

Abstract
Infection of human skin with Mycobacterium ulcerans, the causative agent of Buruli ulcer, is associated with the systemic diffusion of a bacterial macrolide named mycolactone. Patients with progressive disease show alterations in their serum proteome, likely reflecting the inhibition of secreted protein production by mycolactone at the cellular level. Here, we used semi-quantitative metabolomics to characterize metabolic perturbations in serum samples of infected individuals, and human cells exposed to mycolactone. Among the 430 metabolites profiled across 20 patients and 20 healthy endemic controls, there were significant differences in the serum levels of hexoses, steroid hormones, acylcarnitines, purine, heme, bile acids, riboflavin and lysolipids. In parallel, analysis of 292 metabolites in human T cells treated or not with mycolactone showed alterations in hexoses, lysolipids and purine catabolites. Together, these data demonstrate that M. ulcerans infection causes systemic perturbations in the serum metabolome that can be ascribed to mycolactone. Of particular importance to Buruli ulcer pathogenesis is that changes in blood sugar homeostasis in infected patients are mirrored by alterations in hexose metabolism in mycolactone-exposed cells.
Buruli ulcer (BU) is a necrotizing disease of the skin caused by infection with Mycobacterium ulcerans, the third most prevalent mycobacterial pathogen in humans after M. tuberculosis and M. leprae1. How M. ulcerans is transmitted to humans is not fully understood, however there is increasing evidence that breaches in the skin barrier and exposure to contaminated environments are both required2,3,4,5. Since the 1980s, BU has spread in low-income developing countries of West Africa6. If not diagnosed or treated appropriately, it can result in irreversible deformity, functional disability and life-threatening secondary infections. The current diagnosis methods include acid-fast staining, culture or amplification of bacterial DNA from fine needle aspirates, swabs or skin biopsies7. Treatment consists of the daily administration of rifampicin and streptomycin for eight weeks8,9, and excision surgery of large lesions. Although effective, control programs are costly, reactive rather than pro-active, and globally unsuited to field conditions. In order to improve the detection and management of BU, it is essential to improve our understanding of the molecular and cellular mechanisms underpinning BU pathogenesis10.
M. ulcerans is unique amongst human pathogens in its capacity to produce a polyketide-derived macrolide called mycolactone11,12,13,14. Bacterial production of mycolactone is essential for BU formation, as shown by the avirulence of mycolactone-deficient strains of M. ulcerans in rodent models of infection. While bacteria grow primarily in host skin tissues, mycolactone gains access to the peripheral circulation15,16. Foodpad infection of mice with wild-type, but not mycolactone-deficient strains of M. ulcerans, induced intrinsic defects in blood T cells evidenced by their incapacity to produce cytokines upon activation ex vivo15, suggesting that mycolactone modulates the functional biology of T cells at the systemic level. In vitro, mycolactone altered the expression of homing receptors by resting T cells, and the production of cytokines by activated T cells, without altering their viability17,18,19. Mycolactone was shown to operate at the post-transcriptional level, and independently of mTOR17,20. Although its precise mechanism of action remains to be elucidated, there is recent evidence that mycolactone blocks the co-translational translocation of secreted and membrane-bound proteins into the endoplasmic reticulum21,22. In line with this finding, the proteomic profiling of serum samples of patients with BU showed significant reductions in the level of multiple soluble proteins, including T cell cytokines23.
To further explore the physiological consequences of bacterial production of mycolactone in infected hosts, we compared the metabolic perturbations induced by infection with M. ulcerans in human hosts to those induced by mycolactone treatment in human cells. Since bacterially-produced mycolactone diffuses from cutaneous lesions into the peripheral circulation, we focused our analysis on serum samples. Jurkat T cells were selected as a model, because leukocytes are exposed to mycolactone during M. ulcerans infection15,16, and Jurkat T cells display the same functional defects as primary T cells upon exposure to mycolactone in vitro17,18,19. In addition to provide novel insight into the molecular mechanisms underlying BU pathogenesis, our study delineates mycolactone signature in the serum metabolome of infected hosts.
