

Abstract
Nelfinavir mesylate (NFV) is an anti-viral drug, used in the treatment of Acquired Immunodeficiency Syndrome (AIDS). Poor oral bioavailability and shorter half-life (3.5–5 h) remain a major clinical limitation of NFV leading to unpredictable drug bioavailability and frequent dosing. In this context, the objective of the present study was to formulate NFV loaded poly (lactic-co-glycolic acid) (PLGA) nanoparticles (NPs), which can increase the solubility and oral bioavailability along with sustained release of the drug. NFV loaded PLGA-NPs were prepared by nanoprecipitation method using PLGA and Poloxomer 407. The prepared NPs were evaluated for particle size, zeta potential, morphology, drug content, entrapment efficiency (EE) and in vitro dissolution studies. Oral bioavailability studies were carried out in New Zealand rabbits by administering developed NFV PLGA-NPs and pure drug suspension. PLGA-NPs prepared by using 1:4 ratio of drug and PLGA, with a stirring rate of 1500 rpm for 4 h. The prepared NPs were in the size of 185 ± 0.83 nm with a zeta potential of 28.7 ± 0.09 mV. The developed NPs were found to be spherical with uniform size distribution. The drug content and EE of the optimized formulation were found to be 36 ± 0.19% and 72 ± 0.47% respectively. After oral administration of NFV PLGA-NPs, the relative bioavailability was enhanced about 4.94 fold compared to NFV suspension as a control. The results describe an effective strategy for oral delivery of NFV loaded PLGA NPs that helps in enhancing bioavailability and reduce the frequency of dosing.
Keywords: Nelfinavir mesylate, PLGA, Nanoparticles, Sustained release, Bioavailability
1. Introduction
Human Immunodeficiency Virus/Acquired Immunodeficiency Syndrome (HIV/AIDS) is the major health issue among the public world-wide (WHO, 2014b). According to the World Health Organization (WHO), there are about 35 million people globally suffering from HIV/AIDS as of the year 2013. The report also says that around 2.3 million people were infected with HIV among which 260,000 are children (UNAIDS, 2013). The patients with HIV/AIDS are typically treated with High Activity Antiretroviral Therapy (HAART), which involves the chronic administration of at least three combined antiretroviral drugs. However, this therapy can only suppress viral replication temporarily (WHO, 2014a). The prolonged HAART therapy showed a low patient compliance, especially among the children. The approved antiretroviral drugs by regulatory agencies for pediatric use are in fact limited (Sosnik et al., 2012). Nelfinavir mesylate (NFV) is a non-peptidic HIV-1 protease inhibitor, which is the first of its kind recommended for pediatric use (Schuval, 2009). NFV is one of the several currently available protease inhibitors which is used to suppress viral replication and to improve the immune function in HIV-infected individuals. Typically, NFV along with other antiretroviral agents are administered in HAART (Bardsley-Elliot and Plosker, 2000). Based on biopharmaceutical classification, NFV belongs to class IV drug, which has low aqueous solubility and low permeability. It is highly lipophilic (log p = 6.0) and almost insoluble [at pH 7.4 (very low), at pH 3.5 (∼0.5 mg/mL)] and non-ionizable in water. The usual dosing regimen of NFV by conventional dosage form has a shorter half-life of 3.5–5 h, which demands its frequent administration three times a day. By incorporating into Nano particulate carrier and administering it by oral route it might enable the drug to present in system for longer period of time due to its sustained/controlled drug delivery with enhanced bioavailability (Sahoo and Labhasetwar, 2003). Nanoencapsulation promotes controlled drug release and drug targeting, which consecutively improves drug solubility and protects the molecules from premature degradation. Nanoparticles (NPs) enhance specificity, tolerability, drug efficacy and therapeutic index. The pharmacokinetic parameters of the drug are altered when it is formulated as NPs. Improved plasma circulation time by improved absorption and bioavailability is usually seen with reduced clearance. The physicochemical parameters of NPs viz., surface modification charge, particle size and hydrophobicity significantly influences the drug’s pharmacokinetics, impacting the drug’s bioavailability and especially the biodistribution (Khalil et al., 2013). In recent years, nano-based drug delivery systems, such as polymeric NPs, have demonstrated the apparent advantages in improving pharmacological activities and oral bioavailability of poorly soluble drugs (Agüeros et al., 2010). For instance, NPs of synthetic polymers such as PLGA are regularly utilized due to its versatility and biocompatibility in encapsulating variety of drugs (Anderson and Shive, 2012, Jain, 2000, Panyam and Labhasetwar, 2003). Oral delivery of PLGA NPs is also been well studied (Damgé et al., 1996). Encapsulation of drugs in PLGA Nano-carriers reduces the undesirable effects of the drug, such as short circulation half-life and non-site specific targeting, thereby reducing systemic side effects. These drug-loaded PLGA NPs prolong the systemic circulation time by sustained release (Lü et al., 2009). Several disease-related drugs/bioactive molecules are successfully encapsulated using PLGA to control the drug delivery and to improve the bioavailability (Kumari et al., 2010). Thus, these unique nanoparticulate carrier systems have the potential to change the current scenario of research and diagnosis in real time (Ferrari, 2005). Hence, in the present study, an attempt was made to develop nanotechnology based systems, for a poorly water soluble drug NFV using PLGA as polymer, which is expected to improve the dissolution properties of the drug and thereby increasing the bioavailability.
