

ABSTRACT
Perkinsela is an enigmatic early-branching kinetoplastid protist that lives as an obligate endosymbiont inside Paramoeba (Amoebozoa). We have sequenced the highly reduced mitochondrial genome of Perkinsela, which possesses only six protein-coding genes (cox1, cox2, cox3, cob, atp6, and rps12), despite the fact that the organelle itself contains more DNA than is present in either the host or endosymbiont nuclear genomes. An in silico analysis of two Perkinsela strains showed that mitochondrial RNA editing and processing machineries typical of kinetoplastid flagellates are generally conserved, and all mitochondrial transcripts undergo U-insertion/deletion editing. Canonical kinetoplastid mitochondrial ribosomes are also present. We have developed software tools for accurate and exhaustive mapping of transcriptome sequencing (RNA-seq) reads with extensive U-insertions/deletions, which allows detailed investigation of RNA editing via deep sequencing. With these methods, we show that up to 50% of reads for a given edited region contain errors of the editing system or, less likely, correspond to alternatively edited transcripts.
IMPORTANCE
Uridine insertion/deletion-type RNA editing, which occurs in the mitochondrion of kinetoplastid protists, has been well-studied in the model parasite genera Trypanosoma, Leishmania, and Crithidia. Perkinsela provides a unique opportunity to broaden our knowledge of RNA editing machinery from an evolutionary perspective, as it represents the earliest kinetoplastid branch and is an obligatory endosymbiont with extensive reductive trends. Interestingly, up to 50% of mitochondrial transcripts in Perkinsela contain errors. Our study was complemented by use of newly developed software designed for accurate mapping of extensively edited RNA-seq reads obtained by deep sequencing.
INTRODUCTION
Kinetoplastids are a diverse, widespread, and ecologically significant group of protists, some of which are devastating human parasites. Kinetoplastids have been the focus of intense research, mainly because of the medical importance of Leishmania and Trypanosoma species, and have been shown to exhibit a variety of unique cellular and molecular features, including RNA editing, mRNA trans-splicing, and genes arranged in polycistronic arrays (1). However, relatively little is known about the origin and evolution of these features across the full breadth of kinetoplastid diversity, despite the fact that there is tremendous species richness in both terrestrial, obligatory parasitic trypanosomatids (2) and free-living marine bodonids (3).
Insertion/deletion of uridine (U) residues (U-indel) into/from the mitochondrial mRNAs of kinetoplastids was the first type of RNA editing discovered (4). A plethora of posttranscriptional modifications have subsequently been described in organisms ranging from bacteria to plants and humans (for a review, see reference 5). RNA editing events include various insertions and deletions of single or multiple residues as well as base modifications and replacements, and they occur in both noncoding and protein-coding RNAs transcribed from nuclear and/or organellar genomes (6, 7). Numerous types of conversion editing have been implicated in a wide range of cellular processes, including embryonic development of the brain (8) and cancer (9).
While RNA editing in general seems to be particularly abundant in mitochondria and plastids (10, 11), U-indel RNA editing is at present confined to the mitochondria of kinetoplastids (1, 12, 13) and their sister clade, Diplonemea (14–16). U-indel editing is the most complex form of RNA editing known. Multiple sites within most transcripts are edited, with some mRNAs edited over their entire length (so-called pan-editing). In the model kinetoplastid Trypanosoma brucei, more than 70 different proteins have been shown to be incorporated into numerous dynamic editing complexes (12, 13), and up to 1,000 different small RNA molecules, called guide RNAs (gRNAs), act as the templates that define editing sites along a cognate mRNA (17).
Another unusual feature of kinetoplastid mitochondria is the structure and composition of their ribosomes. In T. brucei, 129 mitochondrial ribosomal proteins are encoded in the nucleus and targeted to the organelle posttranslationally (18). Only a single ribosomal protein, RPS12 (19), and two rRNAs are encoded in the mitochondrial genome. The bulk of the mitochondrial DNA (mtDNA) (or kinetoplastid DNA [kDNA]) of kinetoplastids is made up of minicircles encoding gRNA genes (13). The 9S and 12S mitochondrial rRNAs of T. brucei are highly truncated and lack several conserved domains that are functionally significant in other eukaryotes (20). Their transcription is developmentally regulated, and they are 3′-polyuridylylated (21). Determination of the high-resolution three-dimensional structure of a protein-rich, rRNA-poor mitochondrial ribosome of a related species, Leishmania tarentolae, was instrumental in explaining the shrunken mitochondrial rRNAs (22).
We are studying the molecular biology and evolution of the early-branching kinetoplastid Perkinsela. Members of this morphologically divergent, flagellum-lacking genus live as obligate endosymbionts inside marine amoebae (23), which to our knowledge is the only known example of a coevolving endosymbiotic relationship between two nonphotosynthetic eukaryotes. The kinetoplastid-amoeba symbiotic system appears to have emerged early in the evolution of the genus Paramoeba (24). The closest known relative of Perkinsela is the fish ectoparasite Ichthyobodo necator, and both of these kinetoplastids belonging to the Prokinetoplastina clade (25), currently represented by a relatively small number of species in rRNA databases (26). Within the confines of the host amoeba cytoplasm, Perkinsela is sometimes referred to as the “parasome” or “Perkinsela-like organism (PLO).” Amoeba hosts include free-living and facultatively parasitic marine amoebae of the genera Paramoeba and Janickina (24, 27–29). The Perkinsela strains studied here are associated with Paramoeba pemaquidensis, the causative agent of amoebic gill disease, which results in considerable mortality at marine fish farms (30, 31).
Using Perkinsela and Paramoeba genomic and transcriptomic data (GenBank accession number LFNC00000000), we have assembled the mitochondrial genomes of Perkinsela strains CCAP1560/4 and GillNOR1/I and characterized their overall structures and expression with particular attention to RNA editing. Furthermore, we have predicted the composition of their respiratory chain complexes, as well as the proteins involved in RNA editing, processing, and translation. We show that the mitochondrial genome of Perkinsela, which is composed of a huge number of fragments with terminal repeats, has undergone a considerable reduction in gene content and that all detected protein-coding transcripts undergo extensive U-indel RNA editing. While most proteins associated with RNA editing and with mitochondrial ribosomes in T. brucei are recognizable in Perkinsela, mitochondrial rRNAs were not found despite an exhaustive search, suggesting that they are fragmented and/or extremely divergent, similar to the situation observed in the related diplonemid Diplonema papillatum (16).
Importantly, we have conducted, to our knowledge, the first investigation of U-indel-edited mitochondrial transcripts based on deep transcriptome sequencing (RNA-seq), and we have developed software tools for accurate mapping of extensively edited reads. Since the discovery of this type of RNA editing in 1986, editing mechanisms have been unraveled via targeted sequencing on a clone-by-clone basis (32–34). Recently, deep sequencing of gRNA libraries in T. brucei (17, 35) has uncovered an unexpected degree of complexity and disorder inherent in gRNA-mediated editing. By deep sequencing of mRNAs, we have unveiled an even greater level of complexity in the form of “misediting” (36–38), although we have not detected alternative translatable mRNAs with considerable abundance.
RESULTS AND DISCUSSION
Perkinsela mitochondrial genome structure.
Perkinsela can be visualized in the Paramoeba cell (Fig. 1A) via 4′,6-diamidino-2-phenylindole (DAPI) staining of DNA, which shows that the endosymbiont is invariably located in the perinuclear region of amoebae (Fig. 1B and C). Interestingly, based on the intensity of DAPI staining, it appears that Perkinsela harbors a larger amount of DNA in its mitochondrion (kDNA) than in the rather inconspicuously stained nuclei of Perkinsela and Paramoeba (Fig. 1B). High-pressure freezing transmission electron microscopy, which optimally preserves the fine structure, confirmed an earlier observation obtained by standard electron microscopy (23, 39), namely, that the single mitochondrion of Perkinsela is packed with kDNA strands arranged in parallel electron-dense layers (Fig. 1C). This observation, which is further supported by the fact that it is impossible to arrange Perkinsela mitochondrial contigs on one scaffold by using mate-pair reads (data not shown), suggests that the kDNA of Perkinsela is not composed of minicircles and maxicircles, as is the case with the genera Trypanosoma and Leishmania, and other trypanosomatids. Molecules constituting the kDNA of bodonids studied thus far do not fall into distinct categories of mini-circles and maxi-circles, and they also do not form a single catenated kDNA network (40). Both features also seem to be present in Perkinsela, an early-branching bodonid (27). Moreover, since both DAPI staining and electron microscopy show that the kDNA and the single mitochondrion occupy most of the Perkinsela cell volume and that the organellar genome constitutes the most abundant DNA in this endosymbiont-host system, it is likely that this inflated genome is present at an extremely high copy number.
FIG 1 .

Phase-contrast image (A), DAPI staining image (B), and high-pressure freezing transmission electron microscopy image (C) of Perkinsela strain GillNOR1/I. Scale bars, 10 µm (A and B) or 1 µm (C). In panel C, small arrows mark the single membrane separating Perkinsela from the amoeba host cytoplasm, and arrowheads mark the outer mitochondrial membrane of Perkinsela. Abbreviations: ANu, amoeba nucleus; PNu, Perkinsela nucleus; PMt, Perkinsela mitochondrion.
Trypanosomatid mtDNAs invariably have a complement of 18 protein-coding genes and two rRNA genes (1). However, individual flagellate species differ in gene regions at which posttranscriptional U-indel editing takes place (41, 42). Out of this conserved gene set, we identified just six protein-coding genes (cox1, cox2, cox3, cob, atp6, and rps12) on three assembled mitochondrial contigs in Perkinsela that were similar in both studied strains (Fig. 2). Unlike typical trypanosomatid maxicircles, which have a single variable region and a dense protein-coding region (43), these Perkinsela contigs contain short tandem repeats at their ends. No similarity was found among repetitive sequences flanking the three protein-coding contigs (data not shown). Unambiguous connection of the contigs into larger scaffolds proved impossible, despite the usage of mate-pair reads for scaffolding and despite manual analysis of a contig graph produced using the assembly software GS de novo Assembler v.2.9 (Newbler).
FIG 2 .

