

Abstract
Liqui-solid technique and solid dispersion formation are two novel approaches for enhancement of dissolution rate of BCS class II drugs. Liqui-solid compact converts a liquid drug or drug solution into a free flowing powder with enhanced dissolution rate. In case of solid dispersion drug is molecularly dispersed in a hydrophilic polymer in solid state. In the present study, Liqui-solid and solid dispersion techniques were applied to enhance the dissolution of the Hydrochlorothiazide. Three formulations of Hydrochlorothiazide were prepared by liqui-solid technique using micro crystalline cellulose as carrier material and colloidal silicon dioxide as coating material. Water, poly ethylene glycol-400 and Tween-60 were used as solvent system. Solid dispersions of Hydrochlorothiazide were prepared by solvent fusion method using PEG-4000 as carrier polymer. Tablets were subjected to evaluation of various physical and chemical characteristics. Dissolution profiles of tablets prepared by the novel techniques were compared with marketed conventional tablets. Model independent techniques including similarity factor, dissimilarity factor and dissolution efficiency were applied for comparison of dissolution profiles. The results obtained indicated that liqui-solid compact formulations were more effective in enhancing the dissolution rate compared with solid dispersion technique. The liqui-solid compacts improved the dissolution rate up to 95% while the solid dispersion increased it to 88%.
Keywords: Liqui-solid compacts, Hydrochlorothiazide, Solid dispersion, PEG-4000, Dissolution efficiency, Similarity factor
1. Introduction
Dissolution behavior of an active pharmaceutical ingredient (API) from a dosage form is determined by its solubility (Ali, 2005). Nearly 40% of new APIs are poorly water-soluble and lower dissolution rate of the compact solid dosage form is their main limitation (Gowthamarajan and Sachin, 2010). Poor water soluble drugs require more time to dissolve in the gastrointestinal fluid under normal conditions that may delay the absorption of the drug to the systemic circulation (Kavitha et al., 2011). Various approaches as salt formation, size reduction, complexation, microencapsulation and solid dispersion have been applied to improve poor dissolution rate of water insoluble drugs (Appa et al., 2010, Sanjeev and Ravindra, 2010). Solid dispersion systems using hydrophilic polymers have significantly improved the dissolution rate as the drug is dispersed in matrix at molecular level (Ashok and Prabhakar, 2012). Manufacturing problems associated with solid dispersions, such as the use of excessive organic solvents and poor physical characteristics of dosage form development have been overcome using self-emulsifying and surface-active agents supports (Cherukuri et al., 2012). Solid dispersions are dispersion of the API in an inert carrier or a matrix in the solid state. Concept of solid dispersion was introduced first by Sekiguchi and Obi in 1961 (Appa et al., 2010, Tyagi and Dhillon, 2012). Obi and Sekiguchi showed that the eutectic mixture of sulfathiazole and physiologically inert water soluble carrier, urea, exhibited an improved absorption and excretion after oral administration compared with single sulfathiazole. The drug may be dispersed at the molecular level in the amorphous or crystalline matrices. Solid dispersions are prepared by the melt process, solvent process or a melt-solvent method (Santhosh et al., 2011). Supercritical fluid technology has been recently applied in the preparation of solid dispersions. Dispersions obtained by the melting process are often called Melts and those obtained by the solvent method are often referred to as co precipitates or co evaporates. Solid dispersion improves the dissolution rate of the drug due to the extremely small particle size, solubilizing effect of the hydrophilic carrier, an excellent wettability and dispersibility of the drug particles in GIT and the formation of metastable polymorphs. In addition to improving bioavailability, recently solid dispersion systems are being considered for sustained release dosage forms. The only difference between the two uses of solid dispersions is the use of the carrier with different properties. Another new technique for improving the dissolution rate of poor water soluble drugs is the liqui-solid technique. This technique is to turn a liquid into a free flowing powder, apparently dry and compressible (Vijay et al., 2011). The liquid may be a liquid drug, a drug solution or drug dispersion in a non-volatile solvent that is adsorbed on the porous carrier material by physical mixing (Varshney and Chatterjee, 2012). Liquid carriers commonly used are water-miscible organic solvent with a high boiling point such as poly ethylene glycol, propylene glycol and glycerin. Support system is composed of a porous material such as micro crystalline cellulose (Sahil et al., 2011).
Liqui-solid compacts improve the wetting properties and surface area available for dissolution. Liqui-solid technique is a low cost, simple and economical method that can be easily applied commercially. It requires minimal excipients and is less technical compared to other techniques such as improving dissolution by solid dispersions and microencapsulation (Yousef et al., 2007). Formulation of a high dose of lipophilic drug is the main limitation of the solid–liquid technique. Larger dose drugs require large amount of liquid vehicle for the formation of the solution resulting larger amount of carrier and coating material, required to obtain a free flowing powder. This will increase the compression weight of the tablet and will cause problems during administration. Hydrochlorothiazide (HCTZ) is a BCS class II drug with poor water solubility and good permeability. Its molecular weight is 297.74 g/mol. It is commonly used as an antihypertensive agent and as diuretic (Panneer et al., 2010).