2. Materials and methods
2.1. Materials
Nelfinavir mesylate (99.5%) was obtained as a gift sample from Glenmark Pharmaceuticals Ltd (Mumbai, India). Darunavir was obtained as a gift sample from Mylan laboratories (Hyderabad, India). PLGA was purchased from Evonik Degussa India Pvt Ltd (Mumbai, India). Poloxomer 407 was purchased from Sigma life sciences (Mumbai, India). The HPLC grade water was prepared by using Milli-Q Academic, Millipore (Bangalore, India). All other chemicals used in this study were of analytical grade.
2.2. Methods
2.2.1. Preparation of NFV PLGA NPs (Bilati et al., 2005, Kim et al., 2011, Shah et al., 2009)
NPs of PLGA containing NFV were prepared by nanoprecipitation method reported elsewhere with some modifications. 20 mg of the drug and 100 mg of polymer (PLGA) (50:50) were dissolved in 5 ml of acetonitrile (Solution A). 125 mg of Poloxomer F-127 was dissolved in 50 ml of deionized water (Solution B). Solution A was added to Solution B using a syringe at the flow rate of 1 mL/10 min by magnetic stirring (1500 rpm) at room temperature. From above method, five different batches of NFV PLGA-NPs were prepared with various concentrations of PLGA and coded as F1–F5. The obtained nanosuspension was centrifuged, lyophilized with cryoprotectant (2% sucrose) and subjected for various characterization parameters.
2.2.2. Drug loading and Entrapment efficiency (Abou el Ela et al., 2014, Govender et al., 1999, Song et al., 2008)
To separate NPs, the nanosuspension was ultra-centrifuged using Remi Laboratory Centrifuge (REMI – Model R-8C, India) at 93,000g for 1 h. The obtained supernatant was removed and the NPs were collected. The collected NPs were dissolved in 5 mL of acetonitrile and then 10 mL of methanol was added to precipitate the polymer. Then the samples were filtered through 0.22 μ millipore membrane filter and the amount of drug was estimated by HPLC. Lyophilized NFV-PLGA-NPs (1–2 mg) were dissolved in 1 mL of methanol aimed at complete extraction for the loading and encapsulation detection. The samples in methanol were gently shaken on a shaker for 24 h at 37 °C to leach out NFV entirely. Then the solutions were centrifuged at 13,500 rpm for 10 min, and supernatant was collected. The supernatant (100 μL) was diluted to 2 mL. An aliquot of 20 μL was used for HPLC analysis, as described in ‘Section 2.4.2’. The amount of NFV loaded and encapsulated in nanoparticles was expressed as loading efficiency or encapsulation efficiency calculated as follows.
2.2.3. Characterization of NPs
2.2.3.1. Particle size determination (Betancourt et al., 2007)
The developed NPs were washed with double distilled water (filtered through 0.22 μ) several times before particle size analysis. Samples were analyzed using Zeta Sizer 2000 (Malvern instrument, UK) which allows sample measurement in the range of 0.02–2000.00 μm and particle size distribution.
2.2.3.2. Differential Scanning Calorimetry (DSC) (Kashi et al., 2012)
DSC analysis was performed using DSC Q200, (TA Instruments, USA). The samples were heated in sealed aluminum pans at a rate of 10 °C per min in a 30–300 °C temperature under nitrogen flow of 40 mL/min.
2.2.3.3. Scanning electron microscopy (SEM) (Özbaş-Turan and Akbuğa, 2011)
SEM was used to verify the uniformity of particle shape and size. Freeze-dried NPs were re-suspended in distilled water and later dropped into a silicon grid and dried at room temperature. The NPs suspension was vacuum coated with gold for 3 min. The surface morphology of the samples was observed under a scanning electron microscope (JEOL ltd, Japan) operated at 15-keV pulse at different resolutions.
2.2.3.4. Transmission electron microscopy (TEM) (Peter Christoper et al., 2014)
To study the external morphology by TEM (100s, JEOL Ltd, Japan), the prepared NPs were freeze dried and lyophilized. Freeze dried NPs were then diluted with 2 mL of distilled water and evenly mixed by sonication for 2 min. The samples were prepared by placing a drop of the NPs suspension on the Formvar® coated copper grid and air dried.
