to a singlet downfield at δ 4.51 ppm, (iii) the four β-pyrrole protons resonate in the region of δ 8.56–8.81 ppm, and (iv) the eight protons of the β-aryl rings resonate in the region of δ 7.82–8.14 ppm as two pairs of doublets. In summary, the β-substituted Eastern and Western halves enable the rational synthesis of a chlorin building block with synthetic handles at diametrically opposed positions at the perimeter of the macrocycle. Such a building block should be quite valuable for the construction of model systems in biomimetic or materials chemistry.
Scheme 9.
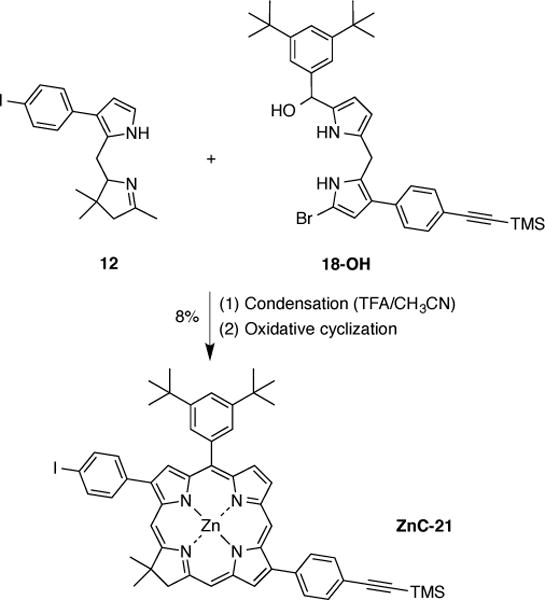
Synthesis of a trans-disubstituted chlorin building block.
III. Wavelength-tunable chlorins
A major finding from the preparation of chlorins that bear diverse substituents is that the position of the long-wavelength absorption band can be tuned across the 600–700 nm region. The chief method for such tuning entails placement of auxochromes at the 3- and 13-positions [13]. The 3- and 13-positions are both in a pyrrole ring that lies along the Qy axis, with the 3-position distal and 13-position proximal to the pyrroline ring. Auxochromes such as ethynyl, phenylethynyl, carboethoxy, acetyl, or formyl at the 3- and/or 13-position result in a bathochromic shift of both the B and Qy bands, as well as an increase in the intensity of the Qy band relative to the B band. Such substituents are regarded as electron-withdrawing groups. By contrast, few studies have concerned the effects of electron-donating groups on the chlorin spectral properties.
This lacuna prompted us to prepare three chlorins with groups that are electron-releasing by resonance (methoxy, methylthio, and dimethylamino) in the 3-position [71], which is described in the first section below. In the second section, we have prepared a collection of 3,13-disubstituted chlorins that bear aryl groups with extended conjugation. The motivation for preparing both classes of chlorins reflects the dual desires to gain a deeper understanding of chlorin electronic/photophysical properties and to understand how to design chlorins with deep-red (or near-IR) absorption and (preferably intense) fluorescent properties. Such chlorins could be used as fluorophores for in vivo bioimaging applications (e.g., fluorescence-guided surgery [23,24]) or as the energy donor in energy-transfer arrays for artificial photosynthesis or in vivo multicolor bioimaging [18,20,25,26].
A. 3-Substituted chlorins
The requisite 3-bromochlorin building block has been prepared previously using 0.20 mmol reactants (Western half 19 [13] and Eastern half 20 [13]), which afforded 45 mg of the chlorin ZnC-22 (37%) [13]. The same synthesis carried out here at 0.57 mmol reactants gave 119 mg of the chlorin (34%) as shown in Scheme 10. Palladium coupling of ZnC-22 with dimethylamine (10 equiv) with Pd(OAc)2 (40 mol%), (tert-Bu)3PHBF4 (40 mol%), and tert-BuONa (3 equiv) in refluxing toluene for 48 h [72] afforded 3-dimethylaminochlorin ZnC-23 in 49% yield. Palladium coupling of ZnC-22 with sodium thiomethoxide under conditions used for aryl thioether formation (20 mol% of Pd2(dba)3 and Xantphos in refluxing xylene) [73] afforded 3-methylthiochlorin ZnC-24 in 95% yield. Similarly, palladium coupling of ZnC-22 with sodium methoxide in the presence of 20 mol% of Pd2(dba)3 and Xantphos in refluxing xylene gave 3-methoxychlorin ZnC-25 in 16% yield. Attempts to mitigate the low yield of the 3-methoxychlorin ZnC-25, either by methoxylation [74] of the tosyl-protected Western half 19-Ts [13] (not shown here) or by use of modified Pd-mediated coupling conditions (as used for the meso-position of a porphyrin) [75] were ineffective [71].
Scheme 10.
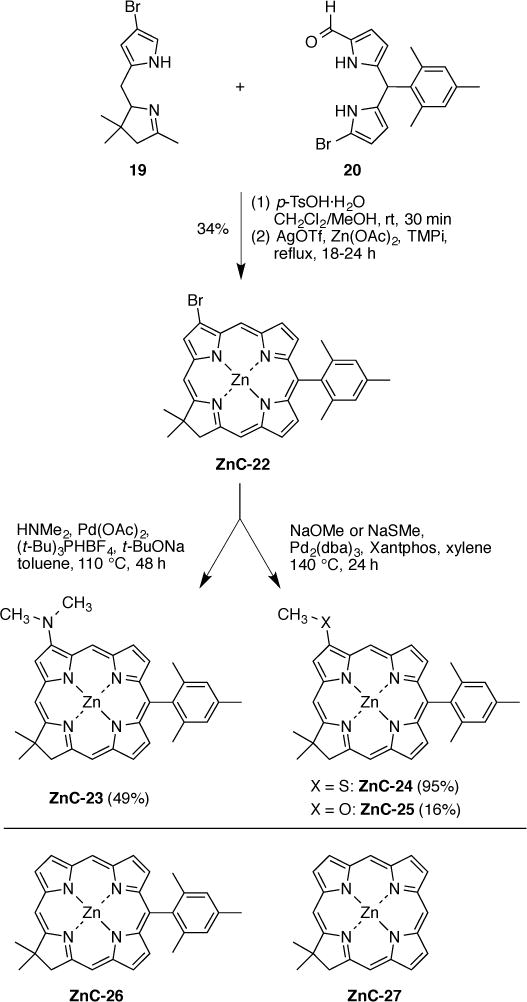
Synthesis of 3-substituted chlorins (top) and benchmark chlorins (bottom).
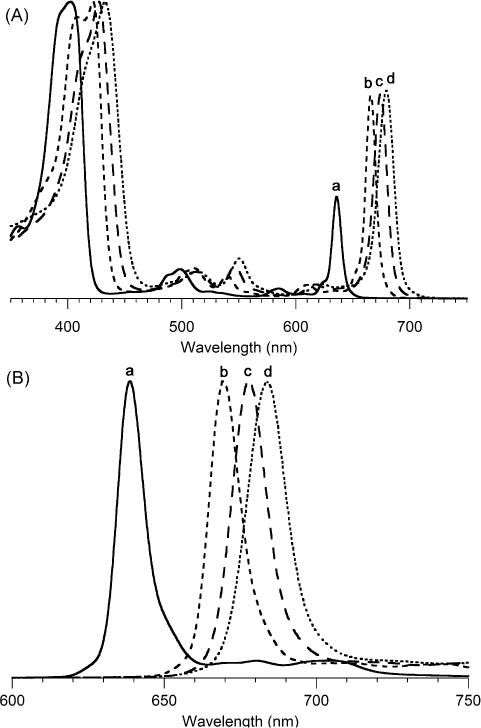
The spectral properties of the zinc chlorins obtained in toluene are listed in Table 2 and displayed in Figure 3. The absorption spectrum in each case is typical of a chlorin, as is the fluorescence emission spectrum, with a stronger Q(0,0) band and weaker Q(0,1) band. The absorption spectral properties of the three 3-substituted chlorins (ZnC-23, ZnC-24, and ZnC-25) can be compared with the benchmark zinc chlorin ZnC-26, which lacks a 3-substituent. The spectral shift of the B band and the Qy band is listed for each compound. While the former is more intense, the position of the latter is of greater interest given that the Qy band position sets an upper limit on the energy of the excited singlet state, and inter alia, that of the excited triplet stated derived therefrom. The Qy band spans the range from 603–616 nm along the series of chlorins with 3-methoxy (ZnC-25), benchmark (3-unsubstituted, ZnC-26), 3-(N,N-dimethylamino) (ZnC-23), and 3-methylthio (ZnC-24) substituents. The Qy band of ZnC-25 is thus shifted hypsochromically with respect to that of the unsubstituted chlorin ZnC-26. A similar effect of the methoxy group has been noted with bacteriochlorins [76]. In parallel with the progressive bathochromic shift of the Qy band position, the relative intensity of the Qy band also increased (i.e., IB/IQ decreased from 4.3 to 2.5). The 603-nm absorption band of the methoxychlorin (ZnC-25) is identical to that of the corresponding zinc chlorin that lacks any substituents, not even the 10-mesityl group (ZnC-27) [77]. We have not determined the molar absorption coefficient of the various chlorins but instead rely on the IB/IQ ratio to indicate the relative intensity of long-wavelength absorption, which can be reliably determined regardless of sample quantity. For perspective, however, the molar absorption coefficient for the benchmark chlorins are as follows [13]: ZnC-26, log ε = 4.81 (606 nm) and ZnC-27, log ε = 4.84 (603 nm). The molar absorption coefficients of some 50 other gem-dimethyl substituted chlorins also have been determined [8, 9,11,13,14]. In summary, the effects of the three substituents examined here, methoxy, dimethylamino, and methylthio are rather insubstantial in altering the position of the Qy band. The extent to which the little-changed spectra stem from intrinsically weak interactions of such groups with the four individual frontier molecular orbitals of the chlorin, or stronger interactions that are muted by cancellation of opposing effects (e.g., induction versus resonance of the substituent) will require in-depth experimental and theoretical studies.
Table 2.
Absorption and fluorescence spectral properties.a
| Compound | λB (nm) | ΔνB (cm−1)b | λQy (nm) | ΔνQy (cm−1)b | IB/IQ | λem (nm) |
|---|---|---|---|---|---|---|
| ZnC-26 | 405 | 0 | 606 | 0 | 4.2 | 609 |
| ZnC-25 | 404 | −61 | 603 | −82 | 4.3 | 606 |
| ZnC-24 | 410 | 301 | 616 | 268 | 3.0 | 621 |
| ZnC-23 | 410 | 301 | 612 | 162 | 2.5 | 617 |
In toluene at room temperature.
Spectral shift versus that of ZnC-26.
Figure 3.
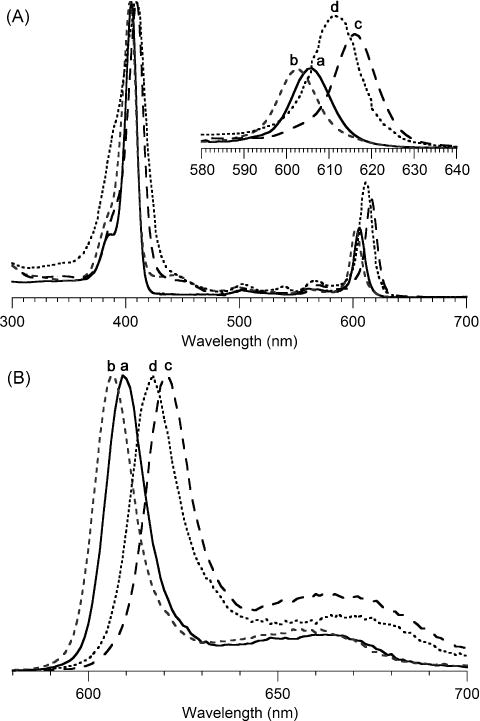
(A) Absorption spectra normalized at the B band in toluene at room temperature of (a) 3-unsubstituted chlorin, ZnC-26, (b) 3-methoxychlorin, ZnC-25, (c) 3-methylthiochlorin, ZnC-24, and (d) 3-(N,N-dimethylamino)chlorin, ZnC-23. (B) Corresponding fluorescence spectra upon excitation at the peak of the B band.
B. 3,13-Disubstituted chlorins
Phenylethynyl substituents, which effectively expand the conjugated system of tetrapyrrolic macrocycles, are known to significantly alter the electronic and optical properties of porphyrins [78], chlorins[15,16], and bacteriochlorins [28,79]. For example, 3,13-bis(phenylethynyl)chlorin C-28 (Chart 2) exhibits a significant bathochromic shift of absorption and emission (λabs = 676 nm, λem = 678 nm), and large increase in fluorescence quantum yield (Φf = 0.37, in toluene), compared to the analogous chlorin that lacks substituents at the 3,13-positions [18].
Chart 2.
A prior 3,13-bis(phenylethynyl)chlorin.
We aimed to determine whether a further bathochromic shift in absorption and emission for chlorins could be achieved by more elaborate substituents at the chlorin 3- and 13-positions. The synthesis makes use of the 3,13-dibromo-10-p-tolylchlorin (C-29) building block (Scheme 11), which has been prepared following a reported procedure [13,21]. Thus, condensation of the 8-bromotetrahydrodipyrrin (19) [29] and 7,8-dibromo-1-formyl-10-p-tolyldipyrromethane (21) [23] in the presence of p-TsOH·H2O followed by oxidative cyclization (mediated by Zn(OAc)2 in the presence of 2,2,6,6-tetramethylpiperidine) and demetalation (dilute TFA in dichloromethane) afforded free base chlorin C-29 in 12% yield. The lower yield of C-29 (compared to that of analogous chlorins reported previously) [13,21] may be due to the poor solubility of Eastern half 21 (and presumably the resulting tetrahydrobiladiene) in the reaction solvents.
Scheme 11.
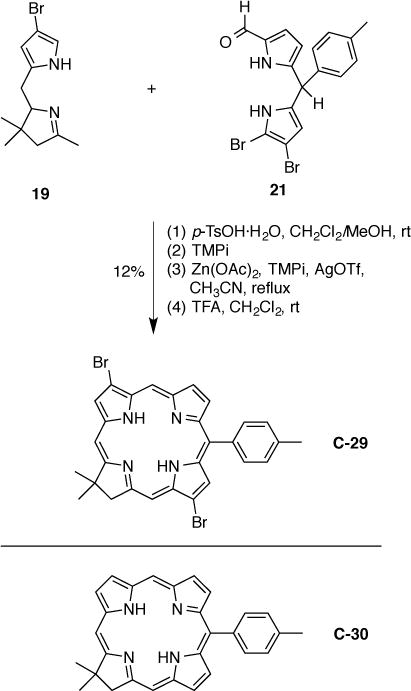
Synthesis of a 3,13-dibromochlorin building block (top) and a chlorin benchmark (bottom).