In current study two methods have been studied for enhancement of dissolution rate of HCTZ i.e.
-
•
Liqui-solid technique.
-
•
Solid dispersion technique.
Different formulations were prepared by the two techniques and evaluated for various official and un-official parameters. The formulations prepared by the two techniques were compared with each other and with the commercially available conventional tablets of HCTZ.
2. Material and methods
2.1. Material
Model drug Hydrochlorothiazide (HCTZ), was received as a kind gift from Ferozsons Laboratories Pvt. Ltd. Nowshera, Pakistan. Rest of the excipients (PEG-400, PEG-4000, Micro crystalline cellulose, colloidal silicon dioxide, magnesium stearate, cross linked carboxy methyl cellulose sodium) were purchased from local market. All the materials were of pharmaceutical grade and were used as received.
2.2. Instrumentation
Digital balance (Precisa, Switzerland), double cone mixer (Morgan Instruments) and rotary compression machine (ZP-21, China) were used during tablet preparation. Pharma Test set of instruments (Pharma Test, Germany) consisting of hardness and thickness tester, disintegration apparatus and dissolution apparatus were used for physical analysis of the tablets.
HPLC system used for analysis consisted of pump (series-200), online degasser (series-200) Peltier column oven (series-200) and UV/visible detector (series-200)..
2.3. Methods
2.3.1. Determination solubility of drug in various non-volatile solvents
The solubility of the Hydrochlorothiazide was determined using three different solvent systems i.e. Poly ethylene glycol (PEG-400), PEG-400 + water and PEG-400 + water + Tween-60 at 25 °C and was calculated by Eq. (1).
| (1) |
2.3.2. Stability of drug in nonvolatile solvent system
The stability of the drug in the solvent system was determined using clear glass bottle with screw cap at 45 ± 2 °C (75% R.H.) for 10 days. Drug content of the solution was determined on 1, 5 and 10 day in triplicate. The drug content, color and odor were evaluated during the specified period.
2.3.3. Designing of liqui-solid compacts
Liqui-solid compacts were designed on the basis of mathematical models proposed by Yousef et al. (2007). On the basis of drug solubility poly ethylene glycol 400 (PEG-400), water and Tween-60 was selected as liquid vehicle. Micro crystalline cellulose was used as carrier material and colloidal silicon dioxide as coating material. The excipients ratio was calculated using Eq. (2):
| (2) |
Where R = Excipients ratio; Q = Weight of carrier and q = Coating material.
The liquid load factor was determined by dissolving the drug in nonvolatile solvent system (PEG-400 + water + Tween 60). Drug solution was then loaded to carrier material and blended with coating material. Liquid load factor was calculated using following equation:
| (3) |
Where W = Weight of liquid medication and Q = Weight of carrier material.
2.3.4. Preparation of tablet on the basis of Liqui-solid technique
Drug was dispersed in solvent system by gently heating at 40 ± 2 °C with constant stirring until uniform dispersion was formed. Drug dispersion was slowly incorporated into carrier material with continuous blending for 15 min. The mixer was then paused for 10 min for complete adsorption of drug into core of the powder. Disintegrant and lubricant were added to the admixture and blended for further 3 min. Same procedure was used for preparation of all the formulations of liquid solid compacts.
All the formulations of liqui-solid compacts were compressed using rotary compression machine (ZP-21, China) fitted with 13.5 mm round shallow concave punches having bisection line. Compression weight of the tablets was 500 mg/tablet.
2.3.5. Preparation of solid dispersion
Solid dispersion of HCTZ was prepared using fusion–solvent method (Santhosh et al., 2011). HCTZ and PEG-4000 were accurately weighed. HCTZ was dissolved in methanol and PEG-4000 was melted on a water bath. Drug solution was added to molten PEG-4000 slowly and stirred vigorously. Solvent was evaporated by heating on water bath. The residue was then dried in desiccators for 24 h and granulated through mesh number 20 (see Table 1).
Table 1.
Composition of liqui-solid compacts.
| Formulation code | Solvent system | Carrier material | Liquid drug concentration | Liquid load factor | Unit dose (mg) |
|---|---|---|---|---|---|
| LSC-01 | PEG + Tween 60 + Water | M.C. Cellulose | 10% w/w | 0.50 | 12.50 |
| LSC-02 | PEG + Tween 60 + Water | M.C. Cellulose | 15% w/w | 0.33 | 12.50 |
| LSC-03 | PEG + Tween 60 + Water | M.C. Cellulose | 20% w/w | 0.25 | 12.50 |
M.C. Cellulose; micro crystalline cellulose.