2.2.3.5. X-ray diffraction (XRD) studies (Kim et al., 2011)
An XRD peak mainly depends on the crystal size as they indicate the crystalline nature at particular value at 2θ range. Molecular arrangements of NFV alone and in nanoparticulate formulations were performed using an X-ray diffractometer (PANalytical X’pert PRO, The Netherlands) using Cu Kα radiation. The data were collected over an angular range from 3 to 50 degrees 2θ in continuous mode using a step size of 0.02 degree 2θ and step-time of 5 s.
2.3. In vitro release studies (Asadi, 2014, Seju et al., 2011)
The dialysis bag diffusion technique was used to study the in vitro drug release of NFV from NPs. The drug loaded NPs were placed in the dialysis bag and immersed into 250 mL of HCl pH (1.2) at 100 rpm for 24 h. The entire system was kept at 37 ± 0.5 °C with continuous magnetic stirring. Samples were withdrawn from the receptor compartment at predetermined intervals (0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 24 h) and replaced with fresh medium to maintain sink conditions. The amount of drug release was analyzed by HPLC. In order to elucidate the mode and mechanism of drug release, the in vitro data were transformed and interpreted by a graphical interface constructed using zero order, First order, Higuchi’s and Peppas’s equation respectively.
2.4. In vivo bioavailability studies (Ghosh, 2007, Ma et al., 2012)
2.4.1. Estimation of drug in rabbit plasma
All the animal investigations were performed as per the requisite protocol approved by the Institutional Animal Ethic Committee of JSS College of Pharmacy, Ooty, India. (Approval/Letter no. JSSCP/IAEC/M.PHARM/PH.ANALYSIS/01/2012-13). Healthy New Zealand rabbits weighing 2.0–2.5 kg were kept in individual cages for 30 days prior to the study in the departmental animal house for the purpose of acclimatization. The animals had free access to water and food. A constant day and night cycle was maintained and the temperature of the animal room was kept constant throughout the period. The animals were divided into 3 groups containing six animals in each group. The dose for the rabbits (35 mg/kg) was selected based on the surface area ratio of rabbit and man. Group 1 animals were treated with pure drug, Group 2 animals were treated with NFV loaded NPs and Group 3 was pertained to be control. Immediately after drug administration the animals were given 5 mL of water. Blood samples each not more than 0.5 ml were withdrawn from the marginal ear vein at 0, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 24 h period using a sterilized syringe. Zero hour fasting blood samples were withdrawn early in the morning. The blood samples were collected in Ria-vials containing anticoagulant (100 μL of 11% sodium citrate) and were centrifuged at 4000 rpm for 15 min to separate plasma and then stored at −20 °C. The plasma samples were deproteinized by mixing the samples with equal volume of 10% perchloric acid and the contents were vortex mixed for 2 min. It was then centrifuged at 4000 rpm for 15 min and the supernatant liquid was separated and analyzed. Estimation of plasma samples by HPLC was carried out at optimized chromatographic conditions ‘Section 2.4.2’. The standard and sample chromatograms are shown in Fig. 1.
Figure 1.
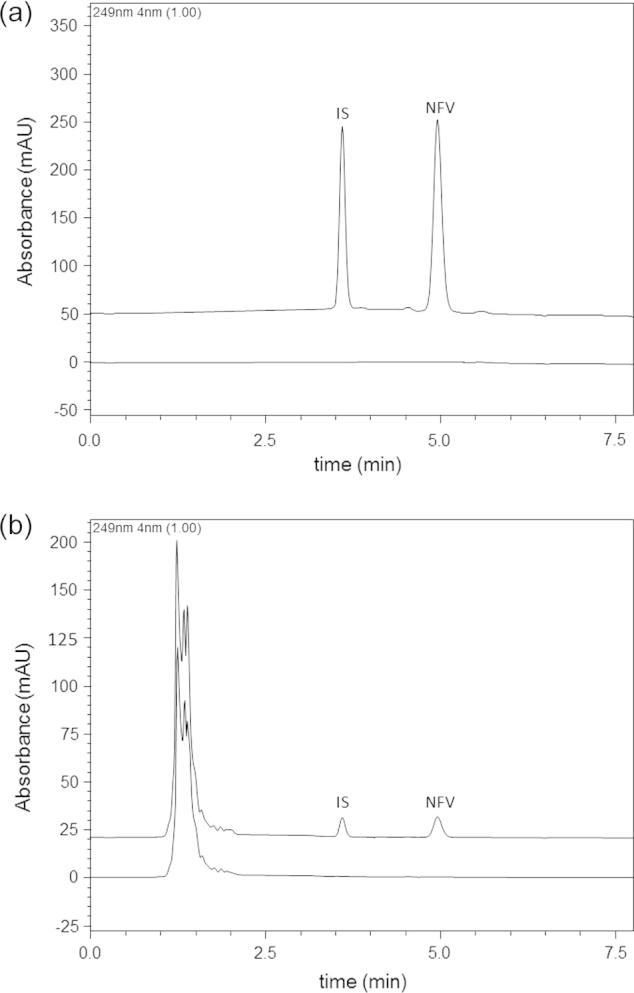
Representative chromatograms corresponding to (a) NFV loaded PLGA NPs with IS (Darunavir) and blank PLGA NPs and (b) NFV spiked in rabbit plasma with IS (Darunavir) and processed blank rabbit plasma.