Gene-bearing mitochondrial scaffolds identified in Perkinsela strains CCAP1560/4 (GenBank accession numbers KT261384 to KT261386) and GillNOR1/I (GenBank accession numbers KT272167 to KT272169). Transcript regions undergoing RNA editing are shown in green. Scaffold 1 contains cox1 and cox2 genes in reverse orientation; scaffold 2 contains cob and closely spaced rps12 and cox3 genes in the same orientation; scaffold 3 contains only the atp6 gene. While most transcripts are edited in separate regions at their ends, rps12 and atp6 are edited over almost their entire lengths, i.e., pan-edited. Gene regions used for detailed mapping of alternatively edited reads (see File S1 in the supplemental material) are shown by teal arrows, which also indicate the direction of RNA editing in these regions. For the GillNOR1/I strain, coverage with strand-specific RNA-seq reads (with U-indel optimized settings) for each transcript is shown in the sense orientation only (for both the sense and antisense reads that were plotted, see Fig. S1 in the supplemental material). For CCAP1560/4, RNA-seq reads were not strand-specific. Coverage (gray blocks) is plotted on a logarithmic scale. Absolute values of coverage were markedly different for the two strains due to the different sequencing approaches used (see File S2 in the supplemental material).
This highly reduced set of a half-dozen genes encodes subunits of three respiratory complexes: cob of complex III (ubiquinone-cytochrome c oxidoreductase); cox1, cox2, and cox3 of complex IV (cytochrome c oxidase); and atp6 of complex V (ATP synthase), suggesting a functional respiratory chain. The apparent absence of respiratory complex I in Perkinsela (in which mtDNA-encoded subunits nad1 through nad9 are missing) is further supported by the absence of the nucleus-encoded subunits of this complex (see below; see also Table S1 in the supplemental material). All six mtDNA protein-coding genes are transcribed (with varied transcript abundance levels) and undergo U-indel editing to slightly different degrees (Fig. 2; Table 1). Long antisense transcripts were undetectable by Northern blotting, at least in the case of cox2 (Fig. 3). Due to the extremely slow growth of Paramoeba in culture, we were not able to accumulate enough RNA for testing antisense transcription of other Perkinsela mitochondrial genes by Northern blotting, but mapping of strand-specific RNA-seq reads revealed no significant antisense transcripts in strain GillNOR1/I (see Fig. S1 in the supplemental material).
TABLE 1 .
Statistics for edited mitochondrial mRNAs in Perkinselaa
| Strain | Gene | Pre-edited size (nt) | Edited size (nt) | % size increase | % U in ORF | Protein length (aa) | Pre-edited region(s) lengthb (nt) | U insertionsb | U deletionsb | No. of edited sitesb |
|---|---|---|---|---|---|---|---|---|---|---|
| CCAP1560/4 | cob | 964 | 1,136 | 18 | 45 | 370 | 35 + 134 | 41 + 146 | 3 + 13 | 14 + 68 |
| GillNOR1/I | 936 | 1,125 | 20 | 47 | 370 | 36 + 134 | 45 + 141 | 4 + 8 | 15 + 66 | |
| CCAP1560/4 | cox1 | 1,374 | 1,567 | 14 | 40 | 521 | 78 + 107 | 77 + 121 | 3 + 3 | 31 + 54 |
| GillNOR1/I | 1,365 | 1,589 | 16 | 42 | 521 | 68 + 103 | 77 + 125 | 2 + 3 | 30 + 53 | |
| CCAP1560/4 | cox2 | 486 | 661 | 36 | 51 | 209 | 55 + 227 | 57 + 151 | 6 + 28 | 23 + 102 |
| GillNOR1/I | 487 | 710 | 46 | 49 | 209 | 57 + 177 | 58 + 148 | 8 + 26 | 24 + 98 | |
| CCAP1560/4 | cox3 | 656 | 814 | 24 | 44 | 256 | 45 + 89 | 69 + 93 | 2 + 2 | 31 + 47 |
| GillNOR1/I | 624 | 801 | 28 | 49 | 255 | 54 + 97 | 71 + 93 | 3 + 2 | 31 + 47 | |
| CCAP1560/4 | rps12c | 157 | 268 | 72 | 55 | 80 | 123 | 123 | 13 | 52 |
| GillNOR1/I | 150 | 257 | 72 | 55 | 80 | 110 | 123 | 12 | 52 | |
| CCAP1560/4 | atp6c | 363 | 651 | 80 | 61 | 197 | 309 | 318 | 30 | 154 |
| GillNOR1/I | 321 | 625 | 94 | 62 | 197 | 298 | 311 | 28 | 152 |
Only the main edited products were taken into account.
Numbers correspond to the 5′ and 3′ regions, respectively.
Pan-edited transcript, i.e., edited throughout most of its length.
FIG 3 .
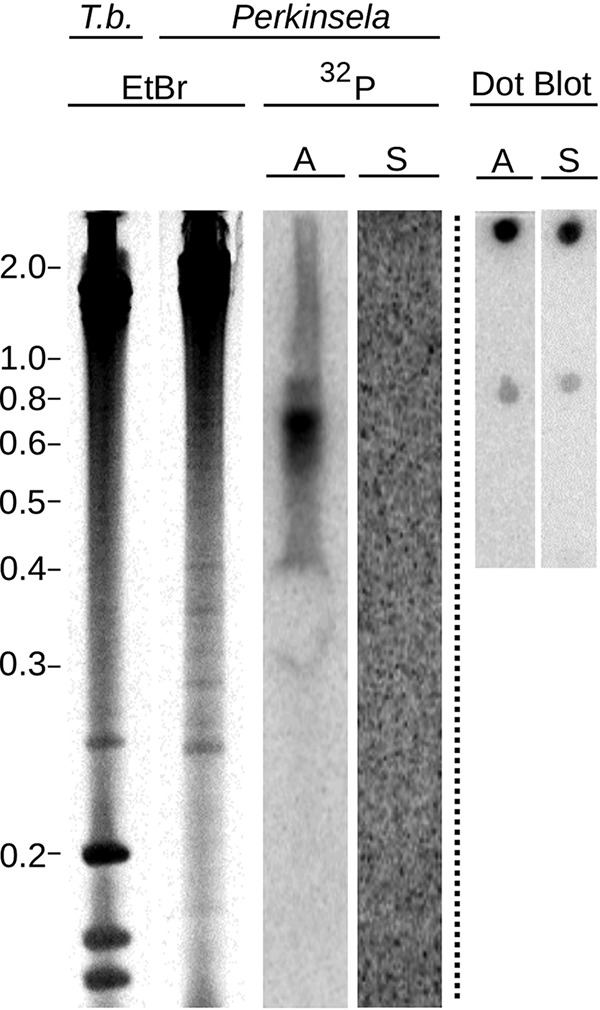
Northern blot with antisense and sense probes for the cox2 transcript, showing that only a single strand of the cox2 gene is transcribed. Total RNA from T. brucei (T.b.) and Perkinsela resolved on a denaturing gel was visualized via ethidium bromide (EtBr) stain. The signal from the antisense cox2 probe is shown in the lane labeled “32P, A,” while the sense-probed Northern membrane is shown in the lane labeled “32P, S.” Dot blots simultaneously probed with antisense (A) and sense (S) probes are shown on the right, with increasing dilutions from top to bottom of a denatured plasmid bearing an insert corresponding to the probed sequence.
Despite extensive searching, rRNA genes could not be identified by BLAST using known kinetoplastid and Diplonema rRNA genes as queries. Further candidate sequences were obtained from transcribed regions of the assembly not assigned to the Perkinsela or host nuclear genomes. In addition, Perkinsela reads containing the peptidyl transferase core motif ACCTCGNTGT conserved even in the highly diverged Diplonema large subunit (LSU) rRNA (16) were assembled, and the resulting contigs were added to the list of putative rRNA-encoding sequences. All top candidates were subjected to a careful manual secondary structure prediction with the help of the Mfold software (terminal hairpin prediction), but no small subunit (SSU) or LSU rRNA-like folds were found (data not shown).
In light of the recent discovery of a split and edited LSU rRNA in Diplonema, a relative of Perkinsela, and the fact that the SSU rRNA of Diplonema remains unidentified (16), it seems likely that extreme divergence and/or fragmentation renders the mitochondrial rRNAs of Perkinsela unrecognizable. We consider it highly improbable that the mitochondrial rRNA is genuinely absent, because upon RNA editing, detected transcripts have evolutionarily conserved open reading frames, implying the requirement of a functional ribosome to translate them into protein. Moreover, both universal and kinetoplastid-specific mitochondrial ribosomal proteins are generally conserved in Perkinsela (see Table S1 in the supplemental material), and a ribosomal subunit gene (rps12) is also present in its organellar genome (Fig. 2).
We searched for putative gRNA genes in the genome assembly of Perkinsela strain CCAP1560/4 that had the following characteristics: (i) at least 25 nucleotides (nt) complementary to the final edited transcript, (ii) up to 5 G-U pairs allowed, and (iii) supported by at least one transcriptomic read. Using this approach, we pinpointed 65 candidate gRNA genes in 29 contigs, matching all protein-coding transcripts except for rps12 (see File S3 in the supplemental material). The average length of matching contigs was 730 bp (from 176 to 3,021 bp). Flanking regions of putative gRNA genes had no common motifs, except for an A- or an A/C-rich tract upstream of the mRNA-complementary region (see File S3). However, we could not obtain robust gRNA candidates for two reasons. First, a dedicated library of short transcripts was lacking, and any standard RNA-seq library is depleted in short transcripts. Second, a strand-specific library is necessary to prove that putative gRNA genes produce transcripts which are antisense relative to the protein gene transcript; however, the strand-specific RNA-seq library we constructed for Perkinsela strain GillNOR1/I had relatively low coverage of mitochondrial transcripts and produced few gRNA candidates (data not shown).
Nucleus- and mitochondrion-encoded respiratory chain subunits.
Using hidden Markov models (HMM) constructed on the basis of trypanosomatid orthologs, the Perkinsela genomic contigs (GenBank accession number LFNC00000000) were searched for mitochondrial proteins (see Materials and Methods for details). Since none of the nucleus-encoded subunits of the respiratory complex I (NADH dehydrogenase) were detected, we considered this component of the respiratory chain missing in Perkinsela (Fig. 4; see also Table S1 in the supplemental material). This inference is in agreement with our failure to detect any of the mtDNA-encoded subunits of complex I in the mitochondrial contigs. Similarly, no alternative NADH dehydrogenases were found. The other respiratory complexes (II through V) that together mediate oxidative phosphorylation are apparently present in Perkinsela (Fig. 4; see also Table S1). We conclude that in the mitochondrion of Perkinsela the respiratory chain is functional, with the missing complex I likely replaced by an as-yet-unidentified alternative NADH dehydrogenase. Although the distantly related T. brucei possesses both mitochondrion- and nucleus-encoded subunits of complex I, its function remains elusive, with a highly active alternative dehydrogenase substituting for the canonical biochemical activity (44, 45). It thus seems that in kinetoplastids, complex I is prone to loss and was eliminated in the early-branching Perkinsela.
FIG 4 .

