

Abstract
We report a dimerization strategy to enhance the antibacterial potency of an otherwise weak cationic amphiphilic polyproline helical (CAPH) peptide. Overall, the dimeric CAPHs were more active against E. coli and S. aureus than the monomeric counterpart, reaching up to a 60-fold increase in potency. At their minimum inhibitory concentration (MIC), the dimeric peptides demonstrated no hemolytic activity or bacterial membrane disruption as monitored by β-galactosidase release in E. coli. At higher concentrations the dimeric agents were found to induce β-galactosidase release, but maintained negligible hemolytic activity, pointing to a potential shift in the mechanism of action at higher concentrations. Thus, discontinuous dimerization of an unnatural proline-rich peptide was a successful strategy to create potent de novo antibacterial peptides without membrane lysis.
Keywords: antimicrobial peptide, proline-rich, dimerization, non-lytic
Graphical abstract
The difficulty in treating multidrug resistant and extensively drug resistant bacteria is a profound problem worldwide.1-6 Pathogens resistant to the “drugs of last resort” such as carbapenem-resistant Enterobacteriaceae (CRE), and methicillinand vancomycin-resistant Staphylococcus aureus (MRSA, VRSA) have undermined almost all usable antibiotics.7-9 Particularly worrisome is the fact that no new antibacterial drugs have been approved in recent years,10 and newly designed antibacterials, such as the boron-based class of aminomethyl benzoxaboroles intended to target CRE, have failed in clinical trials due to the appearance of resistant bacteria.11 Therefore, the need for new drugs for the treatment of pathogenic bacteria is of a very high priority.
Antimicrobial peptides (AMPs) are a diverse group of molecules found in virtually all living organisms.12-15 The majority of AMPs target the bacterial membrane, which leads to rapid bacterial cell death.16-18 A subset of AMPs with a high content of proline residues acts through a non-membrane lytic mechanism.13,17,19-20 These proline-rich AMPs (P-AMPs) are less toxic to mammalian cells, have reduced hemolytic activity, and have shown limited toxicity in animal models19-21 when compared to membrane lytic AMPs.18,22 Thus, P-AMPs are preferred for drug development as anti-infective agents.23-24
P-AMPs such as PR-39, pyrrhocoricin, drosocin, and the synthetic A3-APO peptides contain consensus amino acid triad repeats of PRP or RPP. This high content of proline residues is responsible for the tendency of P-AMPs to adopt a polyproline type II (PPII) helix.25-27 Arginine (R) residues in the motif provide the peptides with an overall cationic character.19,28 Inspired by these motifs, we recently reported the design of an unnatural polyproline-rich peptide (FL-PLPRPR-4), with broad-spectrum activity against Gram-positive and Gram–negative bacteria.29 FL-PLPRPR-4 is composed of repeating units of modified proline residues containing either hydrophobic isobutyl groups (PL) or positively charged guanidinium groups (PR). The peptide forms a cationic amphiphilic PPII helix (CAPH) with a hydrophobic face composed of 5 isobutyl groups, and a hydrophilic face, composed of 8 guanidinium groups (Fig. 1a).29 Interestingly, a shorter CAPH (FL-PLPRPR-3), lacking one PLPRPR repeat (Fig. 1a), demonstrated significantly diminished antimicrobial activity as compared to FL-PLPRPR-4 against E. coli and S. aureus bacteria (Table 1). The difference in antibacterial potency was hypothesized to be due to the lower number of positively charged residues in the FL-PLPRPR-3 peptide, a feature that limited interactions with bacteria.
Figure 1.