2.4.2. Analytical conditions
Quantification of NFV in NPs and plasma samples was performed using Shimadzu HPLC (Kyoto, Japan) with a PDA detector. The analysis was performed on a Luna C18 column (150 × 4.6 mm, 5μ) (Phenomenex, USA) by employing a mobile phase comprising KH2PO4: acetonitrile (40:60) at a pH of 4.0 which was delivered at a flow rate of 1.0 mL/min. The analytes were detected photometrically at 249 nm. Data acquisition and processing were done by using LC Solutions software (SP 1.1). Darunavir was used as internal standard (IS), as it belongs to the same class of drugs and presented acceptable resolution with that of NFV. The retention time of NFV was 5.0 min and IS was 3.1 min. The plotted calibration curves were linear in the concentration range of 1–50 μg/mL (r = 0.999) in NPs and 10–500 ng/ml (r = 0.999) in rabbit plasma. The intra and inter-day accuracy and precision were within R.S.D ⩽ 8%. The extraction efficiency in case of spiked plasma samples was 97.5 ± 2.3%.
2.5. Statistical analysis
Statistical data were analyzed using GraphPad Prism® 6 program (Graph pad Inc., USA). Results are expressed as mean ± SD for the number of observations indicated. Mean values were compared using one-way analysis of variance (ANOVA) followed by Tukey post hoc test to compare means between the different treatment groups. Differences were considered significant at p < 0.05 unless otherwise stated. The amount of drug in the receptor compartment (in vitro) and in plasma (in vivo) was estimated through HPLC. The relative oral bioavailability of NFV was calculated according to the equation:
AUCA and AUCB represent the area under the blood concentration time curve of NFV-PLGA-NPs and NFV suspension, and doseA and doseB mean the dose of NFV-PLGA-NPs and NFV suspension following oral administration.
3. Results and discussion
3.1. Preparation
The NFV loaded PLGA NPs were prepared by employing nanoprecipitation method, as it appeared to be simple and appropriate. The carrier capacity of PLGA relating to NFV was determined by preparing five batches of drug-loaded NPs, by varying the concentration of polymer to a constant amount of drug (Table 1). During the procedure, an increase in polymer concentration increased the size of the NPs and entrapment efficiency. But after reaching a concentration of 200 mg, the saturation rate and entrapment of the drug to that of polymer were decreased. The stirring rate showed a significant influence on the size of NPs, i.e., at higher stirring rate (1500 rpm) the NPs particle size was reduced (185 ± 0.83 nm) whereas at lower stirring rate (500 rpm) the particle size of NPs (842 ± 0.68 nm) was not reduced effectively.
Table 1.
Particle size, entrapment efficiencies and drug loading (%) of Nelfinavir PLGA polymeric nanoparticles.
| Formulation code | Drug: polymer ratio | Particle size (nm) | Entrapment efficiency (%) | Drug loading (%) |
|---|---|---|---|---|
| F1 | 1:1 | 192.8 ± 0.72 | 20 ± 0.09 | 10 ± 0.018 |
| F2 | 1:2 | 140.3 ± 0.28 | 27.5 ± 0.03 | 17.5 ± 0.027 |
| F3 | 1:3 | 167 ± 1.01 | 46 ± 0.87 | 23 ± 0.038 |
| F4 | 1:4 | 185.3 ± 0.83 | 72 ± 0.47 | 36 ± 0.19 |
| F5 | 1:5 | 203.2 ± 0.47 | 58 ± 0.93 | 29 ± 1.25 |
3.2. Drug loading and entrapment efficiency
Drug loading and entrapment efficiency of all NPs made of different concentration were shown in Table 1. Among all the batches, F4 batch was found to have higher entrapment efficiency (72 ± 0.47%) and drug loading (36 ± 0.19%) and hence it was chosen as ideal and preceded for further studies. Drug loading has a very important influence in the polymeric NPs preference over others such as solid lipid NPs. The improper entrapment leads to the initial burst of the NPs, which hinders its sustained release property.