Conservation of respiratory chain subunits, RNA editing and processing factors, and mitochondrial ribosomal proteins in Perkinsela. Each complex is represented as a pie chart, and numbers indicate subunits analyzed in this study. The green color marks proteins identified in the Perkinsela genome (also listed in Table S1 in the supplemental material). Missing proteins are shown in white, and proteins encoded in the mitochondrial genome are shown as bright yellow areas. The left-hand section of the pie chart for respiratory chain complex I represents subunits encoded in the mitochondrial genomes of trypanosomatids but missing in Perkinsela. The following complexes are shown: respiratory chain complexes I to V, the RECC (editosome), mitochondrial RNA-binding complex 1 (MRB1), and the LSU and SSU of the mitochondrial ribosome. A number of other proteins involved in mRNA/gRNA processing are also shown (see Table S1 for definitions of protein acronyms).
RNA editing and processing complexes, mitochondrial ribosomes.
We next verified the presence of nucleus-located genes for proteins imported into the Perkinsela mitochondrion by using T. brucei as a reference. Despite its endosymbiotic lifestyle and large evolutionary distance from other kinetoplastid flagellates, Perkinsela has generally conserved kinetoplastid mitochondrial transcription and translation machineries, as well as a complex RNA editing machinery (Fig. 4; see also Table S1 in the supplemental material). The composition of these protein complexes is described below.
Transcription of the mitochondrial genome is performed by a dedicated single-subunit, phage T3/T7-like RNA polymerase (46) which is present in Perkinsela. In trypanosomatids, the formation of short A-tails on pre-edited mRNAs and long A/U tails on fully edited transcripts is controlled by kinetoplast poly(A) polymerase 1 (KPAP1), 3′-terminal uridylyl transferase (TUTase) KRET1, and their accessory factors, KPAF1 and KPAF2, which together regulate mRNA translatability and stability (47). Except for KPAF2, all these nucleus-encoded and mitochondrion-targeted proteins are present and well conserved in Perkinsela. KRET1 also appends 3′-oligo(U) tails to rRNAs and gRNAs in trypanosomatids (48), so it seems reasonable to assume that this enzyme performs the same function in Perkinsela.
The core set of editing reactions in trypanosomatid mitochondria is executed by the RNA editing core complex (RECC), also called the 20S editosome (13). In the first step of the editing reaction, the cleavage of the mRNA at a mismatch between it and a hybridizing gRNA yields 5′ and 3′ fragments bridged by the gRNA, and this reaction is performed by one of three RECC endonucleases (49). Remarkably, among these three endonucleases, only a homolog of the U-insertion-specific enzyme KREN2 was found in Perkinsela; KREN1 (the deletion-specific endonuclease) and KREN3 were not detected. KREN3 is known to act on the cox2 transcript edited by a cis-gRNA located in its 3′ untranslated region in trypanosomatids (50). Of the KREPB proteins (KREPB6 through 8), which within RECC form dimers with the KREN endonucleases (51), only KREPB6, which in T. brucei interacts with KREN3, was found. KREPB8 and KREPB7, which dimerize with KREN1 and KREN2, respectively, are apparently absent in Perkinsela. With regard to the deletion of extraneous Us from the 5′ mRNA fragment (52), the dedicated exonucleases KREX1 and KREX2 have predicted orthologs in Perkinsela. The KRET2 TUTase, responsible for adding Us to the 5′ mRNA fragment, and the insertion-specific RNA ligase KREL2, which reseals the two RNA fragments, were also found (53, 54). The deletion-specific RNA ligase KREL1 is missing in Perkinsela. Of the accessory and structural RECC subunits (KREPA1 through 6 and KREPB4 and 5), three are present, whereas five seem to be missing (see Table S1 in the supplemental material). The undetected orthologs were presumably replaced or have evolved beyond recognition in Perkinsela, or they are normally essential for editing transcripts encoding the numerous complex I subunits, which have been lost in this kinetoplastid.
In addition to the RECC, which provides the core editing enzymatic activities, various other proteins and macromolecular complexes have been shown to play vital roles in editing. One example is mitochondrial RNA-binding complex 1 (MRB1), a dynamic structure that binds and recruits gRNAs into the editing complex, processes massively edited mRNAs that require several gRNAs, and links RNA editing with mRNA tailing and translation machineries (12). Of six invariably recovered MRB1 subunits (55–58), three are found in Perkinsela, including the crucial gRNA-binding subunit GAP1, which presumably forms a homotetramer, as its paralog GAP2 is missing. Core subunits MRB5390 and MRB8620 are also absent from Perkinsela (see Table S1 in the supplemental material). Of 14 other putative editing complex members, only 5 are found, whereas TbRGG1, TbRGG2, MRB8170, and MRB4160 (58, 59) are missing in Perkinsela (see Table S1). However, TbRGG3, which associates with MRB1 as well as other mitochondrial RNA-binding proteins (60), yields a hit. Hence, the same picture emerges as for the 20S editosome: the functional core of the MRB1 complex is mostly conserved between Perkinsela and its trypanosomatid relatives.
A separate small complex, a heterotetramer of RNA-binding proteins 1 and 2 (MRP1/MRP2) which stimulates annealing of gRNA and mRNA molecules (61, 62), is also present in Perkinsela. The same is true for RNA-binding protein 16 (RBP16), which interacts with both mRNA and gRNA and has a multifunctional role in mitochondrial RNA metabolism (63). However, RNA processing endonuclease mRPN1, which is involved in cleavage of long gRNA precursor transcripts (64), was not detected, suggesting that gRNA transcription patterns may profoundly differ between trypanosomatids and Perkinsela (we did not attempt to identify gRNA genes in the latter). Finally, both RNA-editing helicases KREH1 and KREH2, likely required for unwinding the gRNA:mRNA duplex (55, 65, 66), have been detected in Perkinsela.
Ribosomes in trypanosomatid mitochondria contain extremely reduced rRNAs and have acquired a multitude of novel proteins, apparently to compensate for the loss of RNA domains (22), or through protein “accretion” by a neutral evolutionary mechanism (67). Thus, both ribosomal LSU and SSU contain dozens of trypanosomatid-specific proteins, but they lack some of the universally conserved ones (18). Of 27 mitochondrial LSU proteins conserved throughout eukaryotes, 25 are found in Perkinsela (see Table S1 in the supplemental material). Of 49 trypanosomatid-specific mitochondrial LSU subunits, only 15 could not be detected in Perkinsela (see Table S1). This significant conservation between Perkinsela and trypanosomatids is also seen for the SSU ribosomal proteins: in the case of 10 subunits universally present in mitochondrial ribosomes, only 1 is missing, whereas just 4 out of 43 trypanosomatid-specific proteins could not be detected in Perkinsela (see Table S1).
In summary, proteins incorporated into RNA editing and processing as well as translation machineries are generally conserved in Perkinsela, despite its deep evolutionary separation from T. brucei and other trypanosomatids (26).
Analysis of edited RNA molecules.
We carried out an in-depth analysis of RNA editing based on thousands of Illumina reads per gene for Perkinsela strain CCAP1560/4, greatly surpassing the limits of traditional methods. We also took advantage of lower coverage but longer read sequence data (up to 450 bp long) generated for the GillNOR1/I strain (see File S2 in the supplemental material). Preliminary analyses revealed that read mapping with such a high fraction of U-indels is problematic, as publicly available read mapping software was not designed for such applications. Our initial approach using the Bowtie2 v.2.0.2 mapper with low indel penalties resulted in alignments that required extensive manual improvement due to misalignments in regions with closely spaced U-indel sites (data not shown). In order to improve mapping of U-indel-rich reads, we modified the Bowtie2 v.2.0.2 software, introducing nucleotide-specific gap opening and gap extension penalties into the Smith-Waterman alignment module (see Materials and Methods). Mapping reads with strict penalties for gaps containing A, C, or G but with relaxed penalties for gaps containing only U dramatically reduced the number of misalignments and improved the yield of edited reads (see Fig. S2 in the supplemental material). In the case of pan-edited transcripts or long editing domains, extra runs of mapping on partially edited templates were necessary to reconstruct the final edited product, as reads edited over the entire length lacked seeds long enough for initial mapping.
To overcome the problem of missing seeds, we developed a novel read mapping tool, T-aligner, based on the Smith-Waterman algorithm and designed to mimic the 3′-to-5′ progression of RNA editing in kinetoplastids. Initially, a fixed seed is chosen in a never-edited or universally edited 3′ terminus of the transcript (or editing domain, in appropriate cases), and then reads are mapped and the final edited sequence is reconstructed with the help of T-aligner (see Materials and Methods). At this stage, further iterations of read mapping are possible, shifting the seed in the 5′ direction. Using T-aligner, we identified mature edited transcripts in Perkinsela and investigated the extent to which alternative editing occurs.
RNA editing in Perkinsela resembles the system described in the model Trypanosoma and Leishmania species. However, the general distribution of editing sites (Fig. 2; Table 1), namely, the fact that the 3′ and 5′ regions of genes usually contain separate editing domains, more closely resembles the situation in the bodonid Trypanoplasma borreli, for which only a few genes and transcripts have been sequenced (41). Interestingly, in the case of the Perkinsela cox2 gene, we found that the 5′ domain is edited prior to the 3′ domain, despite the canonical 3′-to-5′ progression of U-indel editing inside these domains (33). Upon inspection of the longest read fraction, we observed no reads in which the 3′ domain was at least partially edited when the 5′ domain was not (see Fig. S3 in the supplemental material).
In total, fully edited versions of six transcripts have 1,196 Us inserted and 103 Us deleted at 576 distinct edited sites in the Perkinsela CCAP1560/4 strain, and there are 1,192 Us inserted and 92 Us deleted at 568 edited sites in the GillNOR1/I strain (Table 1). Alignments of edited and preedited mRNAs, their translation, and the trees built for predicted proteins and their kinetoplastid orthologs are shown for cox2 in Fig. 5 (and for the other five mitochondrial genes in File S4 of the supplemental material). Finding a protein with an expected length and an expected position in a phylogenetic tree constitutes strong in silico evidence that the predicted translation product from a reconstructed edited mRNA sequence is most probably correct. The divergence of editing patterns between the two studied isolates (Fig. 5A and B; see also File S5 in the supplemental material) is similar to that observed among various species of trypanosomatids (68), and sequences become noticeably less divergent following RNA editing, as shown in Fig. 5B for cox2: preedited mRNA sequences of the two Perkinsela strains have 82% identity, while the respective edited molecules have 91% identity (85 and 56 nucleotide differences, respectively). Indeed, this effect is even more pronounced in the case of the pan-edited atp6 transcript, with just 79% identity of preedited mRNAs between the strains but with 94% identity after posttranscriptional modification (see File S4). These results are consistent with the notion that while protein sequences are maintained by selective forces, the sequence of a cryptic gene is able to evolve more freely, with mutations “corrected” by RNA editing (68).