With the 3,13-dibromochlorin building block in hand, we prepared a series of 3,13-disubstituted chlorins to probe the effect of several factors on the optical properties of chlorins (Table 3). One reference compound is a chlorin with no 3,13-substituents (C-30) [17] or a phenylethynyl substituent at both the 3- and 13-positions (C-31). The factors include the length of the conjugated system of substituents (compounds C-32 and C-33), presence of electron-withdrawing (C-33 – C-35) or electron-donating groups (C-36), size of the aromatic portion of a substituent (C-37), and replacement of the aryl group with an alkyl group (C-38), TMS group, or no substituent (C-39, C-40).
Table 3.
Synthesis of 3,13-disubstituted chlorins.
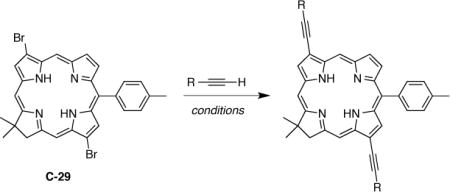
| |||
|---|---|---|---|
| Entry |
|
Conditionsa | Yield |
| C-31 |
|
A | 91% |
| C-32 |
|
A | 80%b |
| C-33 |
|
A | 42% |
| C-34 |
|
A | 43% |
| C-35 |
|
B | 16% |
| C-36 |
|
B | 61% |
| C-37 |
|
A | 27% |
| C-38 |
|
A | 88% |
| C-39 |
|
B | 44% |
| C-40 |
|
c | 79% |
A: (PPh3)2PdCl2, DMF/TEA (2:1), 80 °C. B: Pd2(dba)3, PPh3, toluene/TEA (5:1), 60 °C.
The synthetic product contained a trace amount of blue-fluorescent contaminant, which was removed for fluorescence studies (see text for details).
C-40 was synthesized by deprotection of the TMS groups of C-39 using K2CO3.
The installation of ethynyl substituents was achieved via Sonogashira reaction of the corresponding acetylene derivatives with C-29. Two sets of copper-free conditions, which were previously reported for Sonogashira reaction of hydroporphyrin have been followed; (A) (PPh3)2PdCl2 in DMF/TEA [28] or (B) Pd2(dba)3 and P(o-tol)3 in toluene/TEA [13]. The requisite ethynes were obtained commercially, or for the case of methyl 4-ethynylbenzoate [80], 4-ethynyl-1-(2-phenylethynyl)benzene [81], and methyl 4-[2-(4-ethynylphenyl)ethynyl]benzoate [82], prepared as described in the literature. The yield varied from 16–91% (Table 3). In the case of C-35, the low yield was a consequence of the presence of a small amount of impurities visible in the aromatic region of the 1H NMR spectrum. These impurities were most probably unreacted starting 4-formylphenylacetylene (or product of its homocoupling reaction) and/or dibenzylideneacetone (dba) remnants, the removal of which required two additional column chromatography purifications. A sample of C-32, purified by column chromatography, contained a trace amount of blue-fluorescent contaminant (visible upon TLC analysis and in the fluorescence emission spectrum, but not in the 1H NMR spectrum). This contaminant co-eluted with the product. Conversion of C-32 to the corresponding Zn(II) complex (by reaction with Zn(OAc)2·2H2O in CHCl3/MeOH) caused an increase in polarity and thereby allowed separation from the blue-fluorescent contaminant; subsequent demetalation (using dilute TFA in CH2Cl2) afforded a spectroscopically pure sample. The 3,13-diethynylchlorin (C-40) was obtained by deprotection of the corresponding 3,13-TMS-ethynyl derivative (C-39) using K2CO3 in MeOH/THF. It is noteworthy that attempts to obtain C-40 by deprotection of the corresponding TIPS-ethynyl derivative, using TBAF in THF, led to decomposition of the chlorin.
All new chlorins were characterized by 1H NMR spectrosocopy, LD-MS or ESI-MS; selected chlorins also were characterized by 13C NMR spectroscopy. The characterization data are consistent with the expected structures. In particular, the 1H NMR spectra show two sets of resonances from the 3,13-substituents, as expected given the presence of the geminal dimethyl group at position 18 and the p-tolyl group at the 10-position.
Absorption and emission maxima of the chlorins in toluene are listed in Table 4, and selected spectra are displayed in Figure 4. The benchmark lacking any β-pyrrole substituents in this case is the 10-p-tolylchlorin (C-30). Analysis of these data leads to the following conclusions: (1) The position of the Qy band of the ethynylchlorin C-40 is bathochromically shifted by ~30 nm versus the unsubstituted benchmark chlorin C-30. (2) The position of the Qy band of the phenylethynylchlorin C-31 is bathochromically shifted by ~10 nm versus the ethynylchlorin C-38 or C-40). (3) The further expansion of the size of the π-conjugated substituent attached to the terminus of the ethyne (phenanthryl, C-37) or extension by addition of a second arylethynyl unit (C-32, C-33) has a rather minute bathochromic shift (~5–6 nm) on the position of the Qy band compared to C-31. (4) The largest bathochromic shift occurs with the electron-withdrawing 4-formylphenylethynyl substituent (C-35), though even in that case the shift is 8 nm and 9 nm for absorption and emission, respectively. (5) Attachment of the electron-donating 4-(N,N-dimethylamino)phenylethynyl substituent (C-36) results in the largest relative Qy absorption intensity (IB/IQ = 1.2 ; log ε = 4.97 at 679 nm, to be compared with log ε = 4.90 at 675 nm for a 3,13-bis(phenylethynyl)chlorin constituent of a dyad [20]). At the same time, C-36 exhibits a significant broadening of the B band, compared to the other derivatives in the series. Overall, elaboration of the arylethynyl substituents at the 3,13-positions has a rather small effect on the position of the long-wavelength Qy band and emission maxima.
Table 4.
Absorption and emission maxima for 3,13-disubstituted chlorins.a
| Compound | λB (nm) | λQy | IB/IQ | λem (nm) |
|---|---|---|---|---|
| C-30b | 405 | 635 | 2.8 | 639 |
| C-31 | 427 | 674 | 1.6 | 678 |
| C-32 | 433 | 679 | 1.4 | 684 |
| C-33 | 434 | 680 | 1.5 | 685 |
| C-34 | 431 | 679 | 1.4 | 685 |
| C-35 | 435 | 682 | 1.4 | 687 |
| C-36 | 428 | 679 | 1.3 | 684 |
| C-37 | 431 | 679 | 1.6 | 684 |
| C-38 | 422 | 665 | 1.8 | 668 |
| C-40 | 422 | 666 | 1.5 | 669 |
Figure 4.
(A) Absorption spectra normalized at the B band in toluene at room temperature of (a) benchmark C-30, (b) C-40, (c) C-31, and (d) C-32. (B) Corresponding fluorescence spectra upon excitation at the peak of the B band.
It warrants mention that the IB/IQ ratio can depend on the extent to which the Bx and By bands are spectrally separated or overlapped, which in turn can by affected by environmental factors (e.g., solvent, temperature). Regardless, the difference between the unsubstituted reference chlorin C-30 (IB/IQ = 2.8) and all of the 3,13-diethynylchlorin analogues (C-31 – C-38, C-40; IB/IQ = 1.8–1.3) is pronounced, and reflects the shifting of oscillator strength from the B bands to the Qy band. A more pronounced bathochromic shift than observed herein appears to require more extensive modification of the macrocyclic structure. The research reported here predated and somewhat inspired our recent studies on strongly conjugated hydroporphyrin arrays, which appears to be an efficient way to bathochromically shift the chlorin absorption [27].
OUTLOOK
The ability to create stable chlorins with features of graded polarity, conjugatable substituents, or wavelength tunability provides the foundation for pursuit of diverse applications. While each feature alone can be attained in a fairly straightforward manner, combinations of two or all three features presents challenges:
Chlorins with hydrophobic, hydrophilic, or amphiphilic properties are readily obtained.
Hydrophobic chlorins with one or more conjugatable substituents also are readily obtained, as demonstrated by the synthesis of two such chlorins here as well as by the recent of work of others [26,83,84].
One hydrophilic, bioconjugatable chlorin was prepared herein; the chlorin contains facially encumbering phosphonate groups and a carboxylic acid tether.
The introduction of auxochromes, chiefly at the 3,13-positions, enables the long-wavelength Qy band to be tuned from 603 nm to 715 nm [21,77]. Attempts to employ a previously unexamined set of potential auxochromes (methoxy, dimethylamino, methylthio, phenanthryl, p-substituted phenyl, and oligomeric ethynylphenyl substituents) led to establishment of valuable data concerning structure versus spectral properties but did not enlarge the spectral window for wavelength tuning with chlorins.
The combination of hydrophobic and wavelength tunable presents little challenge, as the chlorin is intrinsically neutral with no polar substituents, and hence is hydrophobic.
The combination of bioconjugatable and wavelength tunable also presents little challenge, as a biconjugatable group can be introduced at the 5- or 10-position via the Eastern half, leaving the 3- and 13-positions open for introduction of auxochromes for wavelength tuning.
The combination of hydrophilic with wavelength tunable presents an unmet challenge, which largely stems from the absence of synthetically versatile and benign groups that impart hydrophilicity. The facially encumbering aryl-bis(phosphonate) group described herein illustrates a viable design, yet whether this approach can be extended alone to encompass wavelength tunability (or together with wavelength tunability without compromising bioconjugatability) remains unclear.
The work described herein has made use of an existing rational synthesis of the chlorin macrocycle to prepare 17 target chlorins along with 6 chlorin intermediates. The molecular architectures prepared illustrate the power of this approach yet also highlight several unmet molecular design challenges.
EXPERIMENTAL SECTION
General
1H NMR and 13C NMR spectra were collected at room temperature unless noted otherwise. Absorption spectra were obtained in CH2Cl2 at room temperature unless noted otherwise. Hydrophobic chlorins were analyzed by fast-atom bombardment mass spectrometry (FAB-MS), laser desorption mass spectrometry (LD-MS) in the absence of a matrix [85], or matrix-assisted laser desorption mass spectrometry (MALDI-MS) with the matrix 1,4-bis-(5-phenyloxazol-2-yl)benzene (POPOP). Water-soluble chlorins were analyzed by direct infusion of water/methanol (60:40) solutions by atmospheric pressure electrospray ionization mass spectrometry (ESI-MS). In FAB-MS, LD-MS, MALDI-MS and ESI-MS analyses, positive ions were detected unless noted otherwise. Melting points are uncorrected. All commercially available materials including compounds I–V were used as received. All palladium-coupling reactions were carried out using standard Schlenk-line techniques. All bromination reactions were performed using freshly recrystallized NBS.
Fluorescence spectroscopy
All chlorins were excited at the peak of the B band where the absorption was set to ~0.1. Spectra were obtained in toluene at room temperature unless noted otherwise. The spectra were corrected for PMT sensitivity and instrument response.
Non-commercial compounds
Dipyrromethane 1 [45–47] was synthesized following a streamlined one-flask procedure [46] whereas dipyrromethane 14 [9] was prepared via a stepwise procedure. The following compounds were prepared via literature procedures: Mukaiyama reagent 2 [48]; Western half precursors 9 [9] and 10 [9]; Western halves 6 [10] and 19 [13]; Eastern halves 20 [13] and 21 [27]; known chlorins as precursors ZnC-4 [10]; C-8 [17], C-9 [17], C-13 [19], and C-14 [19]; and ethynes methyl 4-ethynylbenzoate [80], 4-ethynyl-1-(2-phenylethynyl)benzene [81], and methyl 4-[2-(4-ethynylphenyl)ethynyl]benzoate [82]. Known chlorin ZnC-22 [13] was synthesized at larger scale here. Spectroscopic data for the benchmark chlorins ZnC-26 [13], ZnC-27 [13], and C-30 [17] were taken from the literature.
10-(9-Borabicyclo[3.3.1]non-9-yl)-5-(4-methoxyphenyl)-1-(4-methylbenzoyl)dipyrromethane (4)
Following a standard procedure [49], a solution of EtMgBr (10.0 mL, 10. mmol, 1.0 M in THF) was added slowly to a solution of 1 (1.26 g, 5.00 mmol) in THF (5 mL) under argon. The resulting mixture was stirred at room temperature for 10 min, and then cooled to −78 °C. A solution of S-2-pyridyl 4-methylbenzothioate (2, 1.15 g, 5.00 mmol) in THF (5 mL) was added. The solution was stirred at −78 °C for 10 min, then warmed to room temperature. The reaction mixture was quenched by addition of saturated aqueous NH4Cl (40 mL). The mixture was extracted with ethyl acetate (30 mL). The organic layer was washed (water and brine), dried (Na2SO4), and filtered. The filtrate was concentrated. The crude product (a red-orange oil) thus obtained was dissolved in CH2Cl2 (10 mL) and treated with TEA (1.68 mL, 12.0 mmol) followed by 9-BBN-OTf (20 mL, 10.0 mmol, 0.5 M in hexane) with stirring at room temperature. After 1 h, the mixture was poured onto a pad of silica (4 × 8 cm) eluting with CH2Cl2. The product eluted as a fast-moving yellow band, which upon concentration afforded a yellow-orange solid 1.78 g, 73%): mp 88 °C (dec.); 1H NMR (400 MHz, CDCl3) δ 0.67−0.71 (m, 2H), 1.62−1.83 (m, 6H), 1.95−2.22 (m, 6H), 2.48 (s, 3H), 3.79 (s, 3H), 5.85 (s, 1H), 5.96 (s, 1H), 6.13–6.17 (m, 1H), 6.40–6.43 (m, 1H), 6.69−6.85 (m, 1H), 6.85 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 8.0 Hz, 2H), 7.30–7.38 (m, 4H), 7.80–7.92 (br, 1H), 8.12 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 22.1, 23.9, 25.2, 26.0, 26.5, 30.7, 30.9, 34.7, 44.1, 55.5, 108.0, 108.6, 114.1, 117.4, 118.3, 120.8, 128.3, 129.6, 129.8, 130.0, 132.9, 134.3, 134.9, 145.0, 152.5, 158.7, 174.4. Integration indicated one additional proton in the 1H NMR spectrum (versus formula C32H35BN2O2), and the product was used for the next step without further purification.