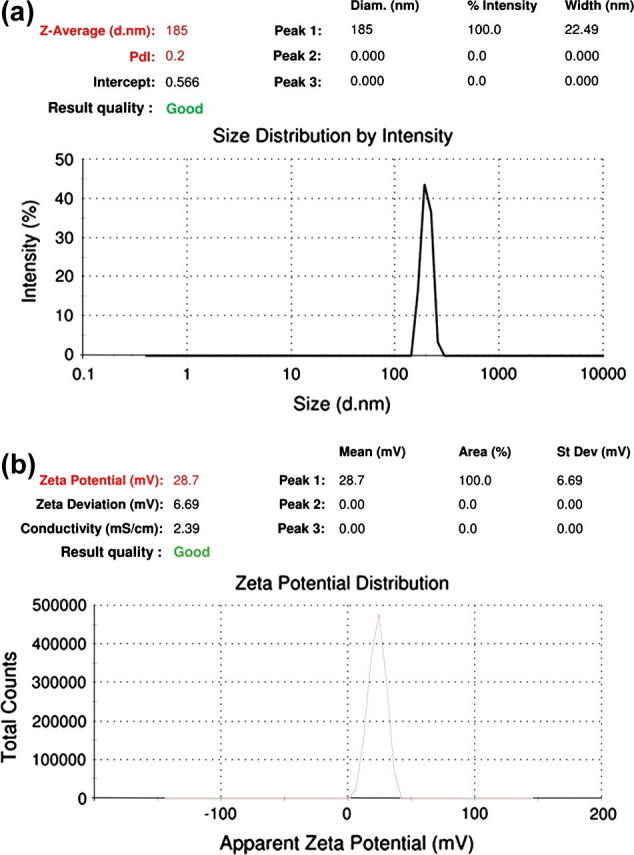
3.3. Determination of particle size and zeta potential (Betancourt et al., 2007)
The mean particle size of NFV NPs was found to be 185 nm (Fig. 2a) and the zeta potential was found to be +28.7 mV (Fig. 2b). The experiment was performed in three replicate (n = 3) in order to ensure reproducibility and minimize the error. Polydispersity index values were found to be less than 0.2, which shows that the system has a relatively narrow distribution. The zeta potential of the prepared NPs is the prominent factor to ensure stability. Highly charged NPs are capable to remain stable as colloidal suspension. Since the prepared NPs have shown high zeta potential, this ensures the stability of the formulation.
Figure 2.
DSC curves of NFV, PLGA, and NFV-PLGA-NPs.
3.4. DSC
DSC studies were done to assess the interaction between the drug and the polymer. Thermogram of NFV, PLGA and NFV PLGA NPs was illustrated in Fig. 3. The DSC curve of NFV showed an endothermic peak at temperature of 97 °C, corresponding to its melting point. PLGA has showed a peak at 54 °C. NFV-PLGA polymeric NPs have shown a major peak at 65 °C and the peak position of NFV was found to be disappearing, which can be attributed to the entrapment of drug in the NPs. This also has the chance of entrapment of drug inside the polymer in polymeric NPs. The DSC studies support the rationale of stability as it is the primary concern, which can affect the formulation in many ways.
Figure 3.

(a) Particle size of NFV-PLGA-NPs and (b) zeta potential of NFV-PLGA-NPs.
3.5. SEM and TEM
SEM and TEM were performed to obtain more information on the particle size, morphology and aggregation phenomena. The image of prepared polymeric NPs has shown that the formulated NPs were of spherical in shape with size ranging from 100 nm to 500 nm (Fig. 4). The NPs were observed with smooth surface, which may contribute to its release of the drug in a sustained manner, when compared to the NPs having rough surface.
Figure 4.

(a) SEM photographs of NFV-PLGA-NPs and (b) TEM photographs of NFV-PLGA-NPs.
3.6. XRD studies
To understand the nature of the nanoparticle-encapsulated NFV, XRD pattern of pure NFV and NFV-loaded PLGA-NPs was studied. In this study, NFV pure drug has shown a sharp, single highest peak at 2θ equals 22.600, which implies its crystalline nature. The diffractogram of NFV loaded PLGA-NPs was found to be different from the pure drug (Fig. 5). The intensity and number of peaks reduced in NFV loaded PLGA-NPs indicate, the maximum NFV was entrapped inside the PLGA-NPs, since the decrease in intensity of the peak can be attributed to lower level of detecting the encapsulated drug which is dispersed in molecular level. This also further indicates the amorphous state of the encapsulated NFV.
Figure 5.

XRD of NFV, PLGA and NFV-PLGA-NPs.
3.7. In vitro drug release
The cumulative percentage release profile of NFV from F4 NPs batch shown in Fig. 6. This was plotted using Graph pad Prism software (v. 5.01) on a Microsoft Windows 7 work station. A slow release of NFV from NPs was observed at pH 1.2 HCl buffer which was sustained up to 24 h at 37 ± 0.5 °C. It was further observed that there was no significant burst release which may be due to the total drug entrapped inside the polymer. The NPs showed a comparatively high percentage drug release (92.80%) over a period of 24 h and the n value was found to be 1 (limits 0.45–1) which shows the mechanism of drug release is by non-Fickian (anomalous) solute diffusion (Korsmeyer et al., 1983). When NPs were kept in a release solution, the PLGA was dissolved gradually to form holes or disintegrated to release the drug. This release of drug from the NPs mainly depends on concentration of the polymer. The compact nature of the polymer coat around the drug sustains the drug release. Particle size is another phenomenon which determines the rate of dissolution and solubility. It has been known that the particle size is inversely proportional to rate of dissolution and hence a higher rate of dissolution was observed with the developed NPs (Asadi, 2014, Ma et al., 2012).