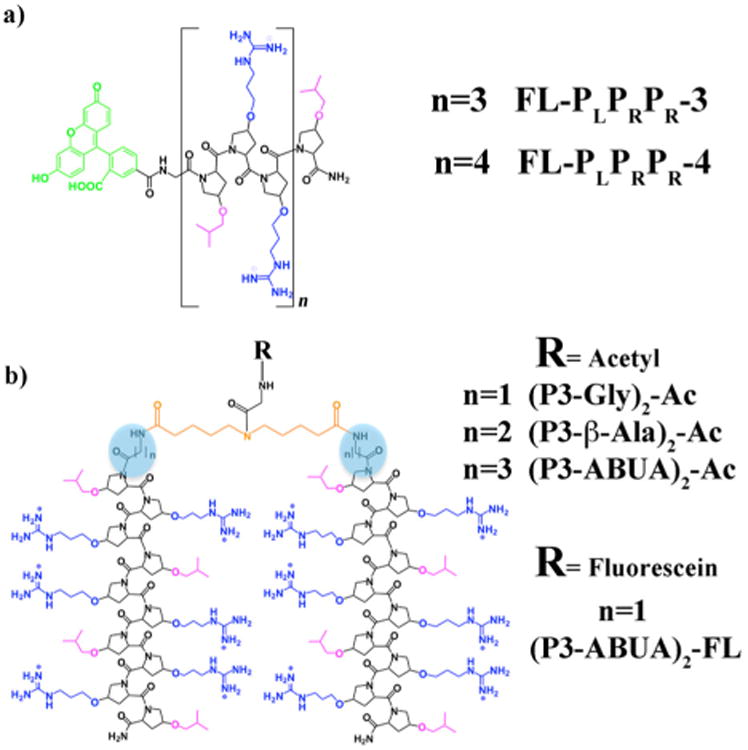
Structure of the cationic amphiphilic polyproline helices (CAPHs) composed of unnatural proline-based amino acids in (a) monomeric and (b) dimeric forms. Hydrophilic residues are shown in blue and hydrophobic residues are shown in pink. The fluorescein moiety is depicted in green, the spacers are highlighted with a blue circle and the linker is shown in orange.
Table 1. Antibacterial activity of designed CAPHs.
| Peptide | E. coli (MICa) [μM] | S. aureus MIC [μM] |
|---|---|---|
| FL-PLPRPR-3 | 6029 | 6029 |
| (P3-Gly)2-Ac | 1 | 2 |
| (P3-β-Ala)2-Ac | 4 | 8 |
| (P3-ABUA)2-Ac | 1 | 4 |
| FL-PLPRPR-4 | 429 | 1229 |
| Melittin | 4.642 | 2.1 42 |
The minimum inhibitory concentration (MIC)
Dimerization has proven to be a successful strategy to enhance the antibacterial activity of naturally occurring AMPs and AMP-inspired analogues.30-33 For instance, pyrrhocoricin dimers are more active against Gram positive bacteria than the monomeric counterpart.34 Likewise the P-AMP-inspired A3-APO dimeric peptide is highly active against clinically isolated pathogenic bacteria and has shown the potential to clear bacterial infection in various animal models.21,35-37 The increased positive charge within the dimers is believed to contribute to the improved potency against bacteria.30 However, linear continuous dimers are less active than branched dimers sharing the same number of positive charges, suggesting that branched dimerization strategies may be more beneficial to increase the antimicrobial activity of a given AMP.38 Thus, we wished to create a potent unnatural antimicrobial agent by covalently linking the amino-termini of two monomeric AMPs in a branched fashion (Fig. 1b). Herein, we describe such a dimerization of PLPRPR-3 to probe whether this modification increases the antibacterial activity of an otherwise weakly active AMP.
The PLPRPR-3 peptide was dimerized at the N-terminus with a dicarboxylic acid linking moiety and an intervening amino acid (glycine, β-alanine and aminobutyric acid – ABUA) to adjust the length of the spacer (Fig. 1b). The crosslinker was designed so as to allow association between the hydrophobic faces of the two PLPRPR-3 peptides. The linker also included an internal secondary amine to allow for a point of attachment for fluorescein (Fig. 1b).39 The synthesis of monomeric and dimeric PLPRPR-3 based peptides was carried out with unnatural proline-based, Fmoc-protected amino acids using a solid-phase strategy as previously described.39-40 The peptides were purified to homogeneity by reverse-phase HPLC and characterized by MALDI mass spectrometry.39 In our initial studies the secondary amine in the linking moiety was acetylated, resulting in dimeric CAPHs (P3-Gly)2-Ac, (P3-β-Ala)2-Ac and (P3-ABUA)2-Ac (Fig. 1b).