10-(9-Borabicyclo[3.3.1]non-9-yl)-1-bromo-5-(4-methoxyphenyl)-9-(4-methylbenzoyl)dipyrromethane (5)
Following a general procedure [48], a solution of 4 (0.491 g, 1.00 mmol) in dry THF (10 mL) was cooled −78 °C under argon. NBS (178 mg, 1.00 mmol) was added and the reaction mixture was stirred for 1 h at −78 °C. Hexanes (10 mL) and water (10 mL) were added and the mixture was allowed to warm to room temperature. The organic phase was extracted with hexanes, dried (Na2SO4) and concentrated under reduced pressure without heating. Chromatography [silica, hexanes/CH2Cl2 (1:1)] afforded a yellow orange powder (0.439 g, 77%): mp 121 °C (dec.); 1H NMR (400 MHz, CDCl3) δ 0.64–0.72 (m, 2H), 1.64–1.88 (m, 6H), 1.94–2.24 (m, 6H), 2.48 (s, 3H), 3.80 (s, 3H), 5.85 (s, 1H), 5.72–5.77 (m, 1H), 5.90 (s, 1H), 6.05–6.08 (m, 1H), 6.40–6.43 (m, 1H), 6.86 (d, J = 7.2 Hz, 2H), 7.11 (d, J = 7.2 Hz, 2H), 7.32–7.39 (m, 4H), 7.70–7. 79 (br, 1H), 8.13 (d, J = 8.0 Hz, 2H). Integration indicated two additional protons in the 1H NMR spectrum (versus formula C32H34BBrN2O2), and the product was used for the next step without further purification.
Zn(II)-17,18-Dihydro-18,18-dimethyl-10-(4-methoxyphenyl)-5-(4-methylphenyl)porphyrin (ZnC-1)
A solution of 5 (0.285 g, 0.500 mmol) in THF/methanol [12.5 mL (4:1)] was treated with NaBH4 (0.190 g, 5.0 mmol). After 30 min (TLC indicated complete reaction), the reaction mixture was quenched with cold water and extracted with CH2Cl2. The organic layer was dried (K2CO3) and concentrated under reduced pressure without heating to afford the dipyrromethane-monocarbinol 5–OH. The foam-like residue was dissolved in CH3CN (5 mL) and Western half (6, 0.094 g, 0.50 mmol) was added followed by TFA (39 μL, 0.50 mmol). The reaction mixture was stirred at room temperature for 30 min, diluted with CH3CN (27 mL). Samples of AgOTf (0.385 g, 1.50 mmol), Zn(OAc)2 (1.38 g, 7.50 mmol), and 2,2,6,6-tetramethylpiperidine (2.53 mL,15.0 mmol) were added. The resulting mixture was refluxed for 21 h. The reaction mixture was concentrated under reduced pressure. The residue thus obtained was chromatographed [silica, CH2Cl2 /ethyl acetate (2:1)] to afford a purple-green solid (0.108 g, 36%): 1 H NMR (400 MHz, CDCl3) δ 2.02 (s, 6H), 2.66 (s, 3H), 4.04 (s, 3H), 4.49 (s, 2H), 7.19 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 7.91–8.00 (m, 4H), 8.37–8.42 (m, 2H), 8.55 (s, 1H), 8.58–8.70 (m, 5H); LD-MS obsd 598.5; FAB-MS obsd 598.1733, calcd 598.1705 (M+, M = C36H30N4OZn); λabs (THF) 412, 609 nm.
Zn(II)-17,18-Dihydro-18,18-dimethyl-10-(4-methoxyphenyl)-17-oxo-5-(4-methylphenyl)porphyrin (ZnC-2)
Following a general procedure [11], a mixture of ZnC-1 (0.060 g, 0.10 mmol) and basic alumina (activity I, 3.0 g) in toluene (4 mL) was stirred at room temperature for 7 h exposed to air. Solvent was removed under reduced pressure and the residue obtained was suspended in CH2Cl2/MeOH (19:1). The suspension was filtered and the filtered material was washed with a mixture of CH2Cl2/MeOH (19:1) until the washings were colorless. The filtrate was concentrated and the residue was dissolved in toluene (40 mL) and treated with DDQ (0.045 g, 0.20 mmol) for 5 min. The reaction was quenched with TEA and the solvent was evaporated. Chromatography [silica, CH2Cl2/MeOH (98:2)] afforded a green-purple solid (0.036 g, 59%): 1H NMR (400 MHz, CDCl3) δ 1.98 (s, 6H), 2.67 (s, 3H), 4.00 (s, 3H), 7.17 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.93–7.99 (m, 4H), 8.55–8.63 (m, 2H), 8.75–8.83 (m, 2H), 8.86–8.92 (m, 3H), 9.44 (s, 1H); LD-MS obsd 612.4; FAB-MS obsd 612.1478, calcd 612.1498 (M+, M = C36H28N4O2Zn); λabs (THF) 420, 608 nm.
Zn(II)-17,18-Dihydro-18,18-dimethyl-10-(4-hydroxyphenyl)-17-oxo-5-(4-methylphenyl)porphyrin (ZnC-3)
Following a literature procedure [50] with slight modification, a mixture of ZnC-2 (0.032 g, 0.050 mmol) and BBr3 (0.20 mL, 0.20 mmol, 1M in CH2Cl2) in CH2Cl2 (5 mL) was stirred at room temperature under argon. After 15 h, the reaction mixture was quenched with saturated aqueous NaHCO3. The organic layer was extracted with CH2Cl2 and dried (Na2SO4) and concentrated. The residue was dissolved in CH2Cl2/MeOH (4:1) and treated with Zn(OAc)2·2H2O (0.055 g, 0.25 mmol) at room temperature for 4 h. The mixture was washed with saturated aqueous NaHCO3, dried (Na2SO4) and concentrated. Chromatography [silica, CH2Cl2/MeOH (9:1)] afforded a green-purple solid (0.026 g, 87%): 1H NMR (400 MHz, CDCl3) δ 2.05 (s, 6H), 2.68 (s, 3H), 5.40–5.44 (br, 1H), 7.15 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.93–7.99 (m, 4H), 8.55–8.63 (m, 2H), 8.75–8.83 (m, 2H), 8.87–8.96 (m, 2H), 9.56 (s, 1H); LD-MS obsd 598.5; FAB-MS obsd 598.1371, calcd 598.1341 (M+, M = C35H26N4O2Zn); λabs (THF) 420, 608 nm.
Zn(II)-10-(4-Carboxyphenyl)-17,18-dihydro-10-mesityl-18,18-dimethyl-5-(4-methylphenyl)porphyrin (ZnC-5)
A solution of ZnC-4 (14.3 mg, 20.0 μmol) in dry DMF (2 mL) was treated with potassium fluoride (23.2 mg, 0.400 mmol) at 75 °C for 18 h. The reaction mixture was concentrated to dryness under reduced pressure. Chromatography [silica, CH2Cl2/methanol (20:1)] followed by filtration through a 0.65 μm filter (CH2Cl2, to remove silica) afforded a greenish purple solid (6.8 mg, 55%): 1H NMR (400 MHz, CDCl3) δ 2.03 (s, 6H), 2.66 (s, 3H), 4.52 (s, 2H), 7.46–7.51 (m, 2H), 7.93–7.97 (m, 2H), 8.17–8.22 (m, 2H), 8.31–8.34 (m, 1H), 8.39–8.45 (m, 3H), 8.58–8.70 (m, 6H); LD-MS obsd 612.5; FAB-MS obsd 612.1531, calcd 612.1498 (M+, M = C36H28N4O2Zn); λabs (CH2Cl2) 413, 608 nm.
Methyl 4-bromo-3,5-bis(diethylphosphonomethoxy)benzoate (7)
A mixture of methyl 4-bromo-3,5-dihydroxybenzoate (I, 741 mg, 3.00 mmol) and K2CO3 (1.65 g, 12.0 mmol) in CH3CN (10 mL) was stirred at room temperature for 30 min. A sample of TfOCH2PO(OEt)2 (II, 2.10 g, 7.00 mmol) was added, and the mixture was refluxed for 4 h. The reaction mixture was allowed to cool to room temperature. The reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, washed (water, brine), dried (Na2SO4) and concentrated. The resulting solid was chromatographed [silica, hexanes/ethyl acetate (1:1) → ethyl acetate → ethyl acetate/MeOH (98:2)] to give a white solid (1.52 g, 92%): mp 62–63 °C; 1H NMR (400 MHz, CDCl3) δ 1.37 (t, J = 7.2 Hz, 12H), 3.92 (s, 3H), 4.24–4.33 (m, 8H), 4.38 (d, J = 10.4 Hz, 4H), 7.32 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 16.5 (d, J = 6.1 Hz), 52.6 (d, J = 3.1 Hz), 63.3 (d, J = 6.1 Hz), 63.5 (d, J = 169.1 Hz, P-C coupling), 107.3, 107.8, 130.3, 156.8, 165.7; ESI-MS obsd 547.0502, calcd 547.0492 [(M + H)+, M = C18H29BrO10P2]. Anal. Calcd for C18H29BrO10P2: C, 39.50; H, 5.34. Found: C, 39.74; H, 5.46.
Methyl 4-bromo-3,5-bis[3-(diethylphosphono)propoxy]benzoate (8)
A mixture of I (1.23 g, 5.00 mmol), K2CO3 (1.38 g, 10.0 mmol) and KI (830 mg, 5.00 mmol) in CH3CN (10 mL) was stirred at room temperature for 30 min. A sample of diethyl (3-bromopropyl)phosphonate (III, 2.59 g, 10.0 mmol) was added, and the mixture was refluxed for 15 h. The reaction mixture was allowed to cool to room temperature. The reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, washed (water, brine), dried (Na2SO4) and concentrated. The resulting solid was chromatographed [silica, hexanes/ethyl acetate (1:1) → ethyl acetate → ethyl acetate/MeOH (97:3)] to give a viscous yellow oil (2.78 g, 92%): 1H NMR (400 MHz, DMSO-d6) δ 1.21 (t, J = 7.2 Hz, 12H), 1.84–2.02 (m, 8H), 3.86 (s, 3H), 3.92–4.07 (m, 8H), 4.15 (t, J = 5.4 Hz, 4H), 7.17 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 16.2 (d, J = 6.1 Hz), 21.0 (d, J = 140.3 Hz, P-C coupling), 22.2, (d, J = 4.6 Hz), 52.4, 60.9 (d, J = 6.1 Hz), 68.5 (d, J = 17.4 Hz), 106.1, 106.5, 130.0, 155.8, 165.5; ESI-MS obsd 603.1115, calcd 603.1118 [(M + H)+, M = C22H37BrO10P2].
4,4,5,5-Tetramethyl-2-(2,6-dimethoxyphenyl)-1,3,2-dioxaborolane
A solution of 2,6-dimethoxyphenylboronic acid (909 mg, 4.99 mmol) in toluene (25 mL) was treated with pinacol (2.95 g, 25.0 mmol) and refluxed with continuous azeotropic removal of water with Dean-Stark trap for 15 h. The reaction mixture was allowed to cool to room temperature and washed with water (5 × 25 mL). The organic layer was separated, dried (Na2SO4) and concentrated. The resulting residue was dried under high vacuum to obtain a white solid (1.30 g, 98%): mp 78 °C; 1H NMR (300 MHz, CDCl3) δ 1.19 (s, 12H), 3.78 (s, 6H), 6.46 (d, J = 8.4 Hz, 2H), 7.23 (t, J = 8.4 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 24.8, 55.7, 83.8, 103.3, 131.4, 163.5; GC-MS obsd 264, calcd 264 (M+, M = C14H21BO4). Anal. Calcd for C14H21BO4: C, 63.66; H, 8.01. Found: C, 63.87; H, 8.03.
17,18-Dihydro-15-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-10)
Following a procedure for Pd-mediated borylation [58], a mixture of C-9 (36.8 mg, 0.0722 mmol) and (PPh3)2PdCl2 (5.0 mg, 0.0072 mmol, 10 mol%) was dried in a Schlenk flask for 1 h. 1,2-Dichloroethane (8.5 mL), 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (89 μL, 0.61 mmol) and TEA (100 μL, 0.722 mmol) were added. The reaction mixture was heated at 90 °C for 3 h. The reaction mixture was allowed to cool to room temperature. The reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, washed (water, brine), dried (Na2SO4) and concentrated. The resulting solid was chromatographed [silica, hexanes → hexanes/CH2Cl2 (1:3)] to afford a purple solid (35.5 mg, 88%): 1H NMR (400 MHz, CDCl3) δ −1.67 (brs, 2H), 1.73 (s, 12H), 2.04 (s, 6H), 2.69 (s, 3H), 4.79 (s, 2H), 7.52 (d, J = 8.0 Hz, 2H), 8.00 (d, J = 8.0 Hz, 2H), 8.55 (d, J = 4.4 Hz, 1H), 8.78 (d, J = 4.4 Hz, 1H), 8.84 (s, 1H), 8.87 (d, J = 4.4 Hz, 1H), 8.89 (d, J = 4.4 Hz, 1H), 9.16 (d, J = 4.4 Hz, 1H), 9.34 (d, J = 4.4 Hz, 1H), 9.71 (s, 1H); LD-MS obsd 556.6; calcd 556.3 (M+, M = C35H37BN4O2).
15-[2,6-Bis(diethylphosphonomethoxy)-4-(methoxycarbonyl)phenyl]-17,18-dihydro-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-11)
Following a reported procedure for Pd-mediated Suzuki-coupling at the 15-position of chlorins [12], samples of C-10 (20.8 mg, 0.0373 mmol), 7 (20.4 mg, 0.0373 mmol), (Pd(PPh3)4 (12.9 mg, 0.0112 mmol) and K2CO3 (41.2 mg, 0.298 mmol) were placed in a Schlenk flask. The flask was pumped-purged with argon three times. A degassed sample of DMF was added. The resulting mixture was stirred at 90 °C for 18 h. The reaction mixture was allowed to cool to room temperature. The reaction mixture was diluted with CH2Cl2 and filtered. The filtrate was concentrated. The resulting crude product was dissolved in CH2Cl2 and chromatographed [silica, hexanes/CH2Cl2 (1:1) → CH2Cl2 → CH2Cl2/MeOH (98:2)] to afford deborylated chlorin (C-8, 14.4 mg, 90%) followed by a trace amount of the title compound admixed with de-brominated 7, which could not be purified. The presence of the product was strongly suggested by ESI-MS: obsd 897.3512, calcd 897.3388 [(M + H)+, M = C47H54N4O10P2].