Figure 6.
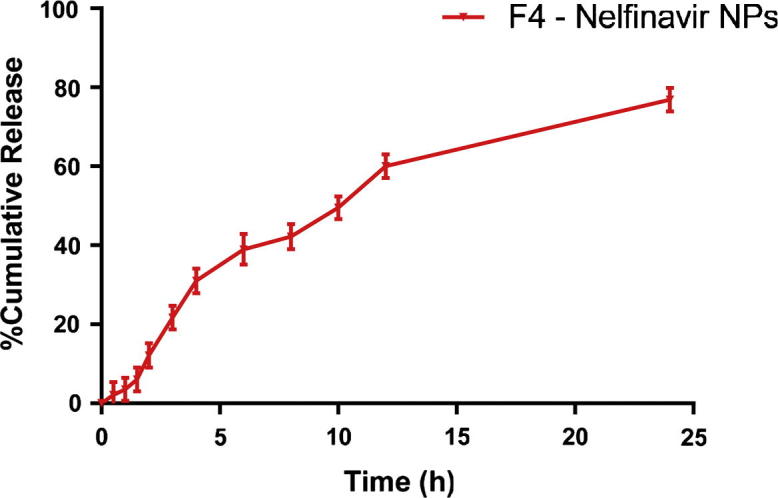
Release rates of NFV from PLGA NPs in vitro in HCl buffer pH 1.2 (mean ± SD, n = 3).
3.8. In vivo data analysis
NFV suspension and NFV loaded PLGA-NPs were orally administered to male New Zealand rabbits. The plasma concentration vs. time curve is shown in Fig. 7. Table 2 shows the pharmacokinetic parameters (peak concentration, time to reach peak concentration, area under the curve 0–24, half-life, and relative bioavailability) of NFV after oral administration in suspension as well as in PLGA-NPs. A remarkable increase in plasma concentration of NFV was observed when NPs group was studied by comparing with control group. The Cmax of pure drug in suspension form was 0.0190 ± 0.055 μg/mL at 1.5 h and for PLGA-NPs it was found to be 0.04590 ± 0.021 μg/mL at 4 h. These results imply a significant difference in bioavailability between NFV suspension and NFV loaded PLGA-NPs. The AUC0–24 value for the developed NPs containing NFV was found to be higher (0.374 ± 0.069 h μg/mL) compared to pure drug in suspension (0.076 ± 0.043 h μg/mL). These results also clearly demonstrate that the PLGA-NPs greatly improved bioavailability for NFV. The reasons are many, but among the most salient is the nano-based drug delivery system. On the one hand, particles that are nanoscale in size are easily absorbed into the folds of the intestinal wall, while larger particle surface area to volume lends itself to rapid drug dissolution (Ma et al., 2012). As shown in Table 2, NFV PLGA-NPs showed the longer half-life of 19.8 ± 2.09 h compared to NFV suspension (6.66 ± 1.25 h) due to the principally associated prolonged absorption phase and sustained release of PLGA-NPs. Finally, the relative bioavailability of NFV-PLGA NPs was calculated to be 494.4% compared to NFV suspension group indicates PLGA-NPs achieved markedly better absorption of NFV compared with the suspension. These results verify that PLGA-NPs were effective in improving the oral bioavailability of NFV. In summary, the developed NFV PLGA-NPs had high plasma concentration, low clearance, long half-life, and higher relative bioavailability compared to NFV in suspension.
Figure 7.
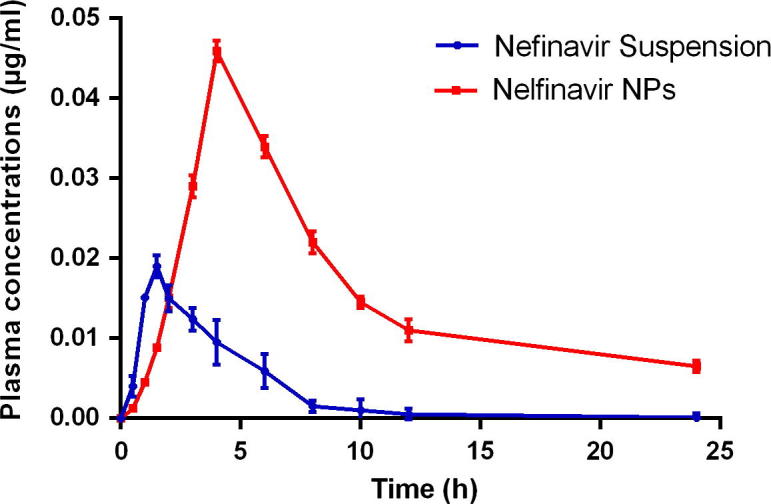
Mean plasma concentration time profiles of NFV in rabbits: (a) NFV suspension and (b) NFV NPs with (mean ± SD, n = 6).