The antibacterial activity of the dimeric and monomeric CAPHs was explored against E. coli and S. aureus bacteria, using a broth micro-dilution assay.41 The antibacterial activity of FLPLPRPR-3 against both bacteria was low, with a MIC of 60 μM, as reported previously.29 In contrast, the dimeric peptides were highly active against both bacteria, reaching a 15- to 60-fold increase in potency with E. coli and a 7.5- to 30-fold increase in potency with S. aureus as compared to their monomeric counterpart (Table 1). When compared to the membrane lytic peptide, melittin, the (P3-Gly)2-Ac and (P3-ABUA)2-Ac dimers were 4-fold more potent against E. coli, while (P3-β-Ala)2-Ac was equipotent with melittin. Overall, the dimers were more potent against E. coli than S. aureus, a trend that has been previously observed with P-AMPs - greater activity against Gram-negative than Gram-positive bacteria.19 Although there was no clear trend in potency versus the length of the dimer spacer - shorter (Gly) and longer (ABUA) spacers were equipotent - there was a significant improvement in activity of the dimers (+12) when compared to the monomeric FL-PLPRPR-3 (+6) and FL-PLPRPR-4 (+8) peptides. Thus increasing the cationic character by means of dimerization led to an increase in potency. Most likely the increase in the positive charge increases the attraction between the negatively charged bacteria and the dimeric CAPHs, which is the first step in the mechanism of AMP-mediated bacteria toxicity.17
A critical feature in the design of AMPs is the ability to preferentially target bacteria without affecting the viability of mammalian cells. To test the safety of the designed dimeric peptides, we performed a hemolysis assay with human red blood cells (hRBCs) and measured the release of hemoglobin upon incubation with the peptides. hRBCs were incubated with the dimeric peptides at varying concentrations. Melittin and 1% Triton X-100 were used as positive controls.43 As expected, melittin and Triton X-100 caused hRBCs lysis and hemoglobin release (Fig. 2), with melittin inducing hemolysis at the lowest concentration tested (1 μM). In contrast, negligible hemolytic activity was observed with the designed dimeric peptides at concentrations up to 32 μM. Thus, the dimeric peptides had an increase in potency in antimicrobial activity without causing damage to hRBCs. These dimeric peptides have also been reported to have minimal toxicity against MCF7 cells.39
Figure 2.
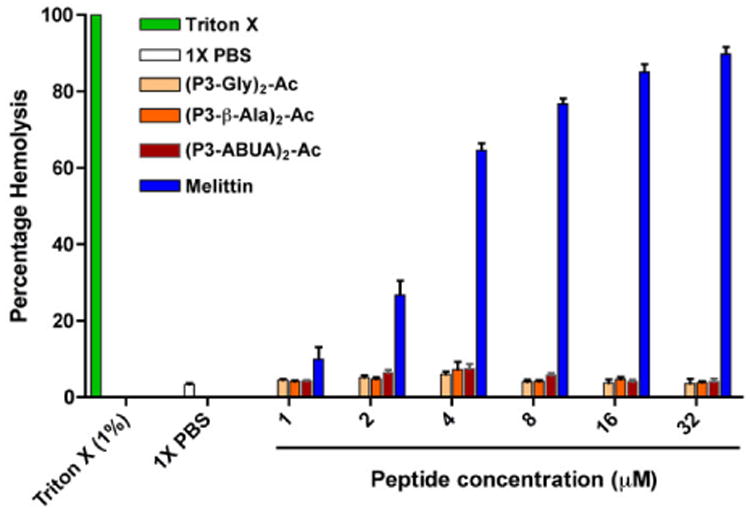
Hemolytic activity of the designed dimeric CAPHs and controls. hRBC were incubated with the peptides or control for 1 h and analyzed for the hemoglobin release at OD405. The data represent the average of two independent experiments.