15-[2,6-Bis[3-(diethylphosphono)propoxy]-4-(methoxycarbonyl)phenyl]-17,18-dihydro-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-12)
Following a reported procedure for Pd-mediated Suzuki-coupling [12], samples of C-10 (7.1 mg, 0.012 mmol), Pd(PPh3)4 (4.4 mg, 0.0038 mmol) and K2CO3 (14 mg, 0.10 mmol) were placed in a Schlenk flask. The flask was pumped-purged with argon three times. A solution of 8 (7.6 mg, 0.012 mmol) in toluene/DMF (1 mL, 2:1) was added. The resulting mixture was stirred at 90 °C for 18 h. The reaction mixture was allowed to cool to room temperature. The reaction mixture was diluted with CH2Cl2 and filtered. The filtrate was concentrated. The crude product was dissolved in CH2Cl2 and chromatographed [silica, hexanes/CH2Cl2 (1:1) → CH2Cl2 → CH2Cl2/MeOH (98:2)] to afford the deborylated chlorin (C-8, 4.8 mg, 87%) followed by a trace amount of the title compound admixed with de-brominated 8, which could not be purified. The presence of the product was strongly suggested by ESI-MS: obsd 953.4008, calcd 953.4013 [(M + H)+, M = C51H62N4O10P2].
15-Bromo-10-[2,6-bis(diethylphosphonomethoxy)phenyl]-17,18-dihydro-18,18-dimethylporphyrin (C-15)
Following a reported procedure for selective bromination of chlorin [14,17], a solution of chlorin C-14 (30. mg, 0.040 mmol) in anhydrous THF (17 mL) was treated with NBS (7.2 mg, 0.040 mmol). The reaction mixture was stirred for 2 h at room temperature. The mixture was diluted with CH2Cl2 (50 mL), washed with (saturated aqueous NaHCO3, H2O, brine). The organic phase was dried (Na2SO4) and filtered. The filtrate was concentrated to obtain a blue solid. Column chromatography [silica, CH2Cl2/MeOH (0 → 5%)] afforded a blue solid (32 mg, 96%): 1H NMR (400 MHz, CDCl3) δ −2.18 (brs, 1H), −2.11 (brs, 1H), −0.20 (m, 12H), 2.08 (s, 6H), 2.5 (m, 8H), 4.15 (s, 2H), 4.18 (s, 2H), 4.70 (s, 2H), 7.07 (d, J = 8.4 Hz, 2H), 7.75 (t, J = 8.4 Hz, 1H), 8.49 (d, J = 4.4 Hz, 1H), 8.68 (d, J = 5.2 Hz, 1H), 8.81 (d, J = 4.4 Hz, 1H), 8.88 (s, 1H), 8.95 (d, J = 4.4 Hz, 1H), 9.18 (m, 2H), 9.68 (s, 1H); ESI-MS obsd 827.1966, calcd 827.1969 [(M + H)+, M = C38H45BrN4O8P2]; λabs (CH2Cl2) 361, 402, 503, 643 nm.
15-[4-(Ethoxycarbonylmethoxy)phenyl]-10-[2,6-bis(diethylphosphonomethoxy)phenyl]-17,18-dihydro-18,18-dimethylporphyrin (C-16)
A mixture of chlorin C-15 (25 mg, 0.030 mmol), Pd(PPh3)4 (5.00 mg, 0.00433 mmol), anhydrous K2CO3 (8.5 mg, 0.060 mmol) and IV (14 mg, 0.045 mmol) was dried in a Schlenk flask for 1 h. Toluene (6 mL) and DMF (3 mL) were degassed for 10 min and then added to Schlenk flask under an inert atmosphere. The mixture was heated to 90 °C for 24 h. After cooling to room temperature, the suspension was diluted with ethyl acetate and filtered over Celite. The flask and the filter cake were rinsed with ethyl acetate until the filtrate was colorless. The filtrate was washed (H2O, brine), dried (Na2SO4) and filtered. The filtrate was concentrated. The resulting residue was subjected to column chromatography [silica, ethyl acetate/MeOH (0 → 5%)] to afford a green solid (14 mg, 50%): 1H NMR (400 MHz, CD3OD) δ −0.31 (m, 12H), 1.39 (t, J = 6.8 Hz, 3H), 2.01 (s, 6H), 2.31 – 2.55 (m, 8H), 4.24 (s, 2H), 4.29 (s, 2H), 4.32 (s, 2H), 4.37 (q, J = 6.8 Hz, 2H), 4.63 (s, 2H), 7.23 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.80 (d, J = 8.8 Hz, 2H), 7.83 (t, J = 8.4 Hz, 1H), 8.29 (d, J = 4.8 Hz, 1H), 8.49 (d, J = 4.4 Hz, 1H), 8.61 (d, J = 4.8 Hz, 1H), 8.94 (d, J = 4.0 Hz, 1H), 9.09 (d, J = 4.8 Hz, 1H), 9.10 (s, 1H), 9.34 (d, J = 4.4 Hz, 1H), 9.86 (s, 1H) (two protons were not observed); ESI-MS obsd 926.3416, calcd 926.3421 [(M + H)+, M = C48H56N4O11P2]; λabs (CH2Cl2) 361, 409, 502, 590, 642 nm.
15-(4-(Carboxymethoxy)phenyl-10-[2,6-bis(phosphonomethoxy)phenyl]-17,18-dihydro-18,18-dimethylporphyrin (C-17)
A solution of chlorin C-16 (14 mg, 0.015 mmol) in THF/H2O (5 mL, 4:1) was treated with aqueous 2M KOH (100 μL, 0.200 mmol). The mixture was stirred for 24 h at room temperature. Then, THF was removed under vacuum. The residue was partitioned between water (5 mL) and CH2Cl2 (5 mL). The aqueous layer was freeze-dried to obtain the potassium salt of the chlorin. The solid was dissolved in CH2Cl2 (5 mL) and few drops of acetic acid were added. The mixture was filtered. The filtrate was concentrated to dryness. The resulting green solid was dried under high vacuum. The solid was dissolved in CH2Cl2 (3 mL), whereupon TMSBr (140 μL, excess) was added. The mixture was stirred for 4 h at room temperature. The solvent and excess TMSBr were removed in vacuo, then methanol/water (5 mL, 4:1) was added. The mixture was stirred for 1 h. The solvent was removed under high vacuum, and the resulting green solid was dried under high vacuum. The residue was triturated with CH2Cl2 (3 × 10 mL) and dried to obtain a green solid (12 mg, quantitative): 1H NMR (400 MHz, CD3OD) δ 2.01 (s, 6H), 4.13 (s, 2H), 4.15 (s, 2H), 5.49 (s, 2H), 7.33 (m, 4H), 7.91 (m, 3H), 8.71 (m, 1H), 8.85 (d, J = 4.8 Hz, 1H), 9.25 (d, J = 4.4 Hz, 1H), 9.36 (d, J = 4.4 Hz, 1H), 9.51 (s, 1H), 9.65 (d, J = 4.4 Hz, 1H), 10.45 (s, 1H), the integration was deficient by 10 protons, of which 7 are exchangeable; ESI-MS obsd 785.1813, calcd 785.1783 [(M – H)−, M = C38H36N4O11P2]; λabs (H2O) 405, 503, 583, 634 nm.
1,3,3-Trimethyl-7-(4-iodophenyl)-2,3,4,5-tetrahydrodipyrrin N10-oxide (11)
A solution of 10 (0.500 g, 1.14 mmol) in acetic acid (10 mL) was treated with zinc dust (1.74 g, 26.5 mmol) all-at-once and the mixture was stirred vigorously for 1 h at room temperature. The crude reaction mixture was filtered (Celite) and concentrated. The residue was dissolved in CH2Cl2, washed (10% aq Na2CO3), dried (Na2SO4) and evaporated to afford a yellow solid. Recrystallization from ethyl acetate gave a white powder (200 mg, 43% yield): mp 173–174 °C; 1H NMR (300 MHz, CDCl3) δ 0.98 (s, 3H), 1.17 (s, 3H), 2.10 (s, 3H), 2.36 (d, J = 17.4 Hz, 1H), 2.52 (d, J = 17.4 Hz, 1H), 3.07 (m, 2H), 3.96 (m, 1H), 6.20 (m, 1H), 6.74 (m, 1H), 7.08 (d, J = 7.8 Hz, 2H), 7.67 (d, J = 7.8 Hz, 2H), 11.31 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 13.2, 22.7, 23.5, 26.8, 37.8, 46.8, 81.0, 90.1, 107.8, 117.0, 120.6, 125.7, 130.2, 137.0, 137.3, 145.5. Anal. Calcd. for C18H21N2IO: C, 52.95; H, 5.18; N, 6.86. Found: C, 52.91; H, 5.23; N, 6.78.
1,3,3-Trimethyl-7-(4-iodophenyl)-2,3,4,5-tetrahydrodipyrrin (12)
Following a procedure for the deoxygenation of N-oxides [10], TiCl4 (1.0 mL, 8.8 mmol) was slowly added to a stirred solution of dry THF (20 mL) under argon at 0 °C. To the resulting yellow solution, LiAlH4 (220 mg, 5.9 mmol) was added slowly and the black mixture was stirred at room temperature for 15 min, then TEA (7.7 mL) was added. The black mixture was then poured into a solution of 11 (500 mg, 1.22 mmol) in THF (20 mL). The mixture was stirred for 2 h at room temperature and then water (20 mL) was added. The mixture was filtered to remove inorganic materials. The filtrate was extracted (CH2Cl2). The organic layer was dried (Na2SO4) and evaporated under reduced pressure. The resulting brown oil was purified by chromatography (silica, ethyl acetate) to give a light brown oil which solidified on cooling (180 mg, 38%): mp 90–91 °C; 1H NMR (300 MHz, CDCl3) δ 0.92 (s, 3H), 1.08 (s, 3H), 2.05 (s, 3H), 2.34 (m, 2H), 2.59 (m, 1H), 2.92 (m, 1H), 3.64 (d, J = 11.7 Hz, 1H), 6.25 (m, 1H), 6.73 (m, 1H), 7.13 (d, J = 6.6 Hz, 2H), 7.66 (d, J = 6.6 Hz, 2H), 10.30 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 21.2, 23.5, 27.0, 27.8, 42.6, 54.9, 80.5, 90.4, 108.4, 117.0, 120.3, 128.9, 130.6, 137.8, 137.9, 175.3; FAB-MS obsd 393.0841, calcd 393.0743 (M+, M = C18H21IN2).
1,3,3-Trimethyl-7-[4-[2-(trimethylsilyl)ethynyl]phenyl]-2,3,4,5-tetrahydrodipyrrin (13)
Samples of 12 (450 mg, 1.15 mmol), Pd2(dba)3 (81 mg, 0.090 mmol), Ph3As (220 mg, 0.72 mmol), and CuI (17 mg, 0.090 mmol) were added to a 50 mL Schlenk flask. The flask was evacuated and purged with argon three times. Then deaerated anhydrous THF/TEA (12 mL, 1:1) was added followed by (trimethylsilyl)acetylene (243 μL, 1.72 mmol). The flask was sealed, immersed in an oil bath (37 °C), and the mixture was stirred overnight. Then CH2Cl2 (30 mL) was added and the mixture was filtered (Celite) and washed (CH2Cl2). The filtrate was concentrated. The resulting residue was purified by flash chromatography (silica, ethyl acetate) to afford a dark brown viscous oil (316 mg, 76%): 1H NMR (300 MHz, CDCl3) δ 0.25 (s, 9H), 0.92 (s, 3H), 1.07 (s, 3H), 2.05 (s, 3H), 2.34 (m, 2H), 2.61 (m, 1H), 2.95 (m, 1H), 3.65 (d, J = 11.7 Hz, 1H), 6.29 (m, 1H), 6.74 (m, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 10.27 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 0.7, 21.1, 23.5, 27.1, 27.7, 42.6, 54.8, 80.5, 94.1, 106.3, 108.4, 117.0, 119.8, 120.9, 128.3, 129.3, 132.7, 138.7, 175.4; FAB-MS obsd 363.2271, calcd 363.2172 (M+, M = C23H30N2Si).
3-(4-Iodophenyl)-9-(3,5-di-tert-butylbenzoyl)-10-N-(tert-butoxycarbonyl)-dipyrromethane (15)
A solution of 14 (0.90 g, 2.0 mmol) in anhydrous THF (30 mL) under argon at 0 °C was treated slowly with EtMgBr (1M in THF, 6 mL, 6.0 mmol). The mixture was stirred for 10 min at 0 °C. Then a solution of 3,5-di-tert-butylbenzoyl chloride (760 mg, 3.0 mmol) in anhydrous THF (5 mL) was added slowly and stirring was continued for 1.5 h at 0 °C. The reaction was quenched with saturated aqueous NH4Cl and extracted (CH2Cl2). The combined organic layers were washed with water and brine, dried (Na2SO4), and concentrated. The product was purified by flash column chromatography [silica, hexanes/ethyl acetate (4:1)] to yield a brown viscous oil which was cooled overnight in the refrigerator. A minimum amount of hexanes was added followed by sonication, affording a white solid (598 mg, 45%): mp 178–179 °C; 1H NMR (300 MHz, CDCl3) δ 1.35 (s, 18H), 1.58 (s, 9H), 4.30 (s, 2H), 5.97 (m, 1H), 6.26 (m, 1H), 6.73 (m, 1H), 7.11 (d, J = 6.6 Hz, 2H), 7.30 (m, 1H), 7.60 (m, 1H), 7.70 (m, 2H), 7.72 (m, 2H), 9.92 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 25.8, 28.5, 32.0, 35.6, 85.5, 93.0, 110.1, 112.3, 120.4, 122.1, 123.9, 126.3, 127.1, 128.2, 131.2, 131.3, 135.4, 138.2, 138.6, 139.2, 150.5, 151.3, 185.7. Anal. Calcd for C35H41IN2O3: C, 63.25; H, 6.22; N, 4.21. Found: C, 64.03; H, 6.35; N, 4.15.