FIG 5 .

U-indel editing in the cox2 mRNA of Perkinsela strains CCAP1560/4 and GillNOR1/I. (A) Alignment of edited and pre-edited transcript sequences. U insertions and deletions are highlighted in light blue and red, respectively. (B) Pairwise percent identities (in the lower left half of the matrix) and numbers of different positions (upper right) between edited/pre-edited sequences of both strains. (C) A maximum likelihood unrooted tree of COX2 proteins of Perkinsela and other kinetoplastids, and of Diplonema papillatum, used as an outgroup. The tree was constructed with the following settings: WAG+Г substitution model, neighbor-joining starting tree, 1,000 bootstrap replicates. Branches supported by bootstrap values of >70% are shown with thicker lines. Scale bar shows the inferred number of amino acid substitutions per site.
Alternative editing and “misediting.”
We observed a certain fraction of alternatively edited reads for each of the 10 edited transcript domains in the Perkinsela mitochondrial genome. We defined “alternative” reads as those containing at least one alternatively edited site that satisfied the following conditions: (i) it was never edited in the main editing product; (ii) the U-indel was longer than in the main product; and (iii) insertion occurred instead of deletion in the main product, or vice versa. Short alternatively edited reads fully contained within longer reads were not considered in further analyses. The fraction of alternatively edited reads (relative to all edited reads), as inferred using T-aligner, was found to vary from 19% in the investigated edited domains of cox2, cox3, and cob to 52% in the pan-edited atp6 transcript (see File S5 in the supplemental material). Absolute numbers of alternative reads in our data set varied from 44 for rps12 to 1,979 for atp6, depending on the level of coverage for a particular strain, transcript abundance, and T-aligner seed selection (see File S5).
Importantly, we observed no cases of a clearly predominant single alternative editing intermediate, and an overwhelming majority of alternative intermediates was represented by single reads, as shown in File S5 in the supplemental material. We used the following edited domains as model cases: (i) cox1, 5′ domain, strain CCAP1560/4; (ii) cox2, 3′ domain, strain GillNOR1/I; (iii) cox3, 5′ domain, strain CCAP1560/4; (iv) cob, 3′ domain, strain GillNOR1/I; (v) the 3′ portion of the pan-edited atp6 transcript, strain CCAP1560/4; and (vi) the pan-edited rps12 transcript, strain GillNOR1/I (see File S1 in the supplemental material). The cox2 3′ domain (Fig. 2) was most informative due to its high coverage with long strand-specific reads (average length, 192 nt; maximum length, ~400 nt; 4,711 edited reads in total) (see File S5 in the supplemental material). A maximum number of 14 alternatively edited sites was observed in the reads available for the 3′ domain of cox2. However, just 10 out of 880 reads mapping to this domain contained 10 or more alternatively edited sites (see File S5). An even larger pool of 1,979 alternatively edited reads mapping to the 3′ part of atp6 contained just 6 reads with 10 or more alternatively edited sites. However, shorter reads were available in this case (see File S5). Taken together, these numbers, the read length distribution (see File S2 in the supplemental material), and the read counts for alternative intermediates (see File S5 in the supplemental material) strongly indicate that alternative final transcripts, comparable with the main transcript in length and abundance, do not occur in this system. A typical selection of alternative intermediates is shown for the 3′ edited domain of cox2 (Fig. 6).
FIG 6 .
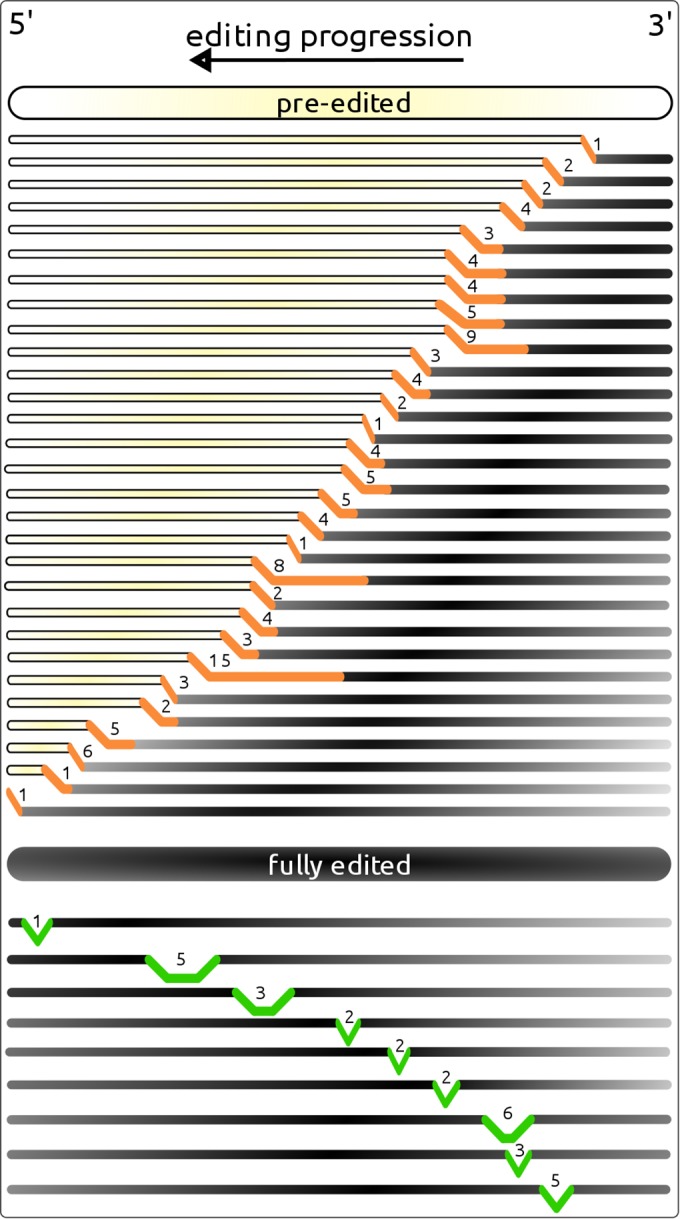
The most abundant alternatively edited intermediates mapped to the cox2 transcript (3′ edited domain) in Perkinsela strain GillNOR1/I. The preedited sequence is shown in yellow, and the main edited product is shown in black. Alternatively edited read fragments are shown in orange if they follow the sequence edited in a standard way or in green if they occur in the middle of such a sequence. The number of alternatively edited sites is shown in each case, and the length of highlighted regions correlates with the number of inserted Us.
The majority of editing intermediates contained one or several alternative sites at the end of an edited stretch of sequence, followed by a preedited sequence. Considering that in T. brucei approximately 45 nucleotides (from nt 24 to 61) are covered by an average gRNA (17), the terminal stretches observed in Perkinsela are probably generated by one or two consecutively acting gRNAs. The paucity of longer terminal stretches (see File S1 in the supplemental material) suggests that we are mainly observing instances of abortive “misediting” (36). As is apparent even from a small selection of alternative intermediates (Fig. 6), editing errors occur almost everywhere along the transcript. However, a wider selection of intermediates (see File S1) revealed a few hot spots.
Another type of editing intermediate contains one to six alternative sites within a sequence corresponding to the main editing pathway. These “internal” intermediates are apparently produced by a single gRNA guiding several editing sites in a noncanonical way, but still generating an anchor sequence for a subsequent gRNA in the main editing pathway. Remarkably, both types of alternative editing have been predicted in T. brucei based on deep sequencing of a gRNA library with a total of ~600 major sequence classes (17); gRNAs were identified that create an alternative sequence not usable as an anchor, as were gRNAs that edit several sites in an alternative way but create an anchor region for the next gRNA in the main editing pathway. For instance, an alternative gRNA might initiate editing at the 3′ end of atp6 (also known as A6) in T. brucei but is also able to create a normal anchor for the next gRNA. The same is true for alternative gRNA editing of the NAD8 transcript (nad8). In Perkinsela, we also observed intermediates containing more than one internal alternatively edited stretch, or intermediates with a combination of terminal and internal alternatively edited stretches (see File S1 in the supplemental material), all of which are of low abundance.
Based on our data, the RNA editing pathway in Perkinsela and probably all kinetoplastids can be viewed as a “tree” with numerous branching points, with only one path in the tree being predominant and the rest probably representing errors of the editing system. In T. brucei, alternative gRNAs were identified for at least five genes, with some being even more abundant than the standard gRNAs for the same site (17). Given a high percentage of alternative reads accumulated for some edited domains in Perkinsela (e.g., 52% for atp6), and the fact that alternative reads that map to the rps12 gene lack stop codons in at least one frame, the mitochondrial transcription-translation system in this organism, in one way or another, has to cope with a large number of “incorrect” transcripts. In plant organelles, only edited translation products appear to accumulate in mitochondrial ribosomes (69). Whether the Perkinsela mitochondrion is able to tolerate “incorrect” protein products, or that some sort of discrimination by the translation machinery is in place, remains an open question.
MATERIALS AND METHODS
Cell culture.
Paramoeba pemaquidensis strain CCAP1560/4 was obtained from the CCAP (Culture Collection of Algae and Protozoa). Cells were grown on MYS medium (0.01% malt extract and 0.01% yeast extract in artificial seawater, solidified with 1.5% agar) (70). P. pemaquidensis strain GillNOR1/l was obtained from the culture collection of the Institute of Parasitology, Czech Academy of Sciences, and was grown on MY75S medium (0.01% malt extract and 0.01% yeast extract in artificial seawater, solidified with 2.0% agar). Both strains were grown in the dark at 20°C, and cells were collected after about 3 months.
Microscopy.
P. pemaquidensis strain GillNOR1/I, carrying Perkinsela and feeding on diverse bacteria, was grown on agar plates, and the cells were collected as described previously (23). Cells were prepared for phase-contrast microscropy, DAPI epifluorescence light microscopy, and high-pressure freezing transmission electron microscopy following protocols described elsewhere (71, 72).
Paramoeba pemaquidensis sequencing.
Two strains of P. pemaquidensis with their respective Perkinsela endosymbionts were used in this study. Strains CCAP1560/4 (73) and GillNOR1/I were isolated from gills of Atlantic salmon captured in the waters of Wales and Tasmania, respectively (74). We prepared and sequenced total genomic and poly(A)-enriched transcriptomic libraries from strain GillNOR1/I (see File S2 in the supplemental material) and genomic, poly(A)-enriched transcriptomic and spliced leader RNA-enriched transcriptomic libraries for strain CCAP1560/4. The latter library was used for assigning contigs to the Perkinsela nuclear genome, as it is currently impossible to physically separate Perkinsela from its amoeba host, or to separate their DNAs (GenBank accession number LFNC00000000), and mRNAs “capped” with a spliced leader RNA do not occur in the amoeba.