AMPs that kill bacteria through membrane disruption have been shown to cause leakage of cytosolic β-galactosidase in E. coli. For instance melittin, a well-know membrane lytic AMP, promotes leakage of β-galactosidase as a consequence of membrane lysis.44-45 To gain further insights into the possible mechanism of antibacterial activity of the dimeric CAPHs, we treated E. coli with the dimers and evaluated the leakage of intracellular β-galactosidase (Fig. 3).46 As expected, melittin caused an 8-fold increase in β-galactosidase release after 1 h of treatment as compared to control buffer. E. coli cells treated with an increasing concentration of the dimeric CAPHs (1- or 5-times the MIC) showed increasing levels of β-galactosidase release after 1 h of incubation, going from no release at the MIC, to 3- to 7-fold increase at 5-times the MIC. For comparison, FL-PLPRPR-4 at 5-times its MIC (20 μM) and FL-PLPRPR-3 also at 20 μM released only low levels of β-galactosidase as compared to the control. These data suggest that the mode of action of the dimeric CAPHs may be dependent on the concentration used, and both modes of action (membrane and non-membrane lytic) may be at play at higher concentration.
Figure 3.
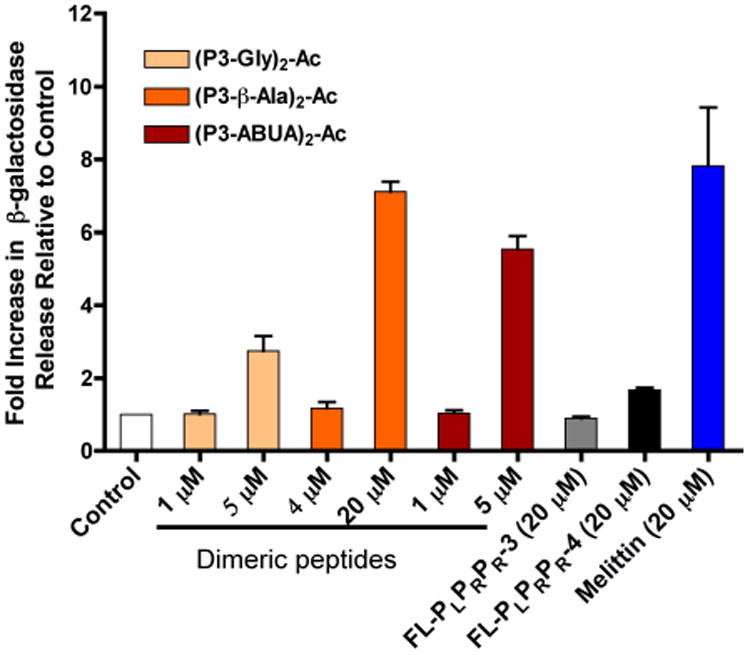
Leakage of β-galactosidase from E. coli treated with peptides for 1h. β-galactosidase activity was monitored by the cleavage of the orthonitrophenyl-β-galactosidase (ONPG) substrate at OD405. The data represent the average of three independent experiments.
To visualize the interaction between dimeric CAPHs and bacteria, a fluorescein moiety was added in place of the acetyl group within the ABUA-containing dimer to generate (P3-ABUA)2-FL (Fig. 1b). E. coli were treated with either FLPLPRPR-3, (P3-ABUA)2-FL or buffer for 10 min, 1 h, and 6 h (Fig. 4).47 FL-PLPRPR-3 was found to poorly interact with E. coli, as evident by low levels of cellular fluorescence at 10 min and 1 h. Longer incubation with FL-PLPRPR-3 (6 h) somewhat increased the levels of cellular fluorescence in bacteria (Fig. 4). E. coli treated with (P3-ABUA)2-FL, however, showed significant levels of green fluorescence as early as 10 min. Longer incubation times (1 h and 6 h) resulted in stronger levels of cellular fluorescence, but changes in bacterial morphology were also observed. At 1 h and 6 h, both filamented and smaller sized bacteria were visible that were not present in bacteria samples treated with FL-PLPRPR-3 or buffer (Fig. 4). Thus the increase in the overall positive charge within the dimeric CAPHs greatly promoted the interaction with bacteria as compared to its monomeric counterpart.