3-(4-Iodophenyl)-9-(3,5-di-tert-butylbenzoyl)dipyrromethane (16)
A solution of 15 (0.60 g, 0.90 mmol) in anhydrous THF (10 mL) under argon at room temperature was treated with methanolic NaOMe (146 mg, 2.70 mmol, in 1.0 mL of anhydrous methanol). After 10 min, the reaction was quenched with a mixture of hexanes and water (20 mL, 1:1) and extracted with ethyl acetate. The combined organic layers were washed with water and brine, dried (Na2SO4), and concentrated. The residue was recrystallized from ethanol to give a pale brown solid (430 mg, 84%): mp 202–203 °C; 1H NMR (300 MHz, CDCl3) δ 1.30 (s, 18H), 4.18 (s, 2H), 6.23 (m, 2H), 6.48 (m, 1H), 6.93 (m, 1H), 7.25 (d, J = 8.1 Hz, 2H), 7.68 (m, 1H), 7.75 (m, 4H), 10.23 (br, 1H), 11.85 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 25.6, 31.9, 35.6, 91.1, 109.0, 111.2, 118.1, 121.6, 124.0, 124.2, 124.8, 127.0, 130.9, 131.5, 137.3, 138.0, 138.2, 141.5, 151.8, 187.5. Anal. Calcd for C30H33IN2O: C, 63.83; H, 5.89; N, 4.96. Found: C, 63.74; H, 6.09; N, 5.00.
3-[4-(2-(Trimethylsilyl)ethynyl)phenyl]-9-(3,5-di-tert-butylbenzoyl)dipyrromethane (17)
Samples of 16 (1.0 g, 1.8 mmol), Pd2(dba)3 (125 mg, 0.140 mmol), Ph3As (334 mg, 1.09 mmol), and CuI (26 mg, 0.14 mmol) were added to a 50 mL Schlenk flask. The flask was evacuated and purged with argon three times. Then deaerated anhydrous THF/TEA (18 mL, 1:1) was added followed by (trimethylsilyl)acetylene (376 μL, 2.66 mmol). The flask was sealed, immersed in an oil bath (37 °C), and the mixture was stirred overnight (16 – 18 h). Then CH2Cl2 (30 mL) was added and the mixture was filtered (Celite) and washed (CH2Cl2). The filtrate was concentrated. The resulting residue was purified by flash chromatography [silica, hexanes/ethyl acetate (3:1)] to afford a yellow oil which solidified on cooling (780 mg, 82%): mp 126–127 °C; 1H NMR (300 MHz, CDCl3) δ 0.26 (s, 9H), 1.27 (s, 18H), 4.20 (s, 2H), 6.20 (m, 2H), 6.44 (m, 1H), 6.87 (m, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.64 (m, 1H), 7.72 (m, 2H), 10.14 (br, 1H), 11.73 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 0.7, 25.7, 31.9, 35.5, 94.4, 106.2, 109.1, 111.1, 118.1, 120.4, 122.0, 123.9, 124.1, 124.9, 126.9, 128.5, 131.5, 132.7, 138.1, 138.2, 141.3, 151.7, 187.45. Anal. Calcd for C35H42N2OSi: C, 78.60; H, 7.92; N, 5.24. Found: C, 78.09; H, 8.03; N, 5.13.
1-Bromo-3-[4-(2-(trimethylsilyl)ethynyl)phenyl]-9-(3,5-di-tert-butylbenzoyl)-dipyrromethane (18)
A solution of 17 (100 mg, 0.19 mmol) in anhydrous THF (6 mL) was cooled to −78 °C under argon. NBS (33 mg, 0.19 mmol) was added and the reaction mixture was stirred for 1 h (−78 °C), then the mixture was quenched with a mixture of hexanes and water (20 mL, 1:1) and allowed to warm to 0 °C. The aqueous portion was extracted with anhydrous ether and the combined organic layers were dried (K2CO3). The solvent was evaporated under vacuum without heating. Purification by flash chromatography [silica, hexanes/ether (2:1)] afforded a pale yellow solid (83 mg, 72%): mp 163–165 °C (dec.); 1H NMR (300 MHz, CDCl3) δ 0.27 (s, 9H), 1.30 (s, 18H), 4.20 (s, 2H), 6.08 (m, 1H), 6.23 (m, 1H), 6.94 (m, 1H), 7.36 (d, J = 7.8 Hz, 2H), 7.52 (d, J = 7.8 Hz, 2H), 7.66 (s, 1H), 7.74 (s, 2H), 10.75 (br, 1H), 12.14 (br, 1H); 13C NMR (75 MHz, CDCl3) δ 0.7, 24.8, 31.2, 34.9, 94.0, 97.9, 105.2, 110.0, 110.7, 120.3, 123.1, 123.7, 123.8, 125.5, 126.5, 127.9, 130.8, 132.0, 136.3, 137.3, 140.5, 151.1, 187.3; FAB-MS obsd 612.2184, calcd 612.2166 (M+, M = C35H41BrN2OSi). Anal. Calcd for C35H41BrN2OSi: C, 68.50; H, 6.73; N, 4.56. Found: C, 68.06; H, 6.64; N, 4.49.
Zn(II)-17,18-Dihydro-18,18-dimethyl-2-(4-iodophenyl)-5-(3,5-di-tert-butylbenzoyl)-12-[4-(2-(trimethylsilyl)ethynyl)phenyl]porphyrin (ZnC-21)
Following the two-flask procedure [10], to a solution of 18 (123 mg, 0.200 mmol) in anhydrous THF/methanol (7.5 mL, 4:1) was added excess NaBH4 (100 mg, 2.60 mmol) in small portions at room temperature. The reaction was monitored by TLC [alumina, hexanes/ethyl acetate (1:1)]. Upon completion, the reaction was quenched with cold water (10 mL), and then the mixture was extracted with distilled CH2Cl2 (3 × 25 mL). The combined organic extract was washed with brine (50 mL), dried (K2CO3) and concentrated in vacuo without heating to leave the resulting dipyrromethane-monocarbinol 18-OH in ~1−2 mL of CH2Cl2. A solution of 12 (78 mg, 0.20 mmol) in a few milliliters of anhydrous CH3CN was combined with the carbinol, then additional anhydrous CH3CN was added to give a total of 20 mL of CH3CN. The solution was stirred at room temperature under argon and TFA (20 μL, 0.26 mmol) was added. The reaction was monitored by TLC [alumina, hexanes/ethyl acetate (3:1)]; after 1 h 18-OH had disappeared. The reaction mixture was quenched with 10% aqueous NaHCO3 and extracted with distilled CH2Cl2 (3 × 25 mL). The combined organic layers were washed with water and brine, then dried (Na2SO4). The solvent was removed in vacuo at room temperature. The residue was dissolved in 14 mL of anhydrous toluene under argon, then AgIO3 (848 mg, 3.00 mmol), piperidine (300. μL, 3.00 mmol) and Zn(OAc)2 (550 mg, 3.00 mmol) were added. The resulting mixture was heated at 80 °C for 3 h. The reaction was monitored by TLC [silica, hexanes/CH2Cl2 (1:1)], which showed a single green spot. The mixture was cooled to room temperature, then passed through a short column (silica, CH2Cl2). The major fraction was concentrated and again chromatographed [silica, hexanes/CH2Cl2 (2:1 then 1:1)] to give a greenish blue solid (15 mg, 8%): 1H NMR (300 MHz, CDCl3) δ 0.35 (s, 9H), 1.51 (s, 18H), 1.97 (s, 6H), 4.51 (s, 2H), 7.42 (s, 1H), 7.82 (d, J = 9.0 Hz, 2H), 7.87 (d, J = 7.8 Hz, 2H), 7.96 (m, 2H), 8.03 (d, J = 8.1 Hz, 2H), 8.14 (d, J = 8.1 Hz, 2H), 8.56 (d, J = 4.5 Hz, 1H), 8.66 (m, 2H), 8.72 (m, 2H), 8.81 (d, J = 4.5 Hz, 1H), 9.61 (s, 1H); LD-MS obsd 964.60; FAB-MS obsd 964.2382, calcd 964.2370 (M+, M = C53H53IN4SiZn); λabs (toluene)/nm 417 (log ε = 5.30, fwhm = 20 nm), 629 (4.86); λem 634, 691 nm.
Zn(II)-3-Bromo-17,18-dihydro-10-mesityl-18,18-dimethylporphyrin (ZnC-22)
Following a streamlined procedure [17], a solution of Western half 19 (153 mg, 0.569 mmol) and Eastern half 20 (210 mg, 0.569 mmol) in distilled CH2Cl2 was treated with a solution of p-TsOH·H2O (541 mg, 2.85 mmol) in anhydrous methanol (6 mL) under argon. The resulting red reaction mixture was stirred at room temperature for 40 min. A sample of 2,2,6,6-tetramethylpiperidine (1.2 mL, 7.11 mmol) was added. The reaction mixture was concentrated. The resulting solid was dissolved in CH3CN (56.9 mL) and subsequently treated with 2,2,6,6-tetramethylpiperidine (2.4 mL, 14 mmol), Zn(OAc)2 (1.62 g, 8.81 mmol), and AgOTf (446 mg, 1.74 mmol). The resulting suspension was refluxed for 14 h exposed to air. The crude mixture was filtered through a pad of silica (CH2Cl2). The filtrate was chromatographed [silica, hexanes/CH2Cl2 (2:1 → 1:1 → 1:2)] to afford a green solid (119 mg, 34%): 1H NMR (300 MHz, CDCl3) δ 1.84 (s, 6H), 2.01 (s, 6H), 2.59 (s, 3H), 4.50 (s, 2H), 7.22 (s, 2H), 8.30 (d, J = 3.4 Hz, 1H), 8.49 (s, 1H), 8.53 (d, J = 3.9 Hz, 1H), 8.60 (d, J = 4.5 Hz, 1H), 8.67 (s, 1H), 8.78 (s, 1H), 8.87 (d, J = 3.9 Hz, 1H), 9.73 (s, 1H); LD-MS obsd 599.8; ESI-MS obsd 598.0710, calcd 598.0705 (M+, M = C31H27BrN4Zn); λabs 410, 614 nm; λem (λex = 410 nm) 617, 647 nm.
Zn(II)-17,18-Dihydro-3-(N,N-dimethylamino)-10-mesityl-18,18-dimethylporphyrin (ZnC-23)
Following a procedure for amination of aryl bromides [72] with slight modification, a mixture of ZnC-22 (10.0 mg, 0.017 mmol), Pd(OAc)2 (1.5 mg, 0.0068 mmol, 40 mol %), (t-Bu)3PHBF4 (1.9 mg, 0.0068 mmol, 40 mol %), and sodium tert-butoxide (4.9 mg, 0.051 mmol, 3 equiv) were dried in a Schlenk flask for 1 h. Degassed toluene (1.6 mL) and dimethylamine (85 μL, 2.0 M in toluene, 0.17 mmol, 10 equiv) were added, and the mixture was purged with argon. The reaction mixture was heated to reflux for 48 h. After cooling to room temperature, the reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, dried (Na2SO4), and concentrated. The resulting residue was chromatographed [silica, hexanes/CH2Cl2 (1:1 → 1:2)], followed by a second column (silica, CH2Cl2 with 1% TEA) to give a green solid (4.6 mg, 49%, 95% purity): 1H NMR (300 MHz, CDCl3) δ 1.87 (s, 6H), 1.95 (s, 6H), 2.58 (s, 3H), 3.61 (s, 6H), 4.36 (s, 2H), 7.19 (s, 2H), 7.69 (s, 1H), 8.18 (s, 1H), 8.22 (d, J = 4.1 Hz, 1H), 8.39 (m, 3H), 8.61 (d, J = 4.1 Hz, 1H), 9.49 (s, 1H); MALDI-MS obsd 563.1; ESI-MS obsd 564.2097, calcd 564.2100 [(M + H)+, M = C33H33N5Zn]; λabs (toluene) 410, 612 nm; λem (λex = 410 nm, toluene) 617, 664 nm.
Zn(II)-17,18-Dihydro-10-mesityl-18,18-dimethyl-3-methylthioporphyrin (ZnC-24)
Following a procedure for aryl thioether formation [73], a mixture of ZnC-22 (45.1 mg, 0.0750 mmol), Pd2(dba)3 (13.7 mg, 0.0150 mmol, 20 mol %), Xantphos (8.7 mg, 0.015 mmol, 20 mol %), and sodium thiomethoxide (15.8 mg, 0.225 mmol, 3 equiv relative to ZnC-22) was dried in a Schlenk flask for 1 h. Degassed xylene (1.5 mL) was added, and the mixture was purged with argon. The reaction mixture was heated to reflux for 24 h. After cooling to room temperature, the reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, dried (Na2SO4), and concentrated. The resulting residue was chromatographed (silica, CH2Cl2) to give a green solid (40.5 mg, 95%): 1H NMR (300 MHz, CDCl3) δ 1.86 (s, 6H), 2.02 (s, 6H), 2.59 (s, 3H), 2.64 (s, 3H), 4.47 (s, 2H), 7.21 (bs, 2H), 8.06 (s, 1H), 8.32 (d, J = 4.2 Hz, 1H), 8.36 (s, 1H), 8.51 (dd, J = 13.5 & 4.2 Hz, 2H), 8.58 (s, 1H), 8.72 (d, J = 4.2 Hz, 1H), 9.25 (s, 1H); MALDI-MS obsd 566.5; ESI-MS obsd 566.1472, calcd 566.1477 (M+, M = C32H30N4SZn); λabs (toluene) 410, 616 nm; λem (λex = 410 nm, toluene) 621, 666 nm.
Zn(II)-17,18-Dihydro-10-mesityl-3-methoxy-18,18-dimethylporphyrin (ZnC-25)
Modifying the procedure used to synthesize ZnC-24 [73], a mixture of ZnC-22 (10.0 mg, 0.017 mmol), Pd2(dba)3 (3.1 mg, 0.0034 mmol, 20 mol %), Xantphos (2.1 mg, 0.0041 mmol, 20 mol %), and sodium methoxide (2.7 mg, 0.051 mmol) was dried in a Schlenk flask for 1 h. Degassed xylene (1.6 mL) was added, and the mixture was purged with argon. The reaction mixture was heated to reflux for 24 h. After cooling to room temperature, the reaction mixture was treated with CH2Cl2 and water. The organic layer was separated, dried (Na2SO4), and concentrated. The resulting residue was chromatographed [silica, hexanes/CH2Cl2 (1:1→1:2)] to give a green solid (1.5 mg, 16%): 1H NMR (300 MHz, CDCl3) δ 1.85 (s, 6H), 1.98 (s, 6H), 2.58 (s, 3H), 4.41 (s, 2H), 4.53 (s, 3H), 7.19 (s, 2H), 7.74 (s, 1H), 8.26 (d, J = 3.9 Hz, 1H), 8.29 (s, 1H), 8.44 (d, J = 3.9 Hz, 1H), 8.46 (d, J = 3.9 Hz, 1H), 8.50 (s, 1H), 8.66 (d, J = 3.9 Hz, 1H), 9.51 (s, 1H); MALDI-MS obsd 551.0; ESI-MS obsd 550.1707, calcd 550.1706 (M+, M = C33H30N4OZn); λabs (toluene) 404, 603 nm; λem (λex = 410 nm, toluene) 606, 656 nm.