Mitochondrial genome assembly.
Raw DNA sequence reads from all sequencing platforms were filtered and trimmed to ensure quality and then depleted of adapter sequences, and paired-end transcriptomic reads were merged using the CLC Genomics Workbench v.6.5 (see File S2 in the supplemental material). The mitochondrial genomes of both Perkinsela strains were assembled from combined next-generation sequencing reads with the Newbler Assembler (GS de novo Assembler v.2.9), from single 454, mate-pair, and paired-end Illumina HiSeq reads in the case of strain CCAP1560/4 and from paired-end Illumina MiSeq reads for strain GillNOR1/I (see File S2). A number of assembly parameters were tested with the goal of maximizing mitochondrial contig size. Manual analysis of a graph of alternative contig connections (produced by Newbler) with an in-house visualizing script was used to close gaps and assemble long repetitive regions. RNA-seq assemblies were performed with Trinity software (75).
Gene identification.
Proteins predicted from the Perkinsela mitochondrial contigs and translated transcriptomic assemblies were initially identified using the HMMER3 software. Available kinetoplastid and diplonemid mitochondrial protein sequences were used for the construction of hidden Markov models (HMMs), which were subsequently used as queries against conceptual translations of the genome and transcriptome assemblies, with an E value cutoff of 10−1. Best-scoring hits were compared to the NCBI(nr) protein database in order to filter out host and bacterial proteins. Additional mitochondrial contigs were then identified via BLASTn, using typical repetitive regions from contigs identified in the first step. Perkinsela nucleus-encoded proteins associated with mitochondrial oxidative phosphorylation, RNA editing and processing machineries, and mitochondrial translation were identified using HMMs (with an E value cutoff of 10−10), based on the corresponding orthologous groups from the OrthoMCL database realigned using MUSCLE (76). We classified as “missing” all Perkinsela hits with an E value of >10−50 that did not recover the corresponding T. brucei ortholog as the best hit in reciprocal BLASTp analysis (with an E value cutoff of 10−3). Perkinsela hits with an E value of <10−50 and without a suitable reciprocal BLASTp hit were aligned with their supposed orthologs in trypanosomatids. All protein alignments were performed using MUSCLE with default settings and checked manually.
Searches for rRNAs.
The following approaches were used to identify mitochondrial rRNA genes in Perkinsela. First, BLAST searches with known kinetoplastid and diplonemid homologs as queries were performed with an E value cutoff of 10−5. Second, transcribed regions on contigs not assigned to the host or Perkinsela nuclear genomes were selected for further inspection. Third, reads containing the LSU peptidyl transferase core sequence (ACCTCGNTGT) conserved in Diplonema (16) were assembled separately using the CLC Genomics Workbench v.6.5. The top candidates from each of these searches were subjected to manual secondary structure folding, with terminal hairpin prediction performed using the Mfold thermodynamic folding application (http://mfold.rit.albany.edu/?q=mfold/RNA-Folding-Form). Default options were used to construct guiding graphs for manual secondary structure prediction (except for the “Loop max” option, which was restricted to 10, 20, and 30 nucleotides). Structures were assessed for similarity to those of Leishmania LSU and SSU and Diplonema LSU rRNAs (16, 22).
Search for gRNA genes.
We implemented a simple search for putative gRNA genes in the following way: we reversed and complemented a final edited sequence of a transcript, cut it into 25-nt seeds with a seed step of 1 nt (taking only transcript regions undergoing RNA editing), searched for exact seed matches in the genome assembly and in its reverse complement, allowing up to 5 A-to-G or C-to-T substitutions in the 25-nt seed, in order to accommodate G-U base pairing. Then, we searched for RNA-seq reads that exactly matched the genomic hits, so that only transcribed hit regions were taken.
Bowtie2 modification.
Bowtie2 is an open-source, fast, and accurate short read mapper written in the C++ programming language (77). It uses a fast multiseeding procedure to find candidate alignment locations and then proceeds with the Smith-Waterman algorithm to create the best gapped alignment. For additional speed, Bowtie2 implements the Smith-Waterman alignment algorithm with SIMD (single instruction, multiple data), allowing it to fill several dynamic programming table cells by executing a single instruction (78). However, Bowtie2 uses a scoring system with equal gap open and extension penalties for the four nucleotides, A, G, T, and C. We modified Bowtie2 to facilitate accurate alignment of U insertion/deletion edited RNA reads, while preserving mapping speed and accuracy. Edited reads of the mitochondrial genomes of kinetoplastids have U-indels only, and therefore they can be aligned correctly when gap penalties for Ts (corresponding to Us in RNA) are different from those for A, G, and C.
We modified the Bowtie2 v.2.0.2 source code and implemented a more complex nucleotide-specific gap-scoring system that allowed separate penalty values for A, G, T, and C by using the −rdg-X and −rfg-X options on the command line (for gaps in the read and reference, respectively, where X can be A, T, G, or C). Source code modifications were made both in the aligner module, which fills the dynamic programming table, and in the backtrack module of the program, which reconstructs the alignment using the filled dynamic programming table. Branch and array access instructions were minimized for each step, ensuring minimal time cost for more complex scoring. Using this scoring matrix, U-indel edited reads can be successfully mapped and accurately aligned with a low T-indel penalty and high penalties for other nucleotides. Additional modifications of the alignment procedure were necessary in order to let reads have a gap/mismatch after the last nucleotide of the read (option gbar 0). This option allows the seeding of more extensively edited reads on a preedited RNA sequence and prevents a significant fraction of edited reads from being discarded.
T-aligner.
T-aligner is a new software program written for the purpose of this study and we used the C++ programming language (source code posted online at https://github.com/jalgard/T-Aligner). T-aligner combines the optimal but time-consuming Smith-Waterman alignment with fast hash-based exact matching. The algorithm is specially designed to map extensively edited RNA-seq reads on pre-edited transcript references, also called cryptogenes. Exact matches between short substrings (seeds) are first found using a hash table. A local optimal alignment is then produced with the Smith-Waterman algorithm, allowing “T, −” and “−, T” gaps with zero penalty, thus taking into account the biological mechanism of U-indel RNA editing. The general T-aligner workflow is as follows (see also Fig. S4 in the supplemental material): a fixed seed is chosen in a never-edited or universally edited 3′-terminus of the transcript (or editing domain, in appropriate cases). Reads are then mapped if they satisfy the following criteria: (i) they contain the seed; (ii) at least part of the read lies 5′ to the seed; (iii) the alignment may contain any number of U-indels of any length; (iv) the alignment contains no other indels and no or few mismatches. After the alignments are produced, T-aligner classifies all editing events (U insertion or U deletion) and clusters the reads into three groups: (i) those matching the reference sequence, (ii) those matching the putative main “editing pathway” (i.e., the user-defined final edited product), and (iii) all other reads containing alternative editing events. Reads matching the main pathway are defined as follows: (i) those with no additional edited sites compared to the main pathway; (ii) reads with insertions/deletions that are shorter or equal in size to those in the main pathway; and (iii) reads in which all sites are edited in the same direction as in the main pathway (e.g., insertion in the main pathway versus insertion in a sequence read). Reads in violation of any of these conditions are placed in the “alternative editing” group. Sequence reads that are exact substrings of other reads are then merged into “editing intermediates.” The support value associated with an editing intermediate can be used to determine the most abundant sequences, which is useful when examining alternative editing. All sequences clustered into the “reference” and “main pathway” groups are assigned a support value equal to the number of reads in each group. For each sequence from the “alternative” group, support is determined as follows: reads falling into the “reference” and “main pathway” groups are excluded; if a read is unique, i.e., can be included as a substring in at most one longer read, it adds 1 to the support value; if a read supports k > 1 alternative sequences, it adds 1/k to a support value for each sequence.
Read mapping and analysis of U-indel RNA editing.
Bowtie2 v.2.0.2 or v.2.1.0 mapping software was used for both DNA and RNA-seq reads, and we utilized the end-to-end mapping mode, the “very sensitive” options, and default alignment scoring. In order to produce precise alignments in extensively edited regions, we used a modification of Bowtie2 v.2.0.2 with the base-specific indel penalties described above. The following set of options was routinely used: (i) high gap opening and extension penalties of 10 for A, G, or C in the reference and individual sequence reads (--rfg 10,10; --rdg 10,10); (ii) minimal gap opening and extension penalties of 1 for T or A (depending on transcript orientation) in the reference and reads (--rfg-T 1,1; --rdg-T 1,1 or --rfg-A 1,1; --rdg-A 1,1); (iii) a high mismatch penalty equal to 18 (--mp 18); (iv) options allowing terminal mismatches (gbar 0; dpad 50), and (v) other options (--end-to-end; -D 20; -R 3; -N 1; -L 14; -i S,1,0.50; --score-min L,0,-2). Reads mapped to the edited regions were manually checked before further processing. Poor-quality alignments, especially those introducing large gaps, were not considered. Alignments made with Bowtie2 were cut into overlapping windows and examined to find sequences appropriate for seeding further read mappings with T-aligner.
One to three iterations of read mapping with T-aligner (with the original seed shifting in the 3′-to-5′ direction) were enough to cover the whole transcript or its edited region and then reconstruct the main editing pathway. Repeating T-aligner-assisted read mapping with prior knowledge of the main edited product allowed us to reveal and quantify alternative editing products.
Northern blotting.
Northern blotting analysis of cox2 was performed as previously described (59). Briefly, 10 µg of RNA isolated from Perkinsela strain GillNOR1/I and T. brucei strain 29-13 was run on a high-resolution 4% acrylamide–7 M urea gel and transferred onto a Zeta-probe membrane (Bio-Rad). The membrane was subsequently probed with 5′-32P-labeled oligonucleotides corresponding to the antisense (5′-CCCTTTCAACACGTCAAAACAAGC-3′) and sense (5′-GCTTGTTTTGACGTGTTGAAAGGGC-3′) pre-edited sequences of the 5′ end, i.e., the last to be processed, of the larger 3′-edited domain. The oligonucleotides were also used to probe dot blots of serially diluted, denatured PCR products amplified from this same region, in order to demonstrate that the two probes were equally sensitive.