2.3.6. Preparation of tablets on the basis of solid dispersion technique
Solid dispersion of HCTZ and PEG-4000 was mixed with diluents, lubricant and disintegrant, the composition of the formulation is shown in Table 2. All the materials, except lubricant, were sifted through mesh number 30 and blended in a laboratory scale double cone mixer for 5 min at 25 rpm. Lubricant was passed through mesh number 60, added to rest of the blend and blended for 2 min. The granules were compressed into tablets using rotary compression machine (ZP-21, China) fitted with 9.5 mm round biconcave punches. Compression weight of the tablets was 200 mg/tablet.
Table 2.
Composition of tablets prepared using solid dispersion.
| Ingredients | SD-01 | SD-02 | SD-03 | SD-04 | SD-05 |
|---|---|---|---|---|---|
| Hydrochlorothiazide | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 |
| Polyethylene glycol 4000 | 6.25 | 12.50 | 25.00 | 31.25 | 37.50 |
| Micro crystalline cellulose | 12.50 | 12.50 | 12.50 | 12.50 | 12.50 |
| Tablettose-80 | 70.50 | 64.25 | 51.75 | 45.5 | 39.25 |
| Cross carmellose sodium | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 |
| Magnesium stearate | 1.50 | 1.50 | 1.50 | 1.50 | 1.50 |
| Ethyl alcohol | Q.S. | Q.S. | Q.S. | Q.S. | Q.S. |
Quantities are given as %w/w.
2.3.7. Post compression evaluation of tablets
2.3.7.1. Physical parameters of tablets
Weight variation of the tablets was performed as per United States Pharmacopoeia (USP-35/NF30, 2012).
Thickness of the tablets was measured using digital tablet hardness and thickness tester (Pharma Test, Germany). Thickness of 20 tablets from each formulation was measured and results were presented as Mean ± S.D.
The crushing strength of the tablets (n = 10) was measured using digital tablet hardness and thickness tester (Pharma Test, Germany) and their mean was calculated.
Friability of the tablets was determined according to United States Pharmacopoeia (USP-35/NF30, 2012), using Rosch friabilator (Faisal Engineering, Pakistan).
Disintegration time of the tablets (n = 6) was determined according to USP (USP-35/NF-30, 2012) using purified water held at 37 ± 2 °C as disintegration medium.
2.3.7.2. Determination of drug content
Drug content of the tablets was determined according to USP (USP-35/NF-30, 2012) using HPLC system. HPLC system was controlled by Perkin–Elmer Totalchrom Workstation Software (version 6.3.1). Phosphate buffer (0.1 M) and acetonitrile (9:1; pH 3.0) were used as mobile phase.
2.3.7.3. In vitro drug release study
Dissolution rate of the tablets was determined according to USP using apparatus-1 at 100 rpm (USP-35/NF-30, 2012). Dissolution media consisted of 900 ml of 0.1 N HCl held at 37 ± 2 °C. Sample (5 ml) was withdrawn at specified time intervals (0, 5, 15, 30, 45 and 60 min), filtered and analyzed for amount of drug released using HPLC.
2.3.8. Comparison of dissolution rate
Model independent approach was applied for comparison of dissolution profiles. Dissolution profile of two types of formulation was compared on the basis of their similarity factor (f2), dissimilarity factor (f1) and dissolution efficiency (D.E.). These parameters were determined for tablets from optimal formulation of both the novel techniques, compared with each other and marketed conventional tablets of HCTZ. Dissimilarity factor was calculated using Eq. (4) (Paulo and Jose, 2001, Bashar et al., 2010) and similarity factor was calculated using Eq. (5) (Yousef et al., 2007).
| (4) |
| (5) |
Where Rt = Dissolution rate of standard product at time “t” and Tt = Dissolution rate of test product at time t.
The “f2” value of 50 or greater ensures sameness or equivalence of the two curves and also the performance of the two products.
Dissolution efficiency is the percentage of the area of rectangle described by 100% dissolution. It was calculated according to the following equation;
| (6) |
where D.E. = Dissolution efficiency; y = Amount of drug released in time “t”; Y100 = 100 percent drug released and T = Total time.
Dissolution efficiency was calculated for optimal formulations of both the novel techniques and marketed product at 30 min and 60 min. Along with various parameters given above comparison of amount of drug released within initial 30 min (Q30min) and maximal drug released within 60 min (Q60min) was also made.
3. Results and discussions
3.1. Solubility of HCTZ in different nonvolatile solvent systems
According to Biopharmaceutical classification system, HCTZ is a class II drug, practically insoluble in water and its reported water solubility is 0.70 mg/ml (Rashmi et al., 2011). The maximum solubility of HCTZ in PEG-400 was 2.53 mg/ml; the addition of water increased the solubility. In PEG:water (3:1), the solubility of HCTZ was increased to 6.50 mg/ml and further increase in water reduced the solubility. The addition of Tween-60 further increased the solubility as shown in Table 3. Maximum solubility (7.18 mg/ml) was observed in solvent system consisting of PEG-400 + water + Tween-60 and was selected for preparation of liqui-solid compacts.
Table 3.