3,13-Dibromo-17,18-dihydro-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-29)
Following a reported procedure [13,21], a mixture of Eastern half 21 (0.284 g, 0.674 mmol) and Western half 19 (0.202 g, 0.750 mmol) in CH2Cl2 (20 mL) was treated with a solution of p-TsOH·H2O (0.640 g, 3.37 mmol) in MeOH (5 mL). The resulting mixture was stirred at room temperature for 30 min. A sample of 2,2,4,4-tetramethylpiperidine (3.21 mL, 18.9 mmol) was added, and the resulting mixture was concentrated. The resulting brown solid was suspended in acetonitrile (67 mL), and samples of Zn(OAc)2 (1.86 g, 10.1 mmol), 2,2,4,4-tetramethylpiperidine (3.25 mL, 19.1 mmol), and AgOTf (0.520 g, 2.00 mmol) were added. The resulting mixture was refluxed for 18 h. The mixture was concentrated and filtered through silica using hexanes/CH2Cl2 as an eluent. Fractions containing chlorin were collected, concentrated, dissolved in CH2Cl2 (20 mL), and treated with TFA (0.400 mL, 5.23 mmol). The resulting mixture was stirred for 4 h and quenched by the addition of saturated aqueous NaHCO3. The organic layer was separated, washed (water and brine), dried (Na2SO4), and concentrated. Column chromatography (silica, hexanes/CH2Cl2) afforded a green solid (47 mg, 12%): 1H NMR (400 MHz, CDCl3) δ −2.02 (bs, 1H), −1.64 (bs, 1H), 2.04 (s, 6H), 2.69 (s, 3H), 4.64 (s, 2H), 7.54 (d, J = 7.8 Hz, 2H), 7.99 (d, J = 7.8 Hz, 2H), 8.60 (d, J = 4.4 Hz, 1H), 8.76 (s, 1H), 8.83 (s, 1H), 8.94 (d, J = 4.4 Hz, 1H), 8.95 (s, 1H), 9.11 (s, 1H), 9.87 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.6, 31.1, 46.5, 52.0, 94.4, 95.3, 105.8, 113.4, 118.5, 122.1, 124.4, 127.9, 129.3, 132.3, 132.7, 132.9, 134.1, 134.6, 137.2, 137.8, 138.0, 140.1, 151.0, 153.2, 163.3, 175.7; ESI-MS obsd 587.0450, calcd 587.0440 [(M + H)+, M = C29H24N4Br2]. (The same reaction on a 2.5 mmol scale provided 11% yield.)
General procedures for Sonogashira reaction with chlorin C-29
Procedure A
Samples of C-29, the appropriate ethyne derivative, and Pd(PPh3)2Cl2 were placed in a Schlenk flask, whereupon solvent (DMF/TEA, 1:1) was added. The reaction mixture was degassed by freeze-pump-thaw (three cycles), after which the reaction flask was filled with nitrogen and placed in preheated oil bath. The reaction mixture was stirred at 80–85 °C with monitoring of the reaction by absorption spectroscopy and TLC [silica, hexanes/CH2Cl2 (2:1)]. Upon consumption of all starting material, the reaction mixture was allowed to cool to room temperature, diluted with ethyl acetate, washed (water, brine), dried (Na2SO4), and concentrated. Subsequent purification is described for each instantiation below.
Procedure B
Samples of C-29, the appropriate ethyne derivative, and tri-o-tolylphosphine were placed in a Schlenk flask, whereupon solvent (toluene/TEA, 5:1) was added. The reaction mixture was degassed by freeze-pump-thaw (two cycles), and then a sample of Pd2(dba)3 was added. The reaction mixture was degassed again by freeze-thaw-pump (three cycles), after which the reaction flask was filled with nitrogen and placed in a pre-heated oil bath. (When TMS-acetylene was used, the addition of TMS-acetylene was done after the final freeze-pump-thaw cycle given the low boiling point of this ethyne.) The reaction mixture was stirred at 60 °C with monitoring of the reaction progress by absorption spectroscopy and TLC. A significant amount of the starting material was typically present after ~4–5 h, whereupon another batch of Pd2(dba)3 and tri-o-tolylphosphine was added under nitrogen. Upon consumption of all starting material, the reaction mixture was allowed to cool to room temperature and concentrated. Subsequent purification is described for each instantiation below.
17,18-Dihydro-18,18-dimethyl-10-(4-methylphenyl)-3,13-bis(2-phenylethynyl)porphyrin (C-31)
Following Procedure A, a mixture of C-29 (24.0 mg, 40.8 μmol), phenylacetylene (20 μL, 200 μmol), and Pd(PPh3)2Cl2 (5.6 mg, 8.0 μmol) in DMF/TEA (4 mL) was allowed to react overnight. Final workup entailed column chromatography [silica, hexanes/CH2Cl2 2 (2:1)] to afford a purple-brown solid (23.4 mg, 91%): H NMR (400 MHz, CDCl3) δ −1.85 (bs, 1H), −1.43 (bs, 1H), 2.06 (s, 6H), 2.69 (s, 3H), 4.66 (s, 2H), 7.45–7.59 (m, 8H), 7.86–7.88 (m, 2H), 7.97–7.99 (m, 2H), 8.02 (d, J = 7.9 Hz, 2H), 8.64 (d, J = 4.3 Hz, 1H), 8.80 (s, 1H), 8.92 (s, 1H), 8.96 (d, J = 4.3 Hz, 1H), 9.02 (s, 1H), 9.29 (s, 1H), 10.03 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.6, 31.2, 46.5, 52.2, 84.3, 84.6, 94.8, 95.5, 96.8, 98.0, 105.4, 117.8, 122.4, 122.7, 123.5, 123.6, 125.0, 127.9, 128.7, 128.8, 129.1, 130.3, 132.0, 132.2, 132.9, 133.1, 134.1, 134.8, 135.4, 137.7, 138.3, 140.15, 140.22, 151.8, 153.7, 163.8, 176.0; LD-MS obsd 631.6, calcd 631.3 [(M + H)+, M = C45H34N4].
17,18-Dihydro-18,18-dimethyl-10-(4-methylphenyl)-3,13-bis[2-(4-(2-phenylethynyl)phenyl)ethynyl]porphyrin (C-32)
Following Procedure A, a mixture of C-29 (24.0 mg, 40.8 μmol), 4-ethynyl-1-(2-phenylethynyl)benzene (41 mg, 0.20 mmol), and Pd(PPh3)2Cl2 (5.6 mg, 8.0 μmol) in DMF/TEA (4 mL) was allowed to react overnight. Final workup entailed column chromatography [silica, hexanes/CH2Cl2 (2:1)] to afford a purple-brown solid (27.2 mg, 80%). The resulting product contained a trace amount of blue-fluorescent material (most probably a trace amount of 4-ethynyl-1-(2-phenylethynyl)benzene or homocoupled byproduct thereof). For fluorescence studies a sample of product (~ 20 mg) was dissolved in CHCl3/MeOH (6 mL, 5:1) and treated with Zn(OAc)2·2H2O (0.180 g, 0.820 mmol). The resulting mixture was stirred for 4 h, diluted with CH2Cl2, washed (aqueous NaHCO3 and brine) dried (Na2SO3), and concentrated. The resulting solid was purified by column chromatography [silica, hexanes/CH2Cl2 (1:1)] to afford a green solid. The resulting solid was dissolved in CH2Cl2 (20 mL) and treated with TFA (0.4 mL). The resulting mixture was stirred for 4 h, diluted with CH2Cl2, treated with aqueous NaHCO3 and subsequently stirred for 5 min. The organic layer was separated, washed (water and brine), dried (Na2SO4), and concentrated. Column chromatography [silica, hexanes/CH2Cl2 (2:1)] afforded spectroscopically pure product (10.5 mg): 1H NMR (400 MHz, CDCl3) δ −1.84 (bs, 1H), −1.40 (bs, 1H), 2.07 (s, 6H), 2.70 (s, 3H), 4.67 (s, 2H), 7.38–7.42 (m, 6H), 7.56 (d, J = 7.8 Hz, 2H), 7.59–7.62 (m, 4H), 7.66 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 8.4 Hz, 2H), 8.04 (d, J = 7.8 Hz, 2H), 8.64 (d, J = 4.3 Hz, 1H), 8.80 (s, 1H), 8.94 (s, 1H), 8.98 (d, J = 4.3 Hz, 1H), 9.01 (s, 1H), 9.27 (s, 1H), 9.99 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.7, 31.2, 46.6, 52.2, 86.2, 86.5, 89.3, 91.7, 91.8, 94.9, 95.6, 96.6, 97.7, 105.4, 117.5, 122.4, 122.6, 123.2, 123.4, 123.6, 123.9, 125.1, 127.9, 128.6, 128.7, 130.4, 131.8, 131.9, 132.0, 132.1, 133.0, 133.2, 134.2, 134.9, 135.4, 137.7, 138.2, 140.17, 140.20, 151.9, 153.8, 163.8, 176.0; ESI-MS obsd 831.3441, calcd 831.3482 [(M + H)+, M = C61H42N4].
17,18-Dihydro-3,13-bis[2-(4-(2-(4-methoxycarbonylphenyl)ethynyl)phenyl)ethynyl]-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-33)
Following Procedure A, a mixture of C-29 (24.0 mg, 40.8 μmol), methyl 4-[2-(4-ethynylphenyl)ethynyl]benzoate (52 mg, 0.20 mmol), and Pd(PPh3)2Cl2 (5.6 mg, 8.0 μmol) in DMF/TEA (4 mL) was allowed to react overnight. Final workup entailed column chromatography (silica, CH2Cl2) to afford a purple-brown solid (16.1 mg, 42%): 1H NMR (400 MHz, CDCl3) δ −1.84 (bs, 1H), −1.40 (bs, 1H), 2.07 (s, 6H), 2.70 (s, 3H), 3.96 (s, 6H), 4.67 (s, 2H), 7.66 (d, J = 7.8 Hz, 2H), 7.63–7.68 (three overlapped doublets, 6H), 7.71 (d, J = 8.3 Hz, 2H), 7.84 (d, J = 8.2 Hz, 2H), 7.95 (d, J = 8.2 Hz, 2H), 8.03 (d, J = 7.8 Hz, 2H), 8.05 (d, J = 8.3 Hz, 2H), 8.07 (d, J = 8.3 Hz, 2H), 8.64 (d, J = 4.4 Hz, 1H), 8.81 (s, 1H), 8.94 (s, 1H), 8.98 (d, J = 4.4 Hz, 1H), 9.02 (s, 1H), 9.27 (s, 1H), 9.99 (s, 1H); LD-MS obsd 947.5, calcd 947.4 [(M + H)+, M = C65H46N4O4].
17,18-Dihydro-3,13-bis[2-(4-methoxycarbonylphenyl)ethynyl]-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-34)
Following Procedure A, a mixture of C-29 (45.5 mg, 77.3 μmol), 4-methoxycarbonylphenylacetylene (37.1 mg, 232 μmol), and Pd(PPh3)2Cl2 (10.7 mg, 15.2 μmol) in DMF/TEA (4.9 mL) was allowed to react overnight. Final workup entailed column chromatography [silica, hexanesCH2Cl2 (1:1)] to afford a purple-brown solid (25.1 mg, 43%): 1H NMR (400 MHz, CDCl3) δ −1.83 (bs, 1H), −1.40 (bs, 1H), 2.07 (s, 6H), 2.70 (s, 3H), 3.99 (s, 6H), 4.67 (s, 2H), 7.56 (d, J = 7.8 Hz, 2H), 7.90 (d, J = 8.3 Hz, 2H), 8.00 (d, J = 8.3 Hz, 2H), 8.03 (d, J =7.8 Hz, 2H), 8.17 (d, J = 8.3 Hz, 2H), 8.20 (d, J = 8.3 Hz, 2H), 8.64 (d, J = 4.3 Hz, 1H), 8.81 (s, 1H), 8.95 (s, 1H), 8.98 (d, J = 4.3 Hz, 1H), 9.03 (s, 1H), 9.26 (s, 1H), 9.98 (s, 1H); ESI-MS obsd 747.2963, calcd 747.2965 [(M + H)+, M = C49H38N4O4].
17,18-Dihydro-3,13-bis[2-(4-formylphenyl)ethynyl]-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-35)
Following Procedure B, a mixture of C-29 (22.3 mg, 39.8 μmol), 4-formylphenylacetylene (12.7 mg, 99.5 μmol), Pd2(dba)3 (7.1 mg, 7.7 μmol), and P(o-tol)3 (15.4 mg, 50.6 μmol) in toluene/TEA (3.9 mL) was allowed to react overnight. TLC analysis showed complete consumption of starting material, and formation of one major product accompanied by a smaller amount of byproducts. Final workup entailed column chromatography (silica, CH2Cl2) to provide a semi-pure product, for which 1H NMR spectroscopy showed a small amount of impurities in the aromatic region (probably dba remnants, and/or a product of homocoupling of 4-formylacetylene). Further purification by column chromatography [silica, hexanes/CH2Cl2 (1:2), twice] afforded a pure product as a purple-brown solid, but with a significant loss in yield (4.4 mg, 16%; no effort has been made to optimize the reaction conditions to improve the yield): 1H NMR (400 MHz, CDCl3) δ −1.79 (bs, 1H), −1.36 (bs, 1H), 2.08 (s, 6H), 2.70 (s, 3H), 4.68 (s, 2H), 7.56 (d, J = 7.7 Hz, 2H), 7.98–8.12 (m, 10H), 8.64 (d, J = 4.3 Hz, 1H), 8.82 (s, 1H), 8.97 (s, 1H), 8.98 (d, overlapped with a singlet, 1H), 9.06 (s, 1H), 9.27 (s, 1H), 9.99 (s, 1H), 10.11 (s, 1H), 10.17 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.7, 31.2, 46.6, 52.2, 88.2, 88.8, 95.2, 95.6, 96.0, 97.0, 105.3, 116.9, 121.8, 122.9, 125.4, 125.7, 127.9, 128.4, 129.2, 129.6, 129.8, 129.98, 129.99, 130.00, 130.91, 132.4, 132.6, 133.3, 134.1, 134.9, 135.5, 135.7, 136.0, 137.9, 138.0, 140.2, 148.0, 152.1, 153.9, 163.9, 176.2, 191.6 (only one signal from the formyl carbon is visible; due to the low quantity of sample, the 13C NMR spectrum shows a low signal-to-noise ratio, in which case identification of several low-intensity signals in aromatic region was ambiguous); LD-MS obsd 686.4, calcd 686.3 (M+, M = C47H34N4O2).