Figure 4.
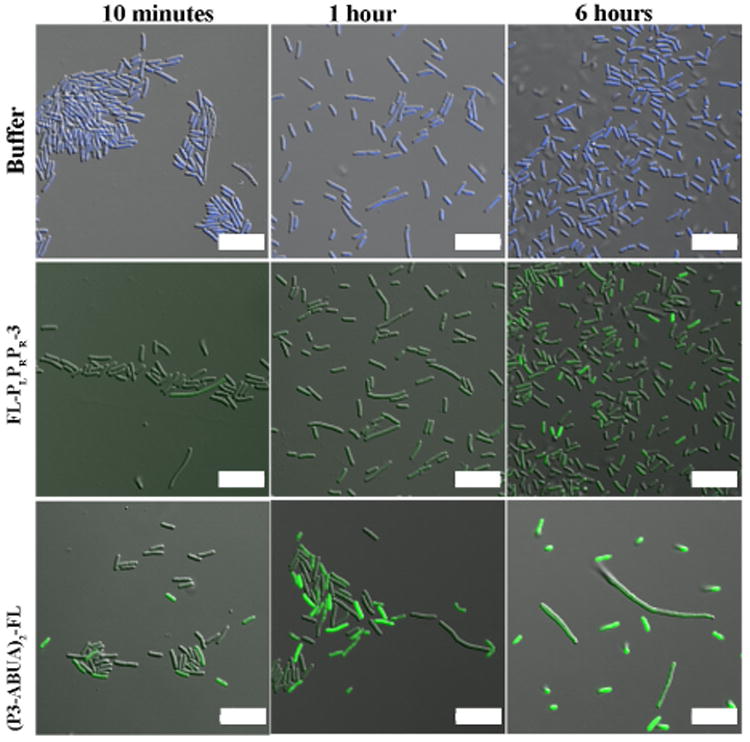
Confocal microscopy images of E. coli incubated with buffer, FLPLPRPR-3 (10 μM) and (P3-ABUA)2-FL (10 μM) for 10 min, 1 h and 6 h. After incubation, bacteria in buffer only were labeled with Hoechst 33342 (blue). The fluorescein (green) in all the peptide-treated samples was detected at the same laser intensity. Scale bars: 10 μm.
In conclusion, we have designed a series of dimeric, discontinuous, unnatural P-AMPs by covalently linking the N-terminus of the monomeric CAPH PLPRPR-3 using different spacer lengths. The dimers overall demonstrated up to a 60-fold increase in antibacterial potency compared to the monomeric peptide, with activity against both Gram positive and negative bacteria. This increase in potency correlated with an increase in the internalization within E. coli as observed by confocal microscopy. Importantly, these potent antimicrobial agents displayed no hemolysis at 8- to 32-times their MIC, a significant improvement over the lytic class of AMPs. At the MIC of the dimeric CAPHs, no β-galactosidase release from bacteria was observed, also supporting a non-lytic mechanism of action. However, at 5-times the MIC, a shift to β-galactosidase release was observed, indicating a possible change in the mode of action of the dimeric CAPHs at higher concentrations. These dimeric antimicrobial peptides are an excellent starting scaffold for the design of more potent antimicrobial agents that function without membrane lysis.