Solubility of HCTZ in various solvents.
| Solvent system | Solubility (mg/ml) |
|---|---|
| PEG-400 | 2.53 |
| PEG-400 + water (50:50) | 1.88 |
| PEG-400 + water (75:25) | 6.49 |
| PEG-400 + water (25:75) | 2.71 |
| PEG-400 + water + Tween-60 | 7.18 |
PEG; poly ethylene glycol.
3.2. Drug stability in non-volatile solvent system
Nonvolatile solvent system selected on the basis of solubility for preparation of liquid solid compacts was composed of PEG-400, water and Tween-60. Drug was dissolved in solvent system up to maximum and subjected to stress condition (45 ± 5 °C and 75 ± 5% R.H.) for ten days. Drug solution was analyzed at day-1, day-5 and day-10 for chemical and physical stability. The data showed that drug was stable in the selected non-volatile solvent system under the stress conditions for 10 days, results are depicted in Table 4. Similarly no changes were observed in physical appearance, color and odor of the samples. These results indicate the physical and chemical stability of the HCTZ in the selected solvent system.
Table 4.
Stability of HCTZ in solvent system.
| Sampling time | Drug content | Physical appearance | Odor |
|---|---|---|---|
| Day-1 | 100.30 ± 0.74 | Un affected | Un affected |
| Day-5 | 101.12 ± 0.29 | Un affected | Un affected |
| Day-10 | 100.80 ± 0.35 | Un affected | Un affected |
Results of drug content are presented as Mean ± S.D. (n = 3).
3.3. Postcompression evaluation of liqui-solid compacts
3.3.1. Physical parameters of liqui-solid compacts
Physical parameters of the tablets include physical appearance, weight variation, mechanical strength, disintegration behavior and drug content. Mechanical strength of the tablets was estimated by crushing strength and friability. Disintegration behavior of the tablets was evaluated by disintegration time determined according to USP (USP-35/NF-30, 2012).
Compression weight of liqui-solid compacts was 500 mg and tablets were compressed on 13.5 mm round shallow concave punches. Physically surface of all the tablets was smooth and shiny without any sticking and picking. The highest weight variation observed was ±3.70% (Table 5) which is within the official range of ±7.50% (USP-35/NF-30, 2012). Low weight variation confirms uniform flow of the material during compression which was expected to be retarded by liquid medication.
Table 5.
Postcompression evaluation of liqui-solid compacts.
| Formulation code | Weight variation (%) | Crushing strengtha (N) | Thicknessa (mm) | Friability (%) | Drug contentb (%) | Disintegration time (sec) |
|---|---|---|---|---|---|---|
| LSC-01 | ±3.70 | 81.69 ± 0.83 | 4.71 ± 0.53 | 0.3 | 99.31 ± 0.09 | 479 ± 39 |
| LSC-02 | ±2.98 | 79.21 ± 1.03 | 4.38 ± 0.10 | 0.48 | 98.19 ± 0.62 | 388 ± 47 |
| LSC-03 | ±3.53 | 94.77 ± 1.29 | 4.55 ± 0.38 | 0.45 | 99.17 ± 0.39 | 395 ± 61 |
Results are presented as Mean ± S.D.
n = 10.
n = 3.
Crushing strength of tablets (n = 10) randomly selected from each formulation, was in the range of 78–96 Newton. The data show that tablets are hard enough to resist stress during handling and transportation.
Friability for all the formulations was within the official range (USP-35/NF-30, 2012). Capping and edging were not observed in any formulation. Inclusion of liquid medication in tablets improved the mechanical strength of tablets prepared by liqui-solid compacts. Strong interlocking produced by liquid component and low porosity may be responsible for higher crushing strength of tablets.
Disintegration time of tablets from all the formulations was within the official (USP-35/NF-30, 2012). Disintegration rate of the liquid solid compacts was expected to be higher due to the presence of organic solvents. Organic solvents tend to enhance compressibility of the tablets by the formation of hydrogen bonds and strong mechanical interlocking (De Jong, 1987). Tablets with high compressibility have high crushing strength and slow disintegration due to lower porosity. PEG-400 is water soluble and does not retard tablet disintegration. The presence of cross linked carboxy methyl cellulose sodium and Tween-80 further contributed to rapid disintegration. CCNa is a super disintegrant, expends to larger extant by water absorption resulting in rapid tablet disintegration and was used in same concentration (3%) in all formulations.
Drug content of all the formulations was in the range of 98–102% of the claimed quantity as shown in Table 5. Uniform drug content confirms proper mixing of drug with rest of the excipients.
3.3.2. Postcompression evaluation of tablet prepared by solid dispersion
Solid dispersion containing tablets was compressed on 9.50 mm round shallow concave punches at compression weight of 200 mg/tablet. The weight variation was within the official limits and highest observed value was ±3.09%. Compared to liqui-solid compact their weight variation was very low.