SUPPLEMENTAL MATERIAL
Perkinsela strain GillNOR1/I mitochondrial scaffolds with both sense and anti-sense transcriptomic reads mapped. Almost no antisense transcription is visible, which is supported by the Northern blot in Fig. 3. The sense transcription profile, showing very low coverage for pan-edited genes rps12 and atp6, is different from that shown in Fig. 2 since “U-indel optimized” settings in Bowtie were not used here. “U-indel optimized” settings may produce strand biases, e.g., favoring U-indels on the forward strand, but not A-indels on the reverse strand. Therefore they were not used for the purpose of inter-strand comparison of transcription profiles. However, regular Bowtie “very sensitive” settings produce especially poor coverage in the case of pan-edited transcripts. Download
Mapping of edited reads with Bowtie2 v.2.0.2 and its modified version. An alignment window shown here covers the 3′ editing region of cox1 in Perkinsela strain GillNOR1/I. Just a few reads are mapped by the standard Bowtie2 algorithm using the “very-sensitive” setting. In contrast, modified Bowtie2 with T-indel-sensitive settings results in 12-fold increase of mapped read count (not all reads are shown in the figure). Moreover, misalignments such as those shown with arrows are missing because gaps containing ACG are penalized. Download
All reads spanning both 5′ and 3′ edited domains of cox2 in Perkinsela strain GillNOR1/I. Only parts of the edited domains adjoining the central non-edited region are shown, and the non-edited region itself is omitted (represented by the black bar in the center of the picture). The pre-edited sequence is shown at the bottom and the final edited sequence is on top. Insertions are shown in light blue, deletions in red, and edits corresponding to the main edited product are boxed (alternative edits are not boxed). We found virtually no reads edited in the 3′ domain but not edited in the 5′ domain, but many examples of the opposite arrangement. Only a single read carries one alternative edit in the 3′ domain and no other edits. Download
Workflow of T-aligner. Download
Alternatively edited and “misedited” products for cox1, cox2, cox3, cob, atp6, and rps12. The figure represents an output of T-aligner, illustrating non-redundant alternatively edited reads (also termed "editing intermediates" in this paper to distinguish them from reads in the raw dataset). Each column represents an editing site irrespective of the U-indel length observed; each line represents a read with its sequence in white. U-insertion and U-deletion sites identical to those in the main edited product are shown in dark green and dark red, respectively. A site is considered alternatively edited if any of the following conditions are met: (i) a site is never-edited in the main editing product; (ii) U indel is longer than in the main product; (iii) U insertion occurs instead of deletion in the main product or vice versa. Alternatively edited sites are shown in bright green and orange for U insertions and U deletions, respectively, and sites with the editing direction reversed (group iii) are denoted in blue. The transcript is schematically shown at the bottom, with edited domains in green and the seed used by T-aligner in yellow. Download
Genomic and transcriptomic NGS reads. Paired transcriptomic reads were merged with the CLC Genomics Workbench v.6.5 prior to mapping. Merged read length distribution is shown for the Perkinsela CCAP1560/4 (A) and GillNOR1/I strains (B). The abrupt edges of the distribution in panel A are due to 100 bp and shorter trimmed reads which produce merged reads of 190 bp or shorter, if a minimum overlap of 10 bp is required. Download
Guide RNA gene candidates revealed in the genome assembly of Perkinsela sp. CCAP1560/4. Aligned stretches of gRNA and mRNA (at least 25 bp in length) are shown in uppercase, and their flanks are shown in lowercase. Contig IDs and start coordinates are shown, along with a number of matching transcriptomic reads (support). The 5′ AC-rich region of gRNAs is underlined. Download
Aligned edited/pre-edited transcripts and trees for the final protein sequences of 6 mitochondrial genes in Perkinsela strains CCAP1560/4 and GillNOR1/I. For each gene (cox1, cox2, cox3, cob, rps12, and atp6) the following information is shown: (i) edited and pre-edited transcript sequences with corresponding translations (U-insertions are denoted in blue and U-deletions are marked with orange circles); (ii) pairwise percent identities (in the lower left part of the matrix) and numbers of different positions (in the upper right part) between edited/pre-edited sequences of both strains; (iii) a maximum likelihood unrooted tree for protein sequences of Perkinsela, other kinetoplastids, and an outgroup (Diplonema papillatum, Euglena gracilis, or Naegleria gruberi, depending on sequence availability). The trees were constructed using the following settings: WAG+Г substitution model, neighbor-joining starting tree, 1,000 bootstrap replicates. Branches supported by bootstrap values >70% are shown with thicker lines. Scale bars show inferred number of amino acid substitutions per site. Download
Alternative editing quantification. Pie charts illustrating counts of reads matching the main editing pathway, the pre-edited sequence, and alternatively edited reads in Perkinsela strains CCAP1560/4 and GillNOR1/I (A). Alternative editing intermediates in Perkinsela sp. Mitochondria, sorted according to their rounded support values (B) and number of editing sites (C) (x-axis) are shown. Read counts in logarithmic scale (y-axis) are given separately for each model edited domain (strain CCAP1560/4 for cox1, cox3, atp6; strain GillNOR1/I for cox2, cob, rps12). Download
Perkinsela nucleus-encoded proteins associated with mitochondrial oxidative phosphorylation, RNA editing and processing machineries, and mitochondrial translation, identified using HMM and reciprocal BLASTp.
ACKNOWLEDGMENTS
We thank Ivan Fiala, Hanka Pecková, and Eva Horáková for technical assistance and Bruce Curtis for bioinformatics support.
This work was supported by the Czech Grant Agency (P305/12/2261), an AMVIS grant (LH 12104), RNPnet FP7 program 289007, and a Praemium Academiae award to J.L. and an operating grant awarded to J.M.A. from the Canadian Institutes of Health Research. J.M.A. also acknowledges support from the Canadian Institute of Advanced Research and Dalhousie University’s Centre for Comparative Genomics and Evolutionary Bioinformatics. We acknowledge the use of research infrastructure funded by the EU 7th Framework Programme (no. 316304). P.F. was supported by the Moravian-Silesian Region Projects MSK2013-DT1, MSK2013-DT2, and MSK2014-DT1; P.F. and E.G. obtained support from the Russian Foundation for Basic Research (project 14-04-31936 to P.F. and E.G. and project 14-04-01717 to E.G.); and M.D.L. was supported by project 14-50-00029 of the Russian Science Foundation.
Footnotes
Citation David V, Flegontov P, Gerasimov E, Tanifuji G, Hashimi H, Logacheva MD, Maruyama S, Onodera NT, Gray MW, Archibald JM, Lukeš J. 2015. Gene loss and error-prone RNA editing in the mitochondrion of Perkinsela, an endosymbiotic kinetoplastid. mBio 6(6):e01498-15. doi:10.1128/mBio.01498-15.
REFERENCES
- 1.Verner Z, Basu S, Benz C, Dixit S, Dobáková E, Faktorová D, Hashimi H, Horáková E, Huang Z, Paris Z, Peña-Diaz P, Ridlon L, Týč J, Wildridge D, Zíková A, Lukeš J. 2015. Malleable mitochondrion of Trypanosoma brucei. Int Rev Cell Mol Biol 315:73–152. doi: 10.1016/bs.ircmb.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Maslov DA, Votýpka J, Yurchenko V, Lukeš J. 2013. Diversity and phylogeny of insect trypanosomatids: all that is hidden shall be revealed. Trends Parasitol 29:43–52. doi: 10.1016/j.pt.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 3.De Vargas C, Audic S, Henry N, Decelle J, Mahé F, Logares R, Lara E, Berney C, le Bescot N, Probert I, Carmichael M, Poulain J, Romac S, Colin S, Aury JM, Bittner L, Chaffron S, Dunthorn M, Engelen S, Flegontova O, Guidi L, Horák A, Jaillon O, Lima-Mendez G, Lukeš J, Malviya S, Morard R, Mulot M, Scalco E, Siano R, Vincent F, Zingone A, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans Coordinators, Acinas SG, Bork P, Bowler C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Not F, Ogata H, Pesant S, Raes J, Sieracki ME, Speich S, Stemmann L, Sunagawa S, Weissenbach J, Wincker P, Karsenti E. 2015. Eukaryotic plankton diversity in the sunlit global ocean. Science 348:1261605. doi: 10.1126/science.1261605. [DOI] [PubMed] [Google Scholar]
- 4.Benne R, van den Burg J, Brakenhoff JPJ, Sloof P, van Boom JH, Tromp MC. 1986. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46:819–826. doi: 10.1016/0092-8674(86)90063-2. [DOI] [PubMed] [Google Scholar]
- 5.Maas S. 2012. Posttranscriptional recoding by RNA editing. Adv Protein Chem Struct Biol 86:193–224. doi: 10.1016/B978-0-12-386497-0.00006-2. [DOI] [PubMed] [Google Scholar]
- 6.Gott JM, Emeson RB. 2000. Functions mechanisms of RNA editing. Annu Rev Genet 34:499–531. doi: 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- 7.Gray M. 2003. Diversity and evolution of mitochondrial RNA editing systems. IUBMB Life 55:227–233. doi: 10.1080/1521654031000119425. [DOI] [PubMed] [Google Scholar]
- 8.Li JB, Church GM. 2013. Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat Neurosci 16:1518–1522. doi: 10.1038/nn.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avesson L, Barry G. 2014. The emerging role of RNA DNA editing in cancer. Biochim Biophys Acta 1845:308–316. doi: 10.1016/j.bbcan.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Dang Y, Green BR. 2009. Substitutional editing of Heterocapsa triquetra chloroplast transcripts and a folding model for its divergent chloroplast 16S rRNA. Gene 442:73–80. doi: 10.1016/j.gene.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Takenaka M, Zehrmann A, Verbitskiy D, Härtel B, Brennicke A. 2013. RNA editing in plants and its evolution. Annu Rev Genet 47:335–352. doi: 10.1146/annurev-genet-111212-133519. [DOI] [PubMed] [Google Scholar]
- 12.Hashimi H, Zimmer SL, Ammerman ML, Read LK, Lukeš J. 2013. Dual core processing: MRB1 is an emerging kinetoplast RNA editing complex. Trends Parasitol 29:91–99. doi: 10.1016/j.pt.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aphasizhev R, Aphasizheva I. 2014. Mitochondrial RNA editing in trypanosomes: small RNAs in control. Biochimie 100:125–131. doi: 10.1016/j.biochi.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marande W, Burger G. 2007. Mitochondrial DNA as a genomic jigsaw puzzle. Science 318:415. doi: 10.1126/science.1148033. [DOI] [PubMed] [Google Scholar]