Acknowledgments
The authors acknowledge NSF for the funding.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Leung E, Weil DE, Raviglione M, Nakatani H. Bull World Health Organ. 2011;89:390. doi: 10.2471/BLT.11.088435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Kraker ME, Wolkewitz M, Davey PG, Koller W, Berger J, Nagler J, Icket C, Kalenic S, Horvatic J, Seifert H, Kaasch A, Paniara O, Argyropoulou A, Bompola M, Smyth E, Skally M, Raglio A, Dumpis U, Melbarde Kelmere A, Borg M, Xuereb D, Ghita MC, Noble M, Kolman J, Grabljevec S, Turner D, Lansbury L, Grundmann H. J Antimicrob Chemother. 2011;66:398. doi: 10.1093/jac/dkq412. [DOI] [PubMed] [Google Scholar]
- 3.Giske CG, Monnet DL, Cars O, Carmeli Y. Antimicrob Agents Chemother. 2008;52:813. doi: 10.1128/AAC.01169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herati RS, Blumberg EA. Curr Opin Infect Dis. 2012;25:445. doi: 10.1097/QCO.0b013e328354f192. [DOI] [PubMed] [Google Scholar]
- 5.Andersson DI, Hughes D. Nat Rev Microbiol. 2010;8:260. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 6.Spellberg B, Blaser M, Guidos RJ, Boucher HW, Bradley JS, Eisenstein BI, Gerding D, Lynfield R, Reller LB, Rex J, Schwartz D, Septimus E, Tenover FC, Gilbert DN. Clin Infect Dis. 2011;52 Suppl. 5:S397. doi: 10.1093/cid/cir153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. Lancet. 2006;368:874. doi: 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- 8.van Duin D, Kaye KS, Neuner EA, Bonomo RA. Diagn Microbiol Infect Dis. 2013;75:115. doi: 10.1016/j.diagmicrobio.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenna M. Nature. 2013;499:394. doi: 10.1038/499394a. [DOI] [PubMed] [Google Scholar]
- 10.Wenzel RP. N Engl J Med. 2004;351:523. doi: 10.1056/NEJMp048093. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez V, Crepin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang YK, Zhou Y, Nieman JA, Anugula MR, Keramane el M, Savariraj K, Reddy DS, Sharma R, Subedi R, Singh R, O'Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MR, Plattner JJ. Antimicrob Agents Chemother. 2013;57:1394. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hassan M, Kjos M, Nes IF, Diep DB, Lotfipour F. J Appl Microbiol. 2012;113:723. doi: 10.1111/j.1365-2672.2012.05338.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang G, Li X, Wang Z. Nucleic Acids Res. 2009;37:D933. doi: 10.1093/nar/gkn823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zasloff M. Nature. 2002;415:389. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 15.van't Hof W, Veerman EC, Helmerhorst EJ, Amerongen AV. Biol Chem. 2001;382:597. doi: 10.1515/BC.2001.072. [DOI] [PubMed] [Google Scholar]
- 16.Hwang PM, Vogel HJ. Biochem Cell Biol. 1998;76:235. doi: 10.1139/bcb-76-2-3-235. [DOI] [PubMed] [Google Scholar]
- 17.Brogden KA. Nat Rev Microbiol. 2005;3:238. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 18.Shai Y. Biopolymers. 2002;66:236. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 19.Scocchi M, Tossi A, Gennaro R. Cell Mol Life Sci. 2011;68:2317. doi: 10.1007/s00018-011-0721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otvos L., Jr Cell Mol Life Sci. 2002;59:1138. doi: 10.1007/s00018-002-8493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szabo D, Ostorhazi E, Binas A, Rozgonyi F, Kocsis B, Cassone M, Wade JD, Nolte O, Otvos L., Jr Int J Antimicrob Agents. 2010;35:357. doi: 10.1016/j.ijantimicag.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 22.Lee YJ, Kang SJ, Kim BM, Kim YJ, Woo HD, Chung HW. Chem Biol Interact. 2007;169:189. doi: 10.1016/j.cbi.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 23.Otvos L, Snyder C, Condie B, Bulet P, Wade JD. Int J Pept Res Ther. 2005;11:29. [Google Scholar]