Crushing strength of the tablets was in the range of 80–105 N. Highest crushing strength was observed for SD-01 (103.41 N ± 0.97) that may be due to small quantity of PEG-4000 in the formulation and Tablettose-80 constituted most of its bulk. Tablettose-80 is a smart excipient having very good flow and compressibility. It contributed to higher crushing strength of the tablets and in the present formulations it constituted the major bulk of the tablets. Thickness of the tablet was within the range of 3–4 mm. Almost all the formulations had same thickness irrespective of their composition.
Friability of all the tablets was within the official limits (USP-35/NF-30, 2012). Tablets from all the formulations were smooth and shiny without any sticking and picking. PEG has good lubrication activity and binding action. Due to the reason all the formulations containing solid dispersion had better mechanical strength as compared to liqui-solid compacts.
Drug content of tablets was determined in triplicate for twenty tablets, randomly selected from each formulation. Drug content for all the formulations was within the range of 99–102% of the claimed quantity.
3.3.3. In vitro drug release
Dissolution rate of HCTZ was studied for both types of formulations (USP-35/NF30, 2012) using 900 ml 0.1 N HCl as dissolution media maintained at 37 ± 2 °C. Results of dissolution rate were presented up to 60 min. The dissolution rate of the marketed conventional tablets was within the official limits (USP-35/NF-30, 2012) however, the drug release was slow and only 64.02% was released in 60 min. During initial 15 min only 19% of the drug was released and Q50% was not achieved during initial 30 min.
Significant increase in dissolution rate was observed with liquid solid compacts. Maximally, 95.38% drug was released in 60 min (LSC-01). During initial 30 min more than 70% of the drug was released. Smallest drug release during initial 30 min was 62.06 showing that Q50% was successfully achieved within initial 30 min (Fig. 1).
Figure 1.
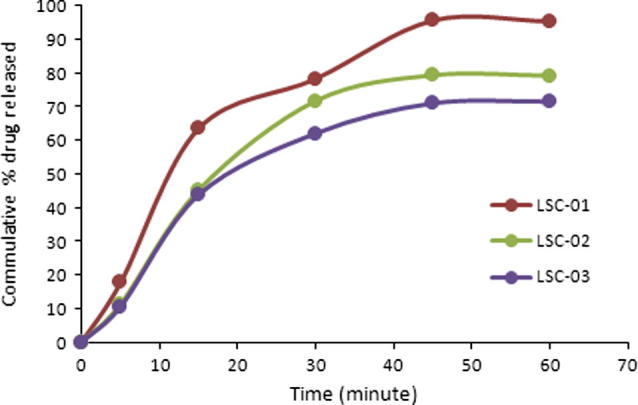
Dissolution profile of liqui-solid compacts.
Concentration of drug in liquid medication has a significant effect on enhancement of dissolution rate. Highest drug release obtained in terms of both rate and extent of drug release when 10% drug concentration in liquid vehicle was used. Maximum drug release was achieved in 60 min when used 10% drug concentration in liquid vehicle as shown in Fig. 1. At lower concentration (10%) most of the drug particles dissolve in liquid system and may present in molecular form that may enhance rate and extent of drug dissolution.
Similarly at higher drug concentration in liquid system, most of the drug remains in dispersed form and ratio of the dissolved particles is lower so the dissolution rate is relatively lower than that of the low drug concentration. Tween-60 is a surfactant and improves wettability that may also have contributed to the enhanced dissolution rate.
In solid dispersion drug and polymers come in close contact with each other at molecular level. Dissolution rate of poor water soluble drug is improved by enhanced drug water interaction. PEG-4000 is water soluble and improves wettability of the drug to a greater extent. Dissolution rate of HCTZ increased with increase in concentration of PEG-4000. When used in 1:1 (drug to polymer ratio) the maximum release was 65.79% in 60 min and Q50% was not achieved during initial 30 min.
Best dissolution profile was obtained when PEG-4000 was used in drug to polymer ratio of 1:6 (SD-05). 88.63% drug was released in 60 min and Q50% was achieved within 15 min (see Fig. 2).
Figure 2.
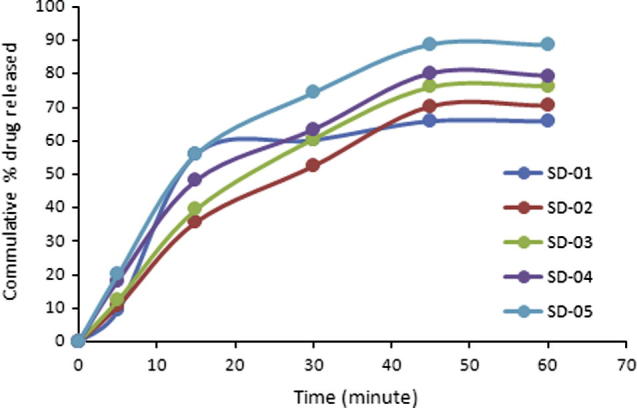
Dissolution profile of tablets containing solid dispersion.