- 15.Kiethega GN, Yan Y, Turcotte M, Burger G. 2013. RNA-level unscrambling of fragmented genes in Diplonema mitochondria. RNA Biol 10:301–313. doi: 10.4161/rna.23340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valach M, Moreira S, Kiethega GN, Burger G. 2014. Trans-splicing and RNA editing of LSU rRNA in Diplonema mitochondria. Nucleic Acids Res 42:2660–2672. doi: 10.1093/nar/gkt1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koslowsky D, Sun Y, Hindenach J, Theisen T, Lucas J. 2014. The insect-phase gRNA transcriptome in Trypanosoma brucei. Nucleic Acids Res 42:1873–1886. doi: 10.1093/nar/gkt973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zíková A, Panigrahi AK, Dalley RA, Acestor N, Anupama A, Ogata Y, Myler PJ, Stuart K. 2008. Trypanosoma brucei mitochondrial ribosomes: affinity purification and component identification by mass spectrometry. Mol Cell Proteom 7:1286–1296. doi: 10.1074/mcp.M700490-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aphasizheva I, Maslov DA, Aphasizhev R. 2013. Kinetoplast DNA-encoded ribosomal protein S12: a possible functional link between mitochondrial RNA editing and translation in Trypanosoma brucei. RNA Biol 10:1679–1688. doi: 10.4161/rna.26733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sloof P, van den Burg J, Voogd A, Benne R, Agostinelli M, Borst P, Gutell R, Noller H. 1985. Further characterization of the extremely small mitochondrial ribosomal RNAs from trypanosomes: a detailed comparison of the 9S 12S RNAs from Crithidia fasciculata and Trypanosoma brucei with rRNAs from other organisms. Nucleic Acids Res 3:4171–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adler BK, Harris ME, Bertrand KI, Hajduk SL. 1991. Modification of Trypanosoma brucei mitochondrial rRNA by posttranscriptional 3′ polyuridine tail formation. Mol Cell Biol 11:5878–5884. doi: 10.1128/MCB.11.12.5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma MR, Booth TM, Simpson L, Maslov DA, Agrawal RK. 2009. Structure of a mitochondrial ribosome with minimal RNA. Proc Natl Acad Sci U S A 106:9637–9642. doi: 10.1073/pnas.0901631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dyková I, Figueras A, Peric Z. 2000. Neoparamoeba Page, 1987: light electron microscopic observations on six strains of different origin. Dis Aquat Org 43:217–223. doi: 10.3354/dao043217. [DOI] [PubMed] [Google Scholar]
- 24.Young ND, Dyková I, Crosbie PBB, Wolf M, Morrison RN, Bridle AR, Nowak BF. 2014. Support for the coevolution of Neoparamoeba and their endosymbionts, Perkinsela amoebae-like organisms. Eur J Protistol 50:509–523. doi: 10.1016/j.ejop.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Moreira D, López-García P, Vickerman K. 2004. An updated view of kinetoplastid phylogeny using environmental sequences and a closer outgroup: proposal for a new classification of the class Kinetoplastea. Int J Syst Evol Microbiol 54:1861–1875. doi: 10.1099/ijs.0.63081-0. [DOI] [PubMed] [Google Scholar]
- 26.Lukeš J, Skalický T, Týč J, Votýpka J, Yurchenko V. 2014. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasitol 195:115–122. doi: 10.1016/j.molbiopara.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Dyková I, Fiala I, Pecková H. 2008. Neoparamoeba spp. and their eukaryotic endosymbionts similar to Perkinsela amoebae (Hollande) 1980: coevolution demonstrated by SSU rRNA gene phylogenies. Eur J Protistol 44:269–277. doi: 10.1016/j.ejop.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Kudryavtsev A, Pawlowski J, Hausmann K. 2011. Description of Paramoeba atlantica n. sp. Amoebozoa, Dactylopodida: a marine amoeba from the eastern Atlantic, with emendation of the dactylopodid families. Acta Protozool 50:239–253. [Google Scholar]
- 29.Feehan C, Johnson-Mackinnon J, Scheibling R, Lauzon-Guay J, Simpson A. 2013. Validating the identity of Paramoeba invadens, the causative agent of recurrent mass mortality of sea urchins in Nova Scotia, Canada. Dis Aquat Org 103:209–227. doi: 10.3354/dao02577. [DOI] [PubMed] [Google Scholar]
- 30.Young ND, Dyková I, Nowak BF, Morrison RN. 2008. Development of a diagnostic PCR to detect Neoparamoeba perurans, agent of amoebic gill disease. J Fish Dis 30:285–295. doi: 10.1111/j.1365-2761.2008.00903.x. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell SO, Rodger HD. 2011. A review of infectious gill disease in marine salmonid fish. J Fish Dis 34:411–432. doi: 10.1111/j.1365-2761.2011.01251.x. [DOI] [PubMed] [Google Scholar]
- 32.Blum B, Bakalara N, Simpson L. 1990. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 60:189–198. doi: 10.1016/0092-8674(90)90735-W. [DOI] [PubMed] [Google Scholar]
- 33.Maslov DA, Simpson L. 1992. The polarity of editing within a multiple gRNA-mediated domain is due to formation of anchors for upstream gRNAs by downstream editing. Cell 70:459–467. doi: 10.1016/0092-8674(92)90170-H. [DOI] [PubMed] [Google Scholar]
- 34.Landweber LF, Fiks AG, Gilbert W. 1993. The boundaries of partially edited transcripts are not conserved in kinetoplastids: implications for the guide RNA model of editing. Proc Natl Acad Sci U S A 90:9242–9246. doi: 10.1073/pnas.90.20.9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madina BR, Kumarumar V, Metz R, Mooers BHM, Bundschuh R, Cruz-Reyes J. 2014. Native mitochondrial RNA-binding complexes in kinetoplastid RNA editing differ in guide RNA composition. RNA 20:1142–1152. doi: 10.1261/rna.044495.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sturm NR, Maslov DA, Blum B, Simpson L. 1992. Generation of unexpected editing patterns in Leishmania tarentolae mitochondrial mRNAs: misediting produced by misguiding. Cell 70:469–476. doi: 10.1016/0092-8674(92)90171-8. [DOI] [PubMed] [Google Scholar]
- 37.Arts GJ, van der Spek H, Speijer D, van den Burg J, van Steeg H, Sloof P, Benne R. 1993. Implications of novel guide RNA features for the mechanism of RNA editing in Crithidia fasciculata. EMBO J 12:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maslov DA, Thiemann O, Simpson L. 1994. Editing and misediting of transcripts of the kinetoplast maxicircle G5 ND3 cryptogene in an old laboratory strain of Leishmania tarentolae. Mol Biochem Parasitol 68:155–159. doi: 10.1016/0166-6851(94)00160-X. [DOI] [PubMed] [Google Scholar]
- 39.Tanifuji G, Kim E, Onodera NT, Gibeault R, Dlutek M, Cawthorn RJ, Fiala I, Lukeš J, Greenwood SJ, Archibald JM. 2011. Genomic characterization of Neoparamoeba pemaquidensis (Amoebozoa) and its kinetoplastid endosymbiont. Eukaryot Cell 10:1143–1146. doi: 10.1128/EC.05027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukeš J, Guilbride D, Votýpka J, Zíková A, Benne R, Englund PT. 2002. The kinetoplast DNA network: evolution of an improbable structure. Eukaryot Cell 1:495–502. doi: 10.1128/EC.1.4.495-502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lukeš J, Arts GJ, van den Burg J, de Haan A, Opperdoes F, Sloof P, Benne R. 1994. Novel pattern of editing regions in mitochondrial transcripts of the cryptobid Trypanoplasma borreli. EMBO J 13:5086–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maslov DA, Simpson L. 2007. Strategies of kinetoplastid cryptogene discovery and analysis. Methods Enzymol 424:127–139. doi: 10.1016/S0076-6879(07)24006-6. [DOI] [PubMed] [Google Scholar]
- 43.Flegontov PN, Guo Q, Ren L, Strelkova MV, Kolesnikov AA. 2006. Conserved repeats in the kinetoplast maxicircle divergent region of Leishmania sp. and leptomonas seymouri. Mol Genet Genomics 276:322–333. doi: 10.1007/s00438-006-0145-5. [DOI] [PubMed] [Google Scholar]
- 44.Verner Z, Čermáková P, Škodová I, Kriegová E, Horváth A, Lukeš J. 2011. Complex I NADH:ubiquinone oxidoreductase is active in but non-essential for procyclic Trypanosoma brucei. Mol Biochem Parasitol 175:196–200. doi: 10.1016/j.molbiopara.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Surve S, Heestand M, Panicucci B, Schnaufer A, Parsons M. 2012. Enigmatic presence of mitochondrial complex I in Trypanosoma brucei bloodstream forms. Eukaryot Cell 11:183–193. doi: 10.1128/EC.05282-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grams J, Morris JC, Drew ME, Wang Z, Englund PT, Hajduk SL. 2002. A trypanosome mitochondrial RNA polymerase is required for transcription replication. J Biol Chem 277:16952–16959. doi: 10.1074/jbc.M200662200. [DOI] [PubMed] [Google Scholar]
- 47.Aphasizheva I, Maslov D, Wang X, Huang L, Aphasizhev R. 2011. Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes. Mol Cell 42:106–117. doi: 10.1016/j.molcel.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aphasizheva I, Aphasizhev R. 2010. RET1-catalyzed uridylylation shapes the mitochondrial transcriptome in Trypanosoma brucei. Mol Cell Biol 30:1555–1567. doi: 10.1128/MCB.01281-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol 28:122–130. doi: 10.1128/MCB.01374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golden DE, Hajduk SL. 2005. The 3′-untranslated region of cytochrome oxidase II mRNA functions in RNA editing of African trypanosomes exclusively as a cis guide RNA. RNA 11:29–37. doi: 10.1261/rna.7170705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carnes J, Soares CZ, Wickham C, Stuart K. 2011. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J Biol Chem 286:19320–19330. doi: 10.1074/jbc.M111.228965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ernst NL, Panicucci B, Carnes J, Stuart K. 2009. Differential functions of two editosome exoUases in Trypanosoma brucei. RNA 15:947–957. doi: 10.1261/rna.1373009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ernst NL, Panicucci B, Igo RP Jr., Panigrahi AK, Salavati R, Stuart K. 2003. TbMP57 is a 3′ terminal uridylyl transferase (TUTase) of the Trypanosoma brucei editosome. Mol Cell 11:1525–1536. doi: 10.1016/S1097-2765(03)00185-0. [DOI] [PubMed] [Google Scholar]
- 54.Aphasizhev R, Aphasizheva I. 2011. Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip Rev RNA 2:669–685. doi: 10.1002/wrna.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hashimi H, Zíková A, Panigrahi AK, Stuart KD, Lukeš J. 2008. TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14:970–980. doi: 10.1261/rna.888808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Panigrahi AK, Zíková A, Halley RA, Acestor N, Ogata Y, Myler PJ, Stuart K. 2008. Mitochondrial complexes in Trypanosoma brucei: a novel complex and a unique oxidoreductase complex. Mol Cell Proteom 7:534–545. doi: 10.1074/mcp.M700430-MCP200. [DOI] [PubMed] [Google Scholar]