Methods
Ethics statement
The ethics committee at the School of Medical Sciences, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana approved the protocol of this study (CHRPE/11/28/06). All adult subjects provided written informed consent, and a parent or guardian of any child participant provided informed consent on their behalf. The review board also gave approval to document informed consent by use of thumbprints for illiterate participants. Studies using human subjects were performed in accordance with the approved guidelines and regulations.
Human studies
Two cohorts of patients and age- and gender-matched healthy controls were recruited for the purposes of this study (Table 1). The first cohort was used to compare the metabolic profiles of BU patients and controls from the same community. The second cohort was recruited subsequently, in order to confirm altered cortisol levels in BU patients, and detect an eventual association with lesion severity. Patients were from the middle forest belt of Ashanti Region of Ghana, from Buruli ulcer endemic villages near Tepa Government Hospital (Ahafo Ano North District), Agogo Presbyterian Hospital and Nkawie Government Hospital (Atwima Nwabiagya district). They were included in the study if they met the WHO clinical case definition of M. ulcerans disease; were not pregnant; were not receiving antibiotic treatment; had no history of tuberculosis, leprosy, or liver, kidney, or hearing impairment. On the day of clinical diagnosis, fine needle aspirates were taken for PCR amplification of IS2404 repeat sequence of M. ulcerans24. Punch biopsy specimens of 4 mm diameter were also stained for acid-fast bacilli and cultured on Lowenstein-Jensen slopes, as previously described25. Patients were started on streptomycin 15 mg/kg and rifampicin 10 mg/kg treatment daily for 8 weeks, as recommended by the WHO, at village health posts under direct observation. Blood samples were also collected at the day of clinical diagnosis of BU, before the initiation of antibiotic therapy. Patients were on empty stomach, in an overnight-fasted state. They were asked if they had taken antibiotic or other medication. Only those who had responded negatively and had confirmed BU were subsequently included in the study. Healthy individuals from the same endemic areas also provided serum samples to serve as a comparator. Serum sampling, freezing and storage were performed in a standardized manner, as follows. Blood samples (8 ml) were collected in the field in BD Vacutainer Serum separator tubes, mixed and left to clot according to the manufacturer’s recommendations. Tubes were then transported within 2 h on ice to the laboratory, for centrifugation and serum separation. The recovered serum (3–4 ml) was aliquoted in Eppendorf SafelockTM tubes and stored at −80oC. Samples of cohort 1 individuals were shipped to Institut Pasteur (Paris, France) on dry ice, thawed and re-aliquoted in 100 μl-containing Eppendorf Safelock tubes prior to shipping to Metabolon Inc. on dry ice. Serum samples of cohort 2 individuals were shipped to St George’s University of London on dry ice, and assayed for cortisol using Siemens Advia Centaur Competitive Immunoassay and Direct Chemiluminescent Technology.
Table 1. Human cohort description.
| Cohort 1 | Healthy controls (n = 20) | Patients with BU (n = 20) |
|---|---|---|
| Age, median (range), years | 12 (6–35) | 13 (7–35) |
| Sex, no. Male/no. Femelle | 12/8 | 12/8 |
| Ulcer category | ||
| I (lesion size ≤5 cm in widest diameter) | 7 | |
| II (lesion size ≤15 cm in widest diameter) | 13 | |
| Cohort 2 | Healthy controls (n = 29) | Patients with BU (n = 38) |
| Age, median (range), years | 13 (5–63) | 13 (5–75) |
| Sex, no. Male/no. Femelle | 14/15 | 18/20 |
| Ulcer category | ||
| I (lesion size ≤5 cm in widest diameter) | 16 | |
| II (lesion size ≤15 cm in widest diameter) | 10 | |
| III (lesion size >15 cm in widest diameter or multiple lesions) | 12 |
Mycolactone
Mycolactone A/B was purified from M. ulcerans bacterial cell pellets (strain 1615, ATCC 35840) as previously described11. Mycolactone was quantified by measure of absorbance (λmax = 362 nm; log ε = 4.29)26, and purity controlled by mass spectrometry. A stock solution (20 μM) was prepared in ethanol solvent that was diluted 1000X for T cell treatments. Controls exposed to the same volume of vehicle were included.