Dissolution rate of liqui-solid compacts and solid dispersion was compared with marketed product at 30 min and 60 min. After 30 min 48.27% drug was released from marketed tablets. Its Q50% was not achieved during initial 30 min. In case of both the novel techniques more than 70% drug was released during initial 30 min. Drug release during initial 30 min was higher for liqui-solid compacts as compared to solid dispersion, as shown in Fig. 3. 78.29% drug was released for liqui-solid compacts while 74.34% drug was released from tablets prepared by solid dispersion. 3.95% more drug released by liqui-solid compacts in comparison with solid dispersion. Amount of dissolved drug in liqui-solid compact system is responsible for higher drug release. When drug concentration in liquid component was increased, amount of dissolved drug reduced and drug was present as dispersion. It resulted in reduced drug release. Formulations of liqui-solid compacts containing 15% and 20% of drug in liquid medium had Q30min lower than that of formulation containing 10% drug (see Fig. 4).
Figure 3.
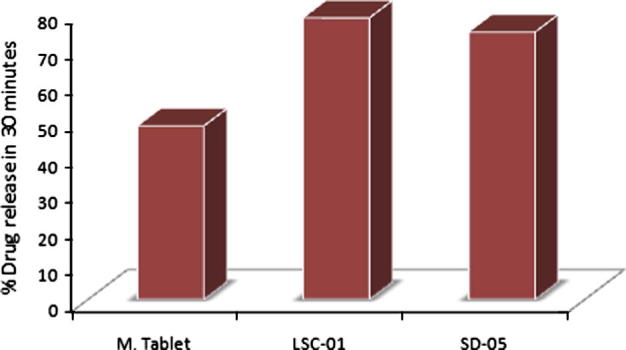
Comparison of amount of drug released during initial 30 min (Q30min). M. Tablet; marketed conventional tablet, LCS-01; optimal formulation prepared by liqui-solid compact, SD-05; optimal formulation containing solid dispersion.
Figure 4.
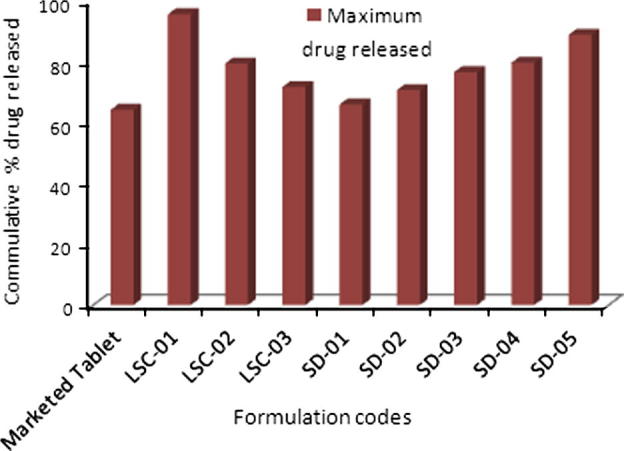
Comparison of maximum drug released in 60 min (Q60min).
Comparison of dissolution profiles of liqui-solid compacts and solid dispersion showed that maximum drug release was observed with liqui–solid system. In liqui–solid system 95.38% (n = 3) of drug was released in 60 min. In case of solid dispersion system 88.63% drug was released in 60 min. Both the novel techniques exhibited a marked increase in maximum release of HCTZ from tablets, as shown in Fig. 5. With tablets prepared by direct compression, 64% of the drug was released maximally while with liqui-solid the percentage was 95.38% and 88.63% with solid dispersion.
Figure 5.
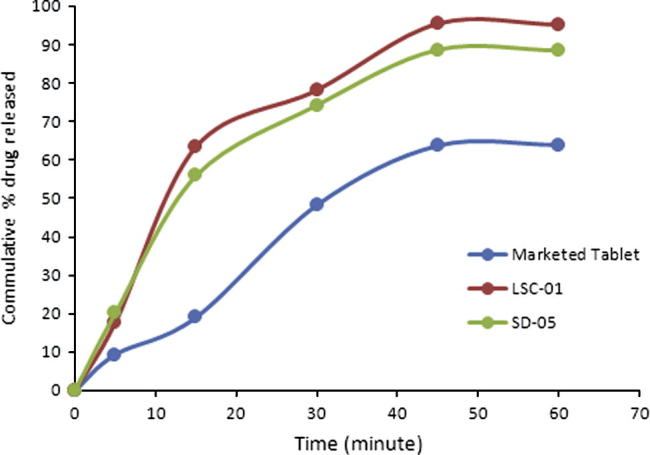
Comparison of dissolution profile of marketed product and optimal formulations. Marketed tablet; conventional tablets available in market, LCS-01; liqui-solid compacts, SD; solid dispersion.
Comparison of the two novel techniques showed that liqui-solid compact released 6.80% more drug as compared to solid dispersion. Its dissolution efficiency was also higher during initial 30 min (39.14%). It proved that liqui-solid compact was more efficient than solid dispersion.