Table 2.
Pharmacokinetic parameters of Nelfinavir after oral administration of NFV loaded PLGA NPs and suspension at the dose of 35 mg/kg.
| Parametersa | NFV suspension (mg/kg) | NFV PLGA-NPs (mg/kg) |
|---|---|---|
| Cmax (μg/mL) | 0.0190 ± 0.055 | 0.04590 ± 0.021 |
| AUC0–24 (h μg/mL) | 0.076 ± 0.043 | 0.374 ± 0.069 |
| t1/2 (h) | 6.66 ± 1.25 | 19.8 ± 2.09 |
| tmax (h) | 1.50 ± 0.056 | 4.12 ± 0.091 |
| Frel (%) | – | 494.4 |
Cmax, peak concentration; AUC, area under the curve; t1/2, half-life; tmax, time to reach peak concentration; Frel, relative bioavailability.
4. Conclusion
NFV loaded PLGA-NPs were successfully prepared by nanoprecipitation method. In this work, the influence of various process parameters on entrapment efficiency and particle size was systemically evaluated. The developed NFV loaded PLGA-NPs were spherical in shape with uniform size distribution and particle size of <200 nm. The prepared NPs were evaluated both in vitro and in vivo. The drug release was sustained up to 24 h from PLGA-NPs. After oral administration of NFV PLGA-NPs, the relative bioavailability was enhanced about 4.94-fold and had a longer half-life compared with that of NFV in suspension. The developed PLGA-NPs also exhibited a longer time to reach the peak concentration (tmax) than the drug in suspension form and appeared to be more consistent in overall performance. Based on these observations, it was concluded that the formulated PLGA-NPs containing NFV showed an increase in oral bioavailability and are capable of exhibiting sustained release over a period of time as compared to pure drug in the suspension form. Hence, PLGA-NPs were found to be a promising drug delivery system in encapsulating hydrophobic drugs like NFV for improved oral bioavailability with sustained release.
Acknowledgment
The authors sincerely thank Glenmark Pharmaceuticals Ltd, Mumbai, India for providing NFV and Mylan Laboratories Limited, Hyderabad, India for providing Darunavir as gift samples.
Footnotes
Peer review under responsibility of King Saud University.
Contributor Information
D. Nagasamy Venkatesh, Email: nagasamyvenkatesh@gmail.com.
Veera Venkata Satyanarayana Reddy Karri, Email: satya.zeal@gmail.com.
Sai Sandeep Mannemala, Email: reddysince1989@gmail.com.
References
- Abou el Ela A.E.S.F., Hassan M.A., El-Maraghy D.A. Ketorolac tromethamine floating beads for oral application: characterization and in vitro/in vivo evaluation. Saudi Pharm. J. 2014;22:349–359. doi: 10.1016/j.jsps.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agüeros M., Zabaleta V., Espuelas S., Campanero M.A., Irache J.M. Increased oral bioavailability of paclitaxel by its encapsulation through complex formation with cyclodextrins in poly(anhydride) nanoparticles. J. Control. Rel. 2010;145:2–8. doi: 10.1016/j.jconrel.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Anderson J.M., Shive M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 2012;64(Supplement):72–82. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- Asadi A. Streptomycin-loaded PLGA-alginate nanoparticles: preparation, characterization, and assessment. Appl. Nanosci. 2014;4:455–460. [Google Scholar]
- Bardsley-Elliot A., Plosker G. Nelfinavir. Drugs. 2000;59:581–620. doi: 10.2165/00003495-200059030-00014. [DOI] [PubMed] [Google Scholar]
- Betancourt T., Brown B., Brannon-Peppas L. Doxorubicin-loaded PLGA nanoparticles by nanoprecipitation: preparation, characterization and in vitro evaluation. Nanomedicine. 2007;2:219–232. doi: 10.2217/17435889.2.2.219. [DOI] [PubMed] [Google Scholar]
- Bilati U., Allémann E., Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur. J. Pharm. Sci. 2005;24:67–75. doi: 10.1016/j.ejps.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Damgé C., Aprahamian M., Marchais H., Benoit J.P., Pinget M. Intestinal absorption of PLAGA microspheres in the rat. J. Anat. 1996;189:491–501. [PMC free article] [PubMed] [Google Scholar]
- Ferrari M. Nanovector therapeutics. Curr. Opin. Chem. Biol. 2005;9:343–346. doi: 10.1016/j.cbpa.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Ghosh, M., 2007. Fundamentals of experimental pharmacology.