- 24.Otvos L, Jr, Wade JD, Lin F, Condie BA, Hanrieder J, Hoffmann R. J Med Chem. 2005;48:5349. doi: 10.1021/jm050347i. [DOI] [PubMed] [Google Scholar]
- 25.Cabiaux V, Agerberth B, Johansson J, Homble F, Goormaghtigh E, Ruysschaert JM. Eur J Biochem. 1994;224:1019. doi: 10.1111/j.1432-1033.1994.01019.x. [DOI] [PubMed] [Google Scholar]
- 26.Bulet P, Urge L, Ohresser S, Hetru C, Otvos L., Jr Eur J Biochem. 1996;238:64. doi: 10.1111/j.1432-1033.1996.0064q.x. [DOI] [PubMed] [Google Scholar]
- 27.Kragol G, Hoffmann R, Chattergoon MA, Lovas S, Cudic M, Bulet P, Condie BA, Rosengren KJ, Montaner LJ, Otvos L., Jr Eur J Biochem. 2002;269:4226. doi: 10.1046/j.1432-1033.2002.03119.x. [DOI] [PubMed] [Google Scholar]
- 28.Cassone M, Vogiatzi P, La Montagna R, De Olivier Inacio V, Cudic P, Wade JD, Otvos L., Jr Peptides. 2008;29:1878. doi: 10.1016/j.peptides.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 29.Kuriakose J, Hernandez-Gordillo V, Nepal M, Brezden A, Pozzi V, Seleem MN, Chmielewski J. Angew Chem, Int Ed Engl. 2013;52:9664. doi: 10.1002/anie.201302693. [DOI] [PubMed] [Google Scholar]
- 30.Liu SP, Zhou L, Lakshminarayanan R, Beuerman RW. Int J Pept Res Ther. 2010;16:199. doi: 10.1007/s10989-010-9230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lorenzon EN, Sanches PR, Nogueira LG, Bauab TM, Cilli EM. Amino Acids. 2013;44:1521. doi: 10.1007/s00726-013-1475-3. [DOI] [PubMed] [Google Scholar]
- 32.Lorenzon EN, Cespedes GF, Vicente EF, Nogueira LG, Bauab TM, Castro MS, Cilli EM. Antimicrob Agents Chemother. 2012;56:3004. doi: 10.1128/AAC.06262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornef MW, Putsep K, Karlsson J, Refai E, Andersson M. Nat Immunol. 2004;5:836. doi: 10.1038/ni1094. [DOI] [PubMed] [Google Scholar]
- 34.Cudic M, Condie BA, Weiner DJ, Lysenko ES, Xiang ZQ, Insug O, Bulet P, Otvos L., Jr Peptides. 2002;23:2071. doi: 10.1016/s0196-9781(02)00244-9. [DOI] [PubMed] [Google Scholar]
- 35.Noto PB, Abbadessa G, Cassone M, Mateo GD, Agelan A, Wade JD, Szabo D, Kocsis B, Nagy K, Rozgonyi F, Otvos L., Jr Protein Sci. 2008;17:1249. doi: 10.1110/ps.034330.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ostorhazi E, Holub MC, Rozgonyi F, Harmos F, Cassone M, Wade JD, Otvos L., Jr Int J Antimicrob Agents. 2011;37:480. doi: 10.1016/j.ijantimicag.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Ostorhazi E, Rozgonyi F, Szabo D, Binas A, Cassone M, Wade JD, Nolte O, Bethel CR, Bonomo RA, Otvos L., Jr Biopolymers. 2011;96:126. doi: 10.1002/bip.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai Y, Liu S, Li J, Lakshminarayanan R, Sarawathi P, Tang C, Ho D, Verma C, Beuerman RW, Pervushin K. J Biol Chem. 2012;287:26606. doi: 10.1074/jbc.M112.363259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geisler IM, Chmielewski J. Pharm Res. 2011;28:2797. doi: 10.1007/s11095-011-0493-7. [DOI] [PubMed] [Google Scholar]
- 40.Fillon YA, Anderson JP, Chmielewski J. J Am Chem Soc. 2005;127:11798. doi: 10.1021/ja052377g. [DOI] [PubMed] [Google Scholar]