3.3.4. Comparison of dissolution profiles
Comparison of dissolution profiles of conventional marketed tablets prepared by direct compression and those prepared by the two novel techniques are presented in Fig. 5. Significant increase was observed in dissolution profile with the two novel techniques.
3.3.4.1. Comparison of dissolution efficiency
Dissolution efficiency is commonly applied for comparison of dissolution profiles to decide better one. Dissolution efficiency was calculated using equation-6, for optimal formulations prepared by the two novel techniques and marketed product at 30 min and 60 min. At 30 min dissolution efficiency of the marketed product was 24.14% which increased to 64.02% at 60 min.
Dissolution efficiency of liqui-solid compact was 39.14% at 30 min and at 60 min became 95.38%. Dissolution efficiency of the two novel techniques was higher as compared to marketed conventional tablets. At 30 min dissolution efficiency of liqui-solid compact was 1.62 times of the marketed product. Similarly at 60 min, it was 1.49 times. Higher dissolution efficiency at both the points indicated that liqui-solid compact has significantly enhanced dissolution rate (see Table 6).
Table 6.
Postcompression evaluation of tablets containing solid dispersion.
| Formulation code | Weight variation (%) | Crushing strengtha (N) | Thicknessa (mm) | Friability (%) | Drug contentb (%) | Disintegration time (sec) |
|---|---|---|---|---|---|---|
| SD-01 | ±2.41 | 103.41 ± 0.97 | 3.42 ± 0.37 | 0.45 | 99.23 ± 0.09 | 398 ± 25 |
| SD-02 | ±2.77 | 82.33 ± 1.10 | 3.57 ± 0.20 | 0.31 | 101.06 ± 0.08 | 391 ± 41 |
| SD-03 | ±3.09 | 96.59 ± 0.38 | 3.51 ± 0.19 | 0.49 | 101.37 ± 0.31 | 386 ± 34 |
| SD-04 | ±2.58 | 89.22 ± 0.13 | 3.69 ± 0.21 | 0.62 | 100.79 ± 0.11 | 395 ± 46 |
| SD-05 | ±2.96 | 90.08 ± 0.62 | 3.65 ± 0.31 | 0.3 | 99.54 ± 0.41 | 391 ± 54 |
Results are presented as Mean ± S.D.
Data have been rounded off to two digits after decimal point.
n = 10.
n = 3.
Solid dispersions had dissolution efficiency of 37.17% which is 1.54 times of the marketed product. Ratio of dissolution efficiency at 60 min was 1.38 times.
From the data presented in Table 7, it was evident that both the novel techniques had improved dissolution efficiency throughout the dissolution profile. In comparison with solid dispersion, liqui-solid technique was more efficient. Its dissolution efficiency was higher at both the points from that of solid dispersions. At 30 min, dissolution efficiency of liqui-solid compacts was 5.31% higher than solid dispersion and at 60 min it was 7.62% higher.
Table 7.
Dissolution efficiency at various points.
| Formulation code | Dissolution efficiency |
|
|---|---|---|
| 30 min | 60 min | |
| Marketed tablet | 24.14 | 64.02 |
| LSC-01 | 39.14 | 95.38 |
| SD-01 | 37.17 | 88.63 |
Data have been rounded off to two digits after decimal point.
LSC-01; optimal formulation of liqui-solid compacts.
SD-01; optimal formulation of tablets containing solid dispersion.
Dissolution efficiency improved directly with amount of liquid medication. Higher dissolution efficiency was exhibited at both test points (30 min and 60 min) by formulations of high liquid load factor. When amount of liquid is high drug is available in the solution form and adsorbed at molecular level on the carrier surface. Adsorption of molecular drug results in improved dissolution efficiency.
3.3.4.2. Dissimilarity factor (f1) and similarity factor (f2) of dissolution profiles
Dissolution profile of the tablets prepared by both the novel techniques was compared with that of marketed product separately by applying similarity factor (f2) and dissimilarity factor (f1). Dissolution profile of both the novel techniques was significantly different from that of the marketed product. Both the techniques showed very low similarity factor and high dissimilarity factor, proving that dissolution profiles are different.
In case of liqui-solid compacts, the difference from dissolution profile of marketed tablets was more prominent. Liqui-solid compact exhibited lower values of similarity factor (27.50, Table 8). Similarly their dissimilarity factor was very high (81.75) as compared to that of solid dispersion. Values of “f1” and “f2” proved that as compared to solid dispersion dissolution profile of liqui-solid compacts was more different from dissolution profile of marketed product. It had improved dissolution rate of HCTZ effectively than solid dispersion.
Table 8.
Similarity (f2) and dissimilarity factor (f1) of dissolution profile.
| Formulation | f1 | f2 |
|---|---|---|
| Liqui-solid Compact | 81.75 | 27.50 |
| Solid dispersion | 70.38 | 37.35 |
Data have been rounded off to two digits after decimal point.
4. Conclusion
The study showed that both the novel techniques enhanced dissolution rate of HCTZ to a larger extent when compared with conventional tablet in terms of dissolution efficiency, similarity factor and dissimilarity factor. Dissolution efficiency of the novel techniques has increased by 40% in comparison with conventional tablets.