- Govender T., Stolnik S., Garnett M.C., Illum L., Davis S.S. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J. Control. Release. 1999;57:171–185. doi: 10.1016/s0168-3659(98)00116-3. [DOI] [PubMed] [Google Scholar]
- Jain R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- Kashi T.S.J., Eskandarion S., Esfandyari-Manesh M., Marashi S.M.A., Samadi N., Fatemi S.M., Atyabi F., Eshraghi S., Dinarvand R. Improved drug loading and antibacterial activity of minocycline-loaded PLGA nanoparticles prepared by solid/oil/water ion pairing method. Int. J. Nanomed. 2012;7:221–234. doi: 10.2147/IJN.S27709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil N.M., Nascimento T.C.F.d., Casa D.M., Dalmolin L.F., Mattos A.C.d., Hoss I., Romano M.A., Mainardes R.M. Pharmacokinetics of curcumin-loaded PLGA and PLGA–PEG blend nanoparticles after oral administration in rats. Colloids Surf. B Biointerfaces. 2013;101:353–360. doi: 10.1016/j.colsurfb.2012.06.024. [DOI] [PubMed] [Google Scholar]
- Kim T.-H., Jeong Y.-I., Jin S.-G., Pei J., Jung T.-Y., Moon K.-S., Kim I.-Y., Kang S.-S., Jung S. Preparation of polylactide-co-glycolide nanoparticles incorporating celecoxib and their antitumor activity against brain tumor cells. Int. J. Nanomed. 2011;6:2621–2631. doi: 10.2147/IJN.S19497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer R.W., Gurny R., Doelker E., Buri P., Peppas N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983;15:25–35. doi: 10.1002/jps.2600721021. [DOI] [PubMed] [Google Scholar]
- Kumari A., Yadav S.K., Yadav S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces. 2010;75:1–18. doi: 10.1016/j.colsurfb.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Lü J.-M., Wang X., Marin-Muller C., Wang H., Lin P.H., Yao Q., Chen C. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev. Mol. Diagn. 2009;9:325–341. doi: 10.1586/erm.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y., Zhao X., Li J., Shen Q. The comparison of different daidzein-PLGA nanoparticles in increasing its oral bioavailability. Int. J. Nanomed. 2012;7:559–570. doi: 10.2147/IJN.S27641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özbaş-Turan S., Akbuğa J. Plasmid DNA-loaded chitosan/TPP nanoparticles for topical gene delivery. Drug Deliv. 2011;18:215–222. doi: 10.3109/10717544.2010.544688. [DOI] [PubMed] [Google Scholar]
- Panyam J., Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003;55:329–347. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- Peter Christoper G.V., Vijaya Raghavan C., Siddharth K., Siva Selva Kumar M., Hari Prasad R. Formulation and optimization of coated PLGA – zidovudine nanoparticles using factorial design and in vitro in vivo evaluations to determine brain targeting efficiency. Saudi Pharm. J. 2014;22:133–140. doi: 10.1016/j.jsps.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo S.K., Labhasetwar V. Nanotech approaches to drug delivery and imaging. Drug Discovery Today. 2003;8:1112–1120. doi: 10.1016/s1359-6446(03)02903-9. [DOI] [PubMed] [Google Scholar]
- Schuval S.J. Pharmacotherapy of pediatric and adolescent HIV infection. Ther. Clin. Risk Manag. 2009;5:469–484. doi: 10.2147/tcrm.s4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seju U., Kumar A., Sawant K.K. Development and evaluation of olanzapine-loaded PLGA nanoparticles for nose-to-brain delivery: In vitro and in vivo studies. Acta Biomater. 2011;7:4169–4176. doi: 10.1016/j.actbio.2011.07.025. [DOI] [PubMed] [Google Scholar]
- Shah N., Chaudhari K., Dantuluri P., Murthy R.S.R., Das S. Paclitaxel-loaded PLGA nanoparticles surface modified with transferrin and Pluronic®P85, an in vitro cell line and in vivo biodistribution studies on rat model. J. Drug Target. 2009;17:533–542. doi: 10.1080/10611860903046628. [DOI] [PubMed] [Google Scholar]
- Song X., Zhao Y., Hou S., Xu F., Zhao R., He J., Cai Z., Li Y., Chen Q. Dual agents loaded PLGA nanoparticles: systematic study of particle size and drug entrapment efficiency. Eur. J. Pharm. Biopharm. 2008;69:445–453. doi: 10.1016/j.ejpb.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Sosnik A., Seremeta K.P., Imperiale J.C., Chiappetta D.A. Novel formulation and drug delivery strategies for the treatment of pediatric poverty-related diseases. Expert Opin. Drug Deliv. 2012;9:303–323. doi: 10.1517/17425247.2012.655268. [DOI] [PubMed] [Google Scholar]
- UNAIDS, 2013. UNAIDS – Global Report – 2013. <http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDSGlobalReport2013en.pdf> (accessed on 09.10.14).
- World Health Organization, 2014a. WHO – HIV – Bhutan. <http://www.who.int/hiv/pub/guidelines/bhutan art.pdf> (accessed on 18.12.14).
- World Health Organization, 2014b. WHO – HIV/AIDS <http://www.who.int/mediacentre/factsheets/fs360/en/index.html> (accessed on 21.08.14).