- 41.Otvos L, Cudic M. In: Peptide Characterization and Application Protocols. Fields G, editor. Vol. 386. Humana Press; 2007. pp. 309–320. E. coli (ATCC 25922) or S. aureus (ATCC 25923) was grown to mid-exponential phase in Tryptic Soy Broth (TSB) at 37 °C and 200 rpm. An aliquot of the bacterial suspension was centrifuged, washed with Muller Hinton Broth (MHB), and then re-suspended in MHB to an OD600 of 0.001. 90 μL of the bacterial suspension were combined with two-fold serial dilutions of the designed peptides in a 96-well plate. The plate was incubated for 6 h at 37 °C and 200 rpm. The final OD590 was determined using a microplate reader (TECAN SpectraFluor Plus). The minimum inhibitory concentration (MIC) was defined as the lowest concentration of peptide at which no growth was observed. [Google Scholar]
- 42.Sovadinova I, Palermo EF, Urban M, Mpiga P, Caputo GA, Kuroda K. Polym. 2011;3:1512. [Google Scholar]
- 43.Fresh human red blood cells (hRBCs) from healthy volunteers were centrifuged at 2000 rpm for 5 min followed to remove the serum. The hRBC pellet was washed with phosphate buffered saline (PBS, pH 7.4). This procedure was repeated three times. Finally, an 8% suspension (v/v) of hRBCs was prepared in 1× PBS and 50 μL of this solution was mixed with 50 mL of two-fold serial dilutions of the designed peptides in a 96-well plate. The plate was incubated at 37 °C for 1 h to allow for hemolysis. The plate was centrifuged at 1000 rpm for 10 min at 4° C. Next 75 μL aliquots of the supernatants in each well were carefully transferred to a new 96-well plate. The release of hemoglobin was monitored by measuring the absorbance at OD405 The hemolysis percentage was calculated based on the 100% release with 0.1 % Triton X-100.
- 44.Porter EA, Weisblum B, Gellman SH. J Am Chem Soc. 2002;124:7324. doi: 10.1021/ja0260871. [DOI] [PubMed] [Google Scholar]
- 45.Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. Antimicrob Agents Chemother. 1998;42:2206. doi: 10.1128/aac.42.9.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.E. coli (ATCC 25922) was grown to mid-exponential phase (OD600≈0.6) in TSB at 37 °C with shaking. β-Galactosidase expression was induced for 1 h using freshly prepared isopropyl-β-D-thiogalactopyranoside (IPTG, 1 μM). At the end of the induction, an aliquot of the bacterial suspension was centrifuged, washed twice with fresh TSB and plated into a sterile 96-well plate (90 μL). Next 10 μL aliquots of the peptides were added. The plate was then incubated for 1 h at 37 °C. At the end of the incubation, the plate was centrifuged at 3000 rpm and 4 °C for 10 min. 80 μL of the supernatant from each well was transferred to a new sterile 96-well plate. Next 20 μL of freshly prepared 2-nitrophenyl-β-D-galactopyranoside (ONPG) was added to each well. The β-galactosidase activity was monitored at OD405 every five minutes for a period of 1 h using a micro-plate reader.
- 47.E. coli (ATCC 25922) was grown to the mid-exponential phase in MHB media at 37 °C and 200 rpm. An aliquot of the bacterial suspension was centrifuged for 5 min at 3000 rpm, the supernatant aspirated and the pellet re-suspended in fresh MHB media. 90 μL of the bacterial suspension was combined with 10 μL of the peptides. The plate was then incubated for 6 h at 37 °C. Aliquots were taken out at 10 min and 6 h time points, centrifuged and washed twice with PBS before being fixed in 10 % formalin solution. Bacterial were labeled with Hoechst dye (1 μM) prior loading on a poly-lysine cover slip. Bacteria were imaged using a Nikon A1R multiphoton inverted confocal microscope equipped with a 60× oil objective. 488 nm and 405nm laser lines for fluorescein and Hoechst excitation were employed, respectively.