17,18-Dihydro-18,18-dimethyl-10-(4-methylphenyl)-3,13-bis[2-(4-(N,N-dimethylamino)phenyl)ethynyl]porphyrin (C-36)
Following Procedure B, a mixture of C-29 (20.0 mg, 34.0 μmol), 4-(N,N-dimethylamino)phenylacetylene (14.8 mg, 102 μmol), Pd2(dba)3 (6.2 mg, 6.7 μmol), and P(o-tol)3 (13.4 mg, 44.1 μmol) in toluene/TEA (6.8 mL) was allowed to react overnight. Final workup entailed column chromatography (silica, CH2Cl2) to provide a semi-pure product, which was washed with MeOH (with sonication, twice) to afford a purple solid (15.0 mg, 61%): 1H NMR (400 MHz, CDCl3) δ −1.87 (bs, 1H), −1.44 (bs, 1H), 2.05 (s, 6H), 2.86 (s, 3H), 3.08 (s, 6H), 3.11 (s, 6H), 4.64 (s, 2H), 6.81 (d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 7.53 (d, J = 7.8 Hz, 2H), 7.74 (d, J = 8.8 Hz, 2H), 7.85 (d, J = 8.9 Hz, 2H), 8.02 (d, J = 7.8 Hz, 2H), 8.62 (d, J = 4.3 Hz, 1H), 8.76 (s, 1H), 8.85 (s, 1H), 8.93 (s, 1H), 8.96 (d, J = 4.3 Hz, 1H), 9.28 (s, 1H), 10.02 (s, 1H); ESI-MS obsd 717.3692, calcd 717.3700 [(M + H)+, M = C49H44N6].
17,18-Dihydro-18,18-dimethyl-10-(4-methylphenyl)-3,13-bis[2-(phenanthren-9-yl)ethynyl]porphyrin (C-37)
Following Procedure A, a mixture of C-29 (19.7 mg, 33.5 μmol), 9-ethynylphenanthrene (16.2 mg, 80.2 μmol), and Pd(PPh3)2Cl2 (4.7 mg, 6.7 μmol) in DMF/TEA (2.3 mL) was allowed to react overnight. Final workup entailed column chromatography [silica, hexanes/CH2Cl2 (3:2)] to afford a violet-brown solid (7.4 mg, 27%): 1H NMR (400 MHz, CDCl3) δ −1.77 (bs, 1H), −1.35 (bs, 1H), 2.10 (s, 6H), 2.72 (s, 3H), 4.71 (s, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.66–7.79 (m, 4H), 7.81–7.94 (m, 4H), 7.99–8.02 (m, 1H), 8.04–8.06 (m, 1H), 8.09 (d, J = 7.8 Hz, 2H), 8.41 (s, 1H), 8.52 (s, 1H), 8.68 (d, J = 4.3 Hz, 1H), 8.74–8.75 (m, 2H), 8.77–8.79 (m, 1H), 8.81–8.83 (m, 1H), 8.85 (s, 1H), 8.96–8.98 (m, 1H), 9.02 (d, J = 4.3 Hz, 1H), 9.05 (s, 1H), 9.05–9.07 (m, 1H), 9.14 (s, 1H), 9.45 (s, 1H), 10.16 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 21.7, 31.3, 45.6, 52.2, 88.8, 89.1, 95.0, 95.2, 95.7, 96.4, 105.5 117.9, 119.9, 120.0, 122.5, 122.8, 122.9, 123.1, 123.2, 125.2, 127.2, 127.3, 127.4, 127.48, 127.54, 127.8, 127.9, 128.9, 129.0, 130.3, 130.4, 130.5, 130.6, 130.7, 131.4, 131.5, 132.4, 132.8, 133.0, 133.2, 134.2, 135.0, 135.5, 137.7, 138.3, 140.3, 140.4, 151.9, 153.8, 163.9, 176.1; LD-MS obsd 830.6, calcd 830.3 (M+, M = C61H42N4).
17,18-Dihydro-18,18-dimethyl-3,13-bis(3-N,N-dimethylamino-1-propyn-1-yl)-10-(4-methylphenyl)porphyrin (C-38)
Following Procedure A, a mixture of C-29 (24.0 mg, 40.8 μmol), N,N-dimethylpropargylamine (21 μL, 0.20 mmol), and Pd(PPh3)2Cl2 (5.6 mg, 8.0 μmol) in DMF/TEA (4 mL) was allowed to react overnight. Final workup entailed column chromatography [silica, CH2Cl2/MeOH (25:1)] to afford a violet-brown solid (21.4 mg, 88%): 1H NMR (400 MHz, CDCl3) δ −1.99 (bs, 1H), −1.58 (bs, 2H), 2.04 (s, 6H), 2.64 (s, 6H), 2.68 (s, 3H), 2.70 (s, 6H), 3.89 (s, 2H), 3.97 (s, 2H), 4.62 (s, 2H), 7.53 (d, J = 7.8 Hz, 2H), 8.00 (d, J = 8.00 Hz, 2H), 8.60 (d, J = 4.3 Hz, 1H), 8.77 (s, 1H), 8.83 (s, 1H), 8.94 (d, overlapped with singlet, 1H), 8.94 (s, 1H), 9.21 (s, 1H), 9.95 (s, 1H); LD-MS obsd 593.0; ESI-MS obsd 593.3406, calcd 593.3387 [(M + H)+, M = C39H40N6]. Note that the most prominent peak in the LD-MS spectrum is m/z = 637, which corresponds to [M + N(CH3)2]+.
17,18-Dihydro-18,18-dimethyl-10-(4-methylphenyl)-3,13-bis[2-(trimethylsilyl)ethynyl]porphyrin (C-39)
Following Procedure B, a mixture of C-29 (45.5 mg, 77.3 μmol), trimethylsilylacetylene (17 μL, 0.12 mmol), Pd2(dba)3 (14.2 mg, 15.5 μmol), and P(o-tol)3 (28.3 mg, 93.0 μmol) in toluene/TEA (30.7 mL) was reacted for 5 h. Another batch of trimethylsilylacetylene (17 μL, 0.12 mmol), Pd2(dba)3 (14.2 mg, 15.5 μmol), and P(o-tol)3 (28.3 mg, 93.0 μmol) was added, whereupon the reaction mixture was allowed to react overnight. Final workup entailed column chromatography [silica, hexanes/CH2Cl2 (4:1)] to provide a purple solid (21.2 mg, 44%): 1H NMR (400 MHz, CDCl3) δ −1.93 (bs, 1H), −1.50 (bs, 1H), 0.54 (s, 9H), 0.61 (s, 9H), 2.06 (s, 6H), 2.70 (s, 3H), 4.67 (s, 2H), 7.55 (d, J = 7.9 Hz, 2H), 8.02 (d, J = 7.9 Hz, 2H), 8.64 (d, J = 4.3 Hz, 1H), 8.78 (s, 1H), 8.91 (s, 1H), 8.98 (d, J = 4.3 Hz, 1H), 8.99 (s, 1H), 9.23 (s, 1H), 9.95 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 0.40, 0.43, 21.6, 31.2, 46.5, 52.2, 94.9, 95.6, 99.4, 99.9, 102.4, 103.9, 105.4, 108.9, 117.6, 122.5, 122.6, 125.8, 127.8, 131.0, 131.9, 132.9, 133.2, 134.1, 134.5, 135.6, 137.7, 138.2, 139.9, 140.4, 151.8, 153.7, 163.8, 176.0; ESI-MS obsd 623.3027, calcd 623.3021 [(M + H)+, M = C39H42N4Si2].
3,13-Diethynyl-17,18-dihydro-18,18-dimethyl-10-(4-methylphenyl)porphyrin (C-40)
A solution of C-29 (11 mg, 18 μmol) in THF/MeOH (2 mL, 1:1) was treated with K2CO3 (7.5 mg, 54 μmol). The resulting mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with CH2Cl2, washed (water, brine), dried (Na2SO4), and concentrated. Column chromatography [silica, hexanes/CH2Cl2 (3:2)] provided a brownish-violet solid (6.8 mg, 79%): 1H NMR (400 MHz, CDCl3) δ −1.97 (bs, 1H), −1.54 (bs, 1H), 2.04 (s, 6H), 2.68 (s, 3H), 3.88 (s, 1H), 4.03 (s, 1H), 4.63 (s, 2H), 7.53 (d, J = 7.7 Hz, 2H), 7.99 (d, J = 7.7 Hz, 2H), 8.62 (d, J = 4.3 Hz, 1H), 8.79 (s, 1H), 8.91 (s, 1H), 8.94 (d, J = 4.3 Hz, 1H), 9.02 (s, 1H), 9.23 (s, 1H), 9.96 (s, 1H); LD-MS obsd 487.8, calcd 478.2 (M+, M = C33H26N4).
X-ray structure determination for 1,3,3-trimethyl-7-(4-iodophenyl)-2,3,4,5-tetrahydrodipyrrin N10-oxide (11)
The sample, mounted on the end of a glass fiber using 5-min epoxy, was transferred to the diffractometer. All X-ray measurements were made on an Enraf-Nonius CAD4 diffractometer at room temperature. The unit cell dimensions were determined by a symmetry-constrained fit of 25 well centered reflections and their Friedel pairs with theta 31° < 2(θ) < 36°. A unique quadrant of data was collected using θ/2θ scan mode. Three standard reflections were measured every 4800 seconds of X-ray exposure time. Scaling the data was accomplished using a 5-point smoothed curved routine fit to the intensity check reflections. The intensity data was corrected for Lorentz and polarization effects. An empirical absorption correction was applied using psi scan data. The data were reduced using routines from the NRCVAX set of programs [86]. The structure was solved using SIR92 [87]. All non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were introduced at calculated positions, and were allowed to ride on the parent carbon or nitrogen atom. The hydrogen atom displacement parameters were also allowed to ride according to the following expression: U(H) = Ueq({C|N}) + 0.01. The calculated structure factors were fit to the data using full matrix least-squares based on F. The calculated structure factors included corrections for anomalous dispersion from the usual tabulation [88]. A secondary extinction correction was included in the final cycles of refinement.
Acknowledgments
The various compounds collectively described herein were prepared over a period of 14 years to realize distinct research objectives. Biomedical sciences studies were supported by a grant from the NIH (GM36238 through 2007), a grant from the National Institute of Allergy and Infectious Diseases (R41AI072854, 2007–2009) to NIRvana Pharmaceuticals, Inc., the University of Maryland, Baltimore County (start-up funds and SRAIS Award), and the National Cancer Institute of the National Institutes of Health (U01CA181628). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH. Fundamental studies of chlorin syntheses, building blocks and photophysics (target compounds ZnC-3, ZnC-5, ZnC-21, ZnC-23, ZnC-24, ZnC-25) have been supported by a grant from the Division of Chemical Sciences, Office of Basic Energy Sciences, Office of Energy Research, U.S. Department of Energy under contract No. DE-FG02-96ER14632. We thank Ms. Dalia Akkad, Ms. Jamie K. Nguyen and Mr. Chirag Pancholi for assistance in preparation of some of the derivatives, and Mr. Ajay Ravichandran and Mr. Mohammed Farooq for assistance in spectroscopic measurements. Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the National Science Foundation. X-ray structure determination was carried out at the X-Ray Structural Facility in the Chemistry Department at North Carolina State University.
Footnotes
Supporting Information
Crystallographic data for 11 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication data no. CCDC-1029006. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 2EZ, UK (email: deposit@ccdc.cam.ac.uk, fax: +44 1223-336033).