Liqui-solid technique was observed to be more effective in enhancing rate and extent of drug release. Due to a large variety of hydrophilic polymers smaller tablets with improved dissolution rate and better physical characteristics can be obtained by solid dispersion technique.
Excipients used in formulation of liqui-solid compact and solid dispersions were commonly used in pharmaceutical industries. All of the excipients are economical and will not affect cost of the final product to a larger extent.
Acknowledgment
We are thankful to Higher Education Commission of Pakistan for providing funds to carry out the study.
Footnotes
Peer review under responsibility of King Saud University.
References
- Ali N. The effect of type and concentration of vehicles on dissolution rate of poorly soluble drugs (indomethacin) from liquisolid compacts. J. Pharm. Pharm. Sci. 2005;8(1):18–25. [PubMed] [Google Scholar]
- Appa R.B., Shivalingam M.R., Kishore R.Y.V., Somesekhara Rao, Rajesh K., Sunitha N. Formulation and evaluation of aceclofenac solid dispersions for dissolution rate enhancement. Int. J. Pharm. Sci. Drug Res. 2010;2:146–150. [Google Scholar]
- Ashok A.H., Prabhakar R.J. Improvement of solubility and dissolution rate of indomethacin by solid dispersion in polyvinyl pyrrolidone K30 and poloxomer 188. Asian J. Pharm. Technol. 2012;2(3):116–122. [Google Scholar]
- Bashar A.A., Hatim S.A., Ayman A.K. Comparative dissolution of diltiazem immediate and extended release products using conventional USP and innovative dissolution paddles. Open Drug Delivery J. 2010;4:48–54. [Google Scholar]
- Cherukuri S., Chappidi S.R., Anilkumar Dindigala, Amrutha Wadla, Anusha Arepalli Leela. Liquisolid technique: a novel approach to enhance solubility and bioavailability of BCS-2 drugs. Int. Res. J. Pharm. 2012;3(7) [Google Scholar]
- De Jong J.A.H. Relations between tablet properties. Pharm. Weekblad Sci. Ed. 1987;9 doi: 10.1007/BF01956488. [DOI] [PubMed] [Google Scholar]
- Gowthamarajan K., Sachin K.S. Dissolution testing for poorly soluble drugs: a continuing perspective. Dissol. Technol. 2010:24–32. [Google Scholar]
- Kavitha K., Kotha N.S., Lova R., Ganesh N.S., Ramesh B. Effect of dissolution rate by liquisolid compact approach: an overview. Pharm. Lett. 2011;3(1):71–83. [Google Scholar]
- Panneer S.M., Natrajan R., Selvaraj S., Rajendran N.N. A novel drug–drug solid dispersion of hydrochlorothiazide losartan potassium. Int. J. Pharma Bio Sci. 2010;1(4):68–80. [Google Scholar]
- Paulo C., Jose M.S.L. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001;13:123–133. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- Rashmi V.T., Pravin S.A., Jayashree B.T., Jayashri G.M., Milind J.U. Solubility enhancement studies of hydrochlorothiazide by preparing solid dispersions using losartan potassium and urea by different methods. Pharm. Lett. 2011;3(6):8–17. [Google Scholar]
- Sahil M.G., Sharad S.P., Shirish V.S., Kisan R.J., Vilasrao J.K. Liquisolid compact: a new technique for enhancement of drug dissolution. Int. J. Res. Pharm. Chem. 2011;1:3. [Google Scholar]
- Sanjeev R.G., Ravindra J. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian J. Pharm. Sci. 2010;5(2):50–60. [Google Scholar]
- Santhosh A.M., Kishore B.A., Thadanki Madhurilatha, Eswara Gupta M. Solid dispersion- An approach to enhance the dissolution rate of Irbesartan. Int. J. Res. Pharm. Chem. 2011;1:4. [Google Scholar]
- The United States Pharmacopoeia (USP-35/NF-30), 2012. United States Pharmacopeial Convention Inc, Rockville.
- Tyagi R., Dhillon V. Solid dispersion: a fruitful approach for improving the solubility and dissolution rate of poorly soluble drugs. J. Drug Delivery Ther. 2012;2(4):5–14. [Google Scholar]
- Varshney H.M., Chatterjee A. Solubility enhancement of poorly hydrophilic drugs by using different techniques: a review. Int. J. Ther. Appl. 2012;6:8–13. [Google Scholar]
- Vijay K.N., Ramarao T., Jayaveera K.N. Liquisolid compacts: a novel approach to enhance bioavailability of poorly soluble drugs. Int. J. Pharm. Biol. Sci. 2011;1(3):89–102. [Google Scholar]
- Yousef J., Baharak J., Ali N. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine) Int. J. Pharm. 2007;341:26–34. doi: 10.1016/j.ijpharm.2007.03.034. [DOI] [PubMed] [Google Scholar]