- 57.Weng J, Aphasizheva I, Etheridge RD, Huang L, Wang X, Falick AM, Aphasizhev R. 2008. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol Cell 32:198–209. doi: 10.1016/j.molcel.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ammerman ML, Downey KM, Hashimi H, Fisk JC, Tomasello DL, Faktorová D, Kafková L, King T, Lukeš J, Read LK. 2012. Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res 40:5637–5650. doi: 10.1093/nar/gks211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kafková L, Ammerman ML, Faktorová D, Fisk JC, Zimmer SL, Sobotka R, Read LK, Lukeš J, Hashimi H. 2012. Functional characterization of two paralogs that are novel RNA binding proteins influencing mitochondrial transcripts of Trypanosoma brucei. RNA 18:1846–1861. doi: 10.1261/rna.033852.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McAdams NM, Ammerman ML, Nanduri J, Lott K, Fisk JC, Read LK. 2015. An arginine-glycine-rich RNA binding protein impacts the abundance of specific mRNAs in the mitochondria of Trypanosoma brucei. Eukaryot Cell 14:149–157. doi: 10.1128/EC.00232-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schumacher MA, Karamooz E, Zíková A, Trantírek L, Lukeš J. 2006. Crystal structures of T. brucei MRP1/MRP2 guide-RNA binding complex reveal RNA matchmaking mechanism. Cell 126:701–711. doi: 10.1016/j.cell.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 62.Zíková A, Kopečná J, Schumacher MA, Stuart K, Trantírek L, Lukeš J. 2008. Structure and function of the native recombinant mitochondrial MRP1/MRP2 complex from Trypanosoma brucei. Int J Parasitol 38:901–912. doi: 10.1016/j.ijpara.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fisk JC, Presnyak V, Ammerman ML, Read LK. 2009. Distinct and overlapping functions of MRP1/2 RBP16 in mitochondrial RNA metabolism. Mol Cell Biol 29:5214–5225. doi: 10.1128/MCB.00520-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Madina BR, Kuppan G, Vashisht AA, Liang Y-, Downey KM, Wohlschlegel JA, Ji X, Sze S-H, Sacchettini JC, Read LK, Cruz-Reyes J. 2011. Guide RNA biogenesis involves a novel RNase III family endoribonuclease in Trypanosoma brucei. RNA 17:1821–1830. doi: 10.1261/rna.2815911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hernandez A, Madina BR, Ro K, Wohlschlegel JA, Willard B, Kinter MT, Cruz-Reyes J. 2010. REH2 RNA helicase in kinetoplastid mitochondria: ribonucleoprotein complexes and essential motifs for unwinding and guide RNA (gRNA) binding. J Biol Chem 285:1220–1228. doi: 10.1074/jbc.M109.051862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li F, Herrera J, Zhou S, Maslov DA, Simpson L. 2011. Trypanosome REH1 is an RNA helicase involved with the 3′-5′ polarity of multiple gRNA-guided uridine insertion/deletion RNA editing. Proc Natl Acad Sci U S A 108:3542–3547. doi: 10.1073/pnas.1014152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lukeš J, Archibald JM, Keeling PJ, Doolittle WF, Gray MW. 2011. How a neutral evolutionary ratchet can build cellular complexity. IUBMB Life 63:528–537. doi: 10.1002/iub.489. [DOI] [PubMed] [Google Scholar]
- 68.Landweber LF, Gilbert W. 1993. RNA editing as a source of genetic variation. Nature 363:179–182. doi: 10.1038/363179a0. [DOI] [PubMed] [Google Scholar]
- 69.Phreaner CG, Williams MA, Mulligan RM. 1996. Incomplete editing of rps12 transcripts results in the synthesis of polymorphic polypeptides in plant mitochondria. Plant Cell 8:107–117. doi: 10.1105/tpc.8.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Page FC. 1973. Paramoeba—common marine genus. Hydrobiologia 41:183–188. doi: 10.1007/BF00016444. [DOI] [Google Scholar]
- 71.Yurchenko VY, Lukeš J, Tesarová M, Jirků M, Maslov DA. 2008. Morphological discordance of the new trypanosomatid species phylogenetically associated with the genus Crithidia. Protist 159:99–114. doi: 10.1016/j.protis.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 72.Votýpka J, Kostygov AY, Kraeva N, Grybchuk-Ieremenko A, Tesařová M, Grybchuk D, Lukeš J, Yurchenko V. 2014. Kentomonas gen. n., a new genus of endosymbiont-containing trypanosomatids of Strigomonadinae subfam. n. Protist 165:825–838. doi: 10.1016/j.protis.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Page FC. 1976. An illustrated key to freshwater and soil amoebae with notes on cultivation and ecology. The ferry house. Far Sawrey, Ambleside, Cumbria, England. [Google Scholar]
- 74.Dyková I, Nowak BF, Crosbie PBB, Fiala I, Pecková H, Adams MB, Macháčková B, Dvořáková H. 2005. Neoparamoeba branchiphila n. sp., related species of the genus Neoparamoeba Page 1987: morphological and molecular characterization of selected strains. J Fish Dis 28:49–64. doi: 10.1111/j.1365-2761.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- 75.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Farrar M. 2007. Striped Smith-waterman speeds database searches six times over other SIMD implementations. Bioinformatics 23:156–161. doi: 10.1093/bioinformatics/btl582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Perkinsela strain GillNOR1/I mitochondrial scaffolds with both sense and anti-sense transcriptomic reads mapped. Almost no antisense transcription is visible, which is supported by the Northern blot in Fig. 3. The sense transcription profile, showing very low coverage for pan-edited genes rps12 and atp6, is different from that shown in Fig. 2 since “U-indel optimized” settings in Bowtie were not used here. “U-indel optimized” settings may produce strand biases, e.g., favoring U-indels on the forward strand, but not A-indels on the reverse strand. Therefore they were not used for the purpose of inter-strand comparison of transcription profiles. However, regular Bowtie “very sensitive” settings produce especially poor coverage in the case of pan-edited transcripts. Download
Mapping of edited reads with Bowtie2 v.2.0.2 and its modified version. An alignment window shown here covers the 3′ editing region of cox1 in Perkinsela strain GillNOR1/I. Just a few reads are mapped by the standard Bowtie2 algorithm using the “very-sensitive” setting. In contrast, modified Bowtie2 with T-indel-sensitive settings results in 12-fold increase of mapped read count (not all reads are shown in the figure). Moreover, misalignments such as those shown with arrows are missing because gaps containing ACG are penalized. Download
All reads spanning both 5′ and 3′ edited domains of cox2 in Perkinsela strain GillNOR1/I. Only parts of the edited domains adjoining the central non-edited region are shown, and the non-edited region itself is omitted (represented by the black bar in the center of the picture). The pre-edited sequence is shown at the bottom and the final edited sequence is on top. Insertions are shown in light blue, deletions in red, and edits corresponding to the main edited product are boxed (alternative edits are not boxed). We found virtually no reads edited in the 3′ domain but not edited in the 5′ domain, but many examples of the opposite arrangement. Only a single read carries one alternative edit in the 3′ domain and no other edits. Download
Workflow of T-aligner. Download
Alternatively edited and “misedited” products for cox1, cox2, cox3, cob, atp6, and rps12. The figure represents an output of T-aligner, illustrating non-redundant alternatively edited reads (also termed "editing intermediates" in this paper to distinguish them from reads in the raw dataset). Each column represents an editing site irrespective of the U-indel length observed; each line represents a read with its sequence in white. U-insertion and U-deletion sites identical to those in the main edited product are shown in dark green and dark red, respectively. A site is considered alternatively edited if any of the following conditions are met: (i) a site is never-edited in the main editing product; (ii) U indel is longer than in the main product; (iii) U insertion occurs instead of deletion in the main product or vice versa. Alternatively edited sites are shown in bright green and orange for U insertions and U deletions, respectively, and sites with the editing direction reversed (group iii) are denoted in blue. The transcript is schematically shown at the bottom, with edited domains in green and the seed used by T-aligner in yellow. Download
Genomic and transcriptomic NGS reads. Paired transcriptomic reads were merged with the CLC Genomics Workbench v.6.5 prior to mapping. Merged read length distribution is shown for the Perkinsela CCAP1560/4 (A) and GillNOR1/I strains (B). The abrupt edges of the distribution in panel A are due to 100 bp and shorter trimmed reads which produce merged reads of 190 bp or shorter, if a minimum overlap of 10 bp is required. Download
Guide RNA gene candidates revealed in the genome assembly of Perkinsela sp. CCAP1560/4. Aligned stretches of gRNA and mRNA (at least 25 bp in length) are shown in uppercase, and their flanks are shown in lowercase. Contig IDs and start coordinates are shown, along with a number of matching transcriptomic reads (support). The 5′ AC-rich region of gRNAs is underlined. Download
Aligned edited/pre-edited transcripts and trees for the final protein sequences of 6 mitochondrial genes in Perkinsela strains CCAP1560/4 and GillNOR1/I. For each gene (cox1, cox2, cox3, cob, rps12, and atp6) the following information is shown: (i) edited and pre-edited transcript sequences with corresponding translations (U-insertions are denoted in blue and U-deletions are marked with orange circles); (ii) pairwise percent identities (in the lower left part of the matrix) and numbers of different positions (in the upper right part) between edited/pre-edited sequences of both strains; (iii) a maximum likelihood unrooted tree for protein sequences of Perkinsela, other kinetoplastids, and an outgroup (Diplonema papillatum, Euglena gracilis, or Naegleria gruberi, depending on sequence availability). The trees were constructed using the following settings: WAG+Г substitution model, neighbor-joining starting tree, 1,000 bootstrap replicates. Branches supported by bootstrap values >70% are shown with thicker lines. Scale bars show inferred number of amino acid substitutions per site. Download
Alternative editing quantification. Pie charts illustrating counts of reads matching the main editing pathway, the pre-edited sequence, and alternatively edited reads in Perkinsela strains CCAP1560/4 and GillNOR1/I (A). Alternative editing intermediates in Perkinsela sp. Mitochondria, sorted according to their rounded support values (B) and number of editing sites (C) (x-axis) are shown. Read counts in logarithmic scale (y-axis) are given separately for each model edited domain (strain CCAP1560/4 for cox1, cox3, atp6; strain GillNOR1/I for cox2, cob, rps12). Download
Perkinsela nucleus-encoded proteins associated with mitochondrial oxidative phosphorylation, RNA editing and processing machineries, and mitochondrial translation, identified using HMM and reciprocal BLASTp.