Electronic version of an article published as Journal of Porphyrins and Phthalocyanines 19, 547, 2015, 547-572, DOI: 10.1142/S1088424615500042, © World Scientific Publishing Company http://www.worldscientific.com/worldscinet/jpp
References
- 1.Hynninen PH. In: Chlorophylls. Scheer H, editor. CRC Press; Boca Raton, FL: 1991. pp. 145–209. [Google Scholar]
- 2.Vicente MGH. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego, CA: 2000. pp. 149–199. [Google Scholar]
- 3.Pavlov VY, Ponomarev GV. Chem Heterocyc Cmpds. 2004;40:393–425. [Google Scholar]
- 4.Balaban TS, Tamiaki H, Holzwarth AR. Top Curr Chem. 2005;258:1–38. [Google Scholar]
- 5.Montforts F-P, Gerlach B, Höper F. Chem Rev. 1994;94:327–347. [Google Scholar]
- 6.Galezowski M, Gryko DT. Curr Org Chem. 2007;11:1310–1338. [Google Scholar]
- 7.de Oliveira KT, Momo PB, de Assis FF, Ferreira MAB, Brocksom TJ. Curr Org Syn. 2014;11:42–58. [Google Scholar]
- 8.Strachan J-P, O’Shea DF, Balasubramanian T, Lindsey JS. J Org Chem. 2000;65:3160–3172. doi: 10.1021/jo991942t. Additions and Corrections: Strachan J-P, O’Shea DF, Balasubramanian T and Lindsey JS. J. Org. Chem.. 2001; 66: 642. [DOI] [PubMed] [Google Scholar]
- 9.Balasubramanian T, Strachan J-P, Boyle PD, Lindsey JS. J Org Chem. 2000;65:7919–7929. doi: 10.1021/jo000913b. [DOI] [PubMed] [Google Scholar]
- 10.Taniguchi M, Ra D, Mo G, Balasubramanian T, Lindsey JS. J Org Chem. 2001;66:7342–7354. doi: 10.1021/jo0104835. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi M, Kim H-J, Ra D, Schwartz JK, Kirmaier C, Hindin E, Diers JR, Prathapan S, Bocian DF, Holten D, Lindsey JS. J Org Chem. 2002;67:7329–7342. doi: 10.1021/jo025843i. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi M, Kim MN, Ra D, Lindsey JS. J Org Chem. 2005;70:275–285. doi: 10.1021/jo048440m. [DOI] [PubMed] [Google Scholar]
- 13.Laha JK, Muthiah C, Taniguchi M, McDowell BE, Ptaszek M, Lindsey JS. J Org Chem. 2006;71:4092–4102. doi: 10.1021/jo060208o. Additions and Corrections: Laha JK, Muthiah C, Taniguchi M, McDowell BE, Ptaszek M and Lindsey JS. J. Org. Chem.. 2009; 74: 5122. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi M, Ptaszek M, McDowell BE, Lindsey JS. Tetrahedron. 2007;63:3840–3849. doi: 10.1016/j.tet.2007.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kee HL, Kirmaier C, Tang Q, Diers JR, Muthiah C, Taniguchi M, Laha JK, Ptaszek M, Lindsey JS, Bocian DF, Holten D. Photochem Photobiol. 2007;83:1110–1124. doi: 10.1111/j.1751-1097.2007.00150.x. [DOI] [PubMed] [Google Scholar]
- 16.Kee HL, Kirmaier C, Tang Q, Diers JR, Muthiah C, Taniguchi M, Laha JK, Ptaszek M, Lindsey JS, Bocian DF, Holten D. Photochem Photobiol. 2007;83:1125–1143. doi: 10.1111/j.1751-1097.2007.00151.x. [DOI] [PubMed] [Google Scholar]
- 17.Muthiah C, Ptaszek M, Nguyen TM, Flack KM, Lindsey JS. J Org Chem. 2007;72:7736–7749. doi: 10.1021/jo701500d. [DOI] [PubMed] [Google Scholar]
- 18.Muthiah C, Kee HL, Diers JR, Fan D, Ptaszek M, Bocian DF, Holten D, Lindsey JS. Photochem Photobiol. 2008;84:786–801. doi: 10.1111/j.1751-1097.2007.00258.x. [DOI] [PubMed] [Google Scholar]
- 19.Borbas KE, Chandrashaker V, Muthiah C, Kee HL, Holten D, Lindsey JS. J Org Chem. 2008;73:3145–3158. doi: 10.1021/jo7026728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kee HL, Nothdurft R, Muthiah C, Diers JR, Fan D, Ptaszek M, Bocian DF, Lindsey JS, Culver JP, Holten D. Photochem Photobiol. 2008;84:1061–1072. doi: 10.1111/j.1751-1097.2008.00409.x. [DOI] [PubMed] [Google Scholar]
- 21.Ptaszek M, Lahaye D, Krayer M, Muthiah C, Lindsey JS. J Org Chem. 2010;75:1659–1673. doi: 10.1021/jo902649d. [DOI] [PubMed] [Google Scholar]
- 22.Liu M, Ptaszek M, Mass O, Minkler DJ, Sommer RD, Bhaumik J, Lindsey JS. New J Chem. 2014;38:1717–1730. [Google Scholar]
- 23.Alexander VM, Sano K, Yu Z, Nakajima T, Choyke PL, Ptaszek M, Kobayashi H. Bioconjugate Chem. 2012;23:1671–1679. doi: 10.1021/bc3002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada T, Sano K, Sato K, Watanabe R, Yu Z, Hanaoka H, Nakajima T, Choyke PL, Ptaszek M, Kobayashi H. Bioconjugate Chem. 2014;25:362–369. doi: 10.1021/bc4005238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ptaszek M, Kee HL, Muthiah C, Nothdurft R, Akers W, Achilefu C, Culver JP, Holten D. Proc SPIE. 2010;7576:1–9. 75760E. [Google Scholar]
- 26.Yu Z, Ptaszek M. J Org Chem. 2013;78:10678–10691. doi: 10.1021/jo4016858. [DOI] [PubMed] [Google Scholar]
- 27.Yu Z, Pancholi C, Bhagavathy GV, Kang HS, Nguyen JK, Ptaszek M. J Org Chem. 2014;79:7910–7925. doi: 10.1021/jo501041b. [DOI] [PubMed] [Google Scholar]
- 28.Yu Z, Ptaszek M. Org Lett. 2012;14:3708–3711. doi: 10.1021/ol3015545. [DOI] [PubMed] [Google Scholar]
- 29.Ptaszek M, Bhaumik J, Kim H-J, Taniguchi M, Lindsey JS. Org Process Res Dev. 2005;9:651–659. doi: 10.1021/op050087e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Battersby AR, Dutton CJ, Fookes CJR, Turner SPD. J Chem Soc Perkin Trans 1. 1988:1557–1567. [Google Scholar]
- 31.Battersby AR, Dutton CJ, Fookes CJR. J Chem Soc Perkin Trans I. 1988:1569–1576. [Google Scholar]
- 32.Jacobi PA, Brielmann HL, Hauck SI. J Org Chem. 1996;61:5013–5023. [Google Scholar]
- 33.Jacobi PA, Lanz S, Ghosh I, Leung SH, Löwer F, Pippin D. Org Lett. 2001;3:831–834. doi: 10.1021/ol006983m. [DOI] [PubMed] [Google Scholar]
- 34.O’Neal WG, Roberts WP, Ghosh I, Wang H, Jacobi PA. J Org Chem. 2006;71:3472–3480. doi: 10.1021/jo060041z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Neal WG, Jacobi PA. J Am Chem Soc. 2008;130:1102–1108. doi: 10.1021/ja0780075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ben Dror S, Bronshtein I, Garini Y, O’Neal WG, Jacobi PA, Ehrenberg B. Photochem Photobiol Sci. 2009;8:354–361. doi: 10.1039/b814970d. [DOI] [PubMed] [Google Scholar]
- 37.Montforts F-P. Angew Chem Int Ed Engl. 1981;20:778–779. [Google Scholar]
- 38.Montforts F-P, Schwartz UM. Angew Chem Int Ed Engl. 1985;24:775–776. [Google Scholar]
- 39.Schmidt W, Montforts FP. Synlett. 1997:903–904. [Google Scholar]
- 40.Abel Y, Montforts F-P. Tetrahedron Lett. 1997;38:1745–1748. [Google Scholar]
- 41.Montforts F-P, Kutzki O. Angew Chem Int Ed. 2000;39:599–601. [PubMed] [Google Scholar]
- 42.Könekamp T, Ruiz A, Duwenhorst J, Schmidt W, Borrmann T, Stohrer W-D, Montforts F-P. Chem Eur J. 2007;13:6595–6604. doi: 10.1002/chem.200700222. [DOI] [PubMed] [Google Scholar]
- 43.Könekamp T, Borrmann T, Montforts F-P. J Porphyrins Phthalocyanines. 2009;13:166–170. [Google Scholar]
- 44.Thanh NTV, Könekamp T, Hanke D, Löwer F, Borrmann T, Montforts F-P. J Porphyrins Phthalocyanines. 2012;16:626–632. [Google Scholar]
- 45.Littler BJ, Miller MA, Hung C-H, Wagner RW, O’Shea DF, Boyle PD, Lindsey JS. J Org Chem. 1999;64:1391–1396. [Google Scholar]
- 46.Laha JK, Dhanalekshmi S, Taniguchi M, Ambroise A, Lindsey JS. Org Process Res Dev. 2003;7:799–812. [Google Scholar]
- 47.Khoury RG, Jaquinod L, Aoyagi K, Olmstead MM, Fisher AJ, Smith KM. Angew Chem Int Ed Engl. 1997;36:2497–2500. [Google Scholar]
- 48.Zaidi SHH, Muthukumaran K, Tamaru S-I, Lindsey JS. J Org Chem. 2004;69:8356–8365. doi: 10.1021/jo048587d. [DOI] [PubMed] [Google Scholar]
- 49.Muthukumaran K, Ptaszek M, Noll B, Scheidt WR, Lindsey JS. J Org Chem. 2004;69:5354–5364. doi: 10.1021/jo0492620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iovine PM, Kellett MA, Redmore NP, Therien MJ. J Am Chem Soc. 2000;122:8717–8727. [Google Scholar]
- 51.Thamyongkit P, Speckbacher M, Diers JR, Kee HL, Kirmaier C, Holten D, Bocian DF, Lindsey JS. J Org Chem. 2004;69:3700–3710. doi: 10.1021/jo049860e. [DOI] [PubMed] [Google Scholar]
- 52.Wagner RW, Lindsey JS, Turowska-Tyrk I, Scheidt WR. Tetrahedron. 1994;50:11097–11112. [Google Scholar]
- 53.Fürstner A, Stelzer F, Rumbo A, Krause H. Chem Eur J. 2002;8:1856–1871. doi: 10.1002/1521-3765(20020415)8:8<1856::AID-CHEM1856>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 54.Johnson MG, Foglesong RJ. Tetrahedron Lett. 1997;38:7001–7002. [Google Scholar]
- 55.Sivanandan K, Aathimanikandan SV, Arges CG, Bardeen CJ, Thayumanavan S. J Am Chem Soc. 2005;127:2020–2021. doi: 10.1021/ja043356+. [DOI] [PubMed] [Google Scholar]
- 56.Jurecka P, Vojtisek P, Novotny K, Rohovec J, Lukes I. J Chem Soc Perkin Trans 2. 2002:1370–1377. [Google Scholar]
- 57.Phillion DP, Andrew SS. Tetrahedron Lett. 1986;27:1477–1480. [Google Scholar]
- 58.Chng LL, Chang CJ, Nocera DG. J Org Chem. 2003;68:4075–4078. doi: 10.1021/jo026610u. [DOI] [PubMed] [Google Scholar]
- 59.Sinclair DJ, Sherburn MS. J Org Chem. 2005;70:3730–3733. doi: 10.1021/jo050105q. [DOI] [PubMed] [Google Scholar]
- 60.Weimar M, Dürner G, Bats JW, Göbel MW. J Org Chem. 2010;75:2718–2721. doi: 10.1021/jo100053j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou X, Zhou Z-Y, Mak TCW, Chan KS. J Chem Soc Perkin Trans 1. 1994:2519–2520. [Google Scholar]
- 62.Yu L, Lindsey JS. Tetrahedron. 2001;57:9285–9298. [Google Scholar]
- 63.Fan D, Taniguchi M, Lindsey JS. J Org Chem. 2007;72:5350–5357. doi: 10.1021/jo070785s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holten D, Bocian DF, Lindsey JS. Acc Chem Res. 2002;35:57–69. doi: 10.1021/ar970264z. [DOI] [PubMed] [Google Scholar]
- 65.Mass O, Ptaszek M, Taniguchi M, Diers JR, Kee HL, Bocian DF, Holten D, Lindsey JS. J Org Chem. 2009;74:5276–5289. doi: 10.1021/jo900706x. [DOI] [PubMed] [Google Scholar]
- 66.Lindsey JS, Taniguchi M, Ra D, Mo G, Balasubramanian T. US. 2005;6552:946. doi: 10.1021/jo0104835. [DOI] [PubMed] [Google Scholar]
- 67.Krayer M, Balasubramanian T, Ruzié C, Ptaszek M, Cramer DL, Taniguchi M, Lindsey JS. J Porphyrins Phthalocyanines. 2009;13:1098–1110. [Google Scholar]
- 68.Balasubramanian T, Lindsey JS. Tetrahedron. 1999;55:6771–6784. [Google Scholar]
- 69.Hasan I, Marinelli ER, Lin L-CC, Fowler FW, Levy AB. J Org Chem. 1981;46:157–164. [Google Scholar]
- 70.Sonogashira K, Tohda T, Hagihara N. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]
- 71.Gauger KA. MS Thesis. North Carolina State University; 2010. [Google Scholar]
- 72.Thanh NC, Ikai M, Kajioka T, Fujikawa H, Taga Y, Zhang Y, Ogawa S, Shimada H, Miyahara Y, Kuroda S, Oda M. Tetrahedron. 2006;62:11227–11239. [Google Scholar]
- 73.Mispelaere-Canivet C, Spindler J-F, Perrio S, Beslin P. Tetrahedron. 2005;61:5253–5259. [Google Scholar]
- 74.Aalten HL, van Koten G, Grove DM, Kuilman T, Piekstra OG, Hulshof LA, Sheldon RA. Tetrahedron. 1989;45:5565–5578. [Google Scholar]
- 75.Gao G-Y, Colvin AJ, Chen Y, Zhang XP. Org Lett. 2003;5:3261–3264. doi: 10.1021/ol035081t. [DOI] [PubMed] [Google Scholar]
- 76.Kim H-J, Lindsey JS. J Org Chem. 2005;70:5475–5486. doi: 10.1021/jo050467y. [DOI] [PubMed] [Google Scholar]
- 77.Taniguchi M, Ptaszek M, McDowell BE, Boyle PD, Lindsey JS. Tetrahedron. 2007;63:3850–3863. doi: 10.1016/j.tet.2007.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.LeCours SM, DiMagno SG, Therien MJ. J Am Chem Soc. 1996;118:11854–11864. [Google Scholar]
- 79.Yang E, Kirmaier C, Krayer M, Taniguchi M, Kim H-J, Diers JR, Bocian DF, Lindsey JS, Holten D. J Phys Chem B. 2011;115:10801–10816. doi: 10.1021/jp205258s. [DOI] [PubMed] [Google Scholar]
- 80.Li Q, Rukavishnikov AV, Petukhov PA, Zaikova TO, Jin C, Keana JFW. J Org Chem. 2003;68:4862–4869. doi: 10.1021/jo026923p. [DOI] [PubMed] [Google Scholar]
- 81.Dirk SM, Tour JM. Tetrahedron. 2003;59:287–293. [Google Scholar]
- 82.Khairul WM, Albesa-Jové D, Yufit DS, Al-Haddad MR, Collings JC, Hartl F, Howard JAK, Marder TB, Low PJ. Inorg Chim Acta. 2008;361:1646–1658. [Google Scholar]
- 83.Laakso J, Rosser GA, Szíjjártó C, Beeby A, Borbas KE. Inorg Chem. 2012;51:10366–10374. doi: 10.1021/ic3015354. [DOI] [PubMed] [Google Scholar]
- 84.Pershagen E, Borbas KE. Coord Chem Rev. 2014:273–274. 30–46. [Google Scholar]
- 85.Srinivasan N, Haney CA, Lindsey JS, Zhang W, Chait BT. J Porphyrins Phthalocyanines. 1999;3:283–291. [Google Scholar]
- 86.Gabe EJ, Le Page Y, Charland J-P, Lee FL, White PS. J Appl Cryst. 1989;22:384–387. [Google Scholar]
- 87.Altomare A, Cascarano G, Giacovazzo C, Guagliardi A, Burla MC, Polidori G, Camalli G. J Appl Cryst. 1994;27:435. [Google Scholar]
- 88.International Tables for X-ray Crystallography. IV. Kynoch Press: Birmingham England; 1974. [Google Scholar]