Cellular studies
Jurkat E6.1 (ATCC TIB-152TM) T cells were cultured in RPMI GlutamaxTM (Life Technologies), supplemented with 10% heat-inactivated fetal calf serum (FCS) (Invitrogen) and penicillin/streptomycin (100 U/ml, 100 μg/ml). Cells in exponential phase of growth were exposed to 20 nM mycolactone (n = 6) or ethanol (n = 5) for 16 h. Cells were recovered and dried by two rounds of centrifugation at 750 g for 3 min, flash-frozen, and stored at −80 °C until analysis.
Metabolomic profiling
Semi-quantitative metabolomic analyses were performed by Metabolon Inc., as described (http://www.metabolon.com/). On the day of extraction, serum samples (100 μl) or cell pellets (50 μl) were thawed on ice. Proteins were precipitated with methanol, using an automated liquid handler (Hamilton LabStar). The methanol contained four standards, which permitted the monitoring of extraction efficiency. The resulting extract was divided into three fractions that were placed briefly on a TurboVap® (Zymark) to remove the organic solvent, frozen and dried under vacuum. Samples destined to LC/MS analysis were reconstituted in acidic or basic LC-compatible solvents, each of which contained 11 or more injection standards at fixed concentrations. One aliquot was analyzed using acidic positive ion optimized conditions and the other using basic negative ion optimized conditions in two independent injections using separate dedicated columns. Extracts reconstituted in acidic conditions were gradient eluted using water and methanol both containing 0.1% formic acid, while the basic extracts, which also used water/methanol, contained 6.5 mM ammonium bicarbonate. The samples destined for GC/MS analysis were re-dried under vacuum desiccation for a minimum of 24 h prior to being derivatized under dried nitrogen using bistrimethyl-silyl-triflouroacetamide. Technical replicates created from a homogenous pool containing a small amount of all study samples were included. The UPLC-MS/MS platform used a Waters Acquity UPLC with Waters UPLC BEH C18 columns (2.1 × 100 mm, 1.7 μm) and a ThermoFisher LTQ mass spectrometer. GC-MS was performed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole MS. Metabolites were identified by automated comparison of the ion features in the experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, and in-source fragments as well as associated MS spectra. Peaks were quantified by area under the curve measurements. Raw area counts for each metabolite in each sample were normalized to correct for variation resulting from instrument inter-day, tuning differences by the median value for each run-day, therefore setting the medians to 1.0 for each run. Metabolites missing more than one value were excluded from the analysis.
Statistical analyses
Following log transformation and normalization, Principal Component Analysis (PCA) was used to identify the biochemicals discriminating patients from controls with a false discovery rate (q-value) inferior to 0.2. We then used Welch’s two-sample t-test to identify biochemicals differing significantly between the two groups (p ≤ 0.05). The metabolomic analysis of Jurkat T cells being part of a larger study including multiple treatments, two-way ANOVA with contrasts was used to identify biochemicals differing significantly between mycolactone- and vehicle-treated groups. In both human and cell studies, q-values were calculated for each metabolite to take into account multiple comparisons. The GraphPad Prism software (v5.0d, La Jolla, CA) was used for box-and-whisker plot representation, with outliers identified by Tukey’s test.
Results and Discussion
Metabolomic profiling of BU
Serum samples were harvested from 20 patients with newly diagnosed BU lesions and 20 age- and gender-matched healthy controls from the same endemic community (Cohort 1, Table 1). Following solvent extraction, samples were split for analysis on liquid or gas chromatography platforms coupled with mass spectrometry. A total of 430 metabolites were identified, whose spectrometric signals were normalized and compared across patients and controls. PCA revealed a separate clustering between the patient and control populations (Fig. 1), showing that BU disease is associated with significant metabolic alterations. Nineteen (4%) metabolites were discriminative (p-value < 0.01, q-value < 0.2). Among them, 11 were upregulated in patients relative to controls, whereas 8 were downregulated. Intermediates of glycolysis, pentose-phosphate pathway (PPP) and tricarboxylic acid cycle (TCA) were modulated, indicating that energy-generating pathways had been perturbed. Alterations in the peptide, lipid and nucleotide metabolic pathways were also observed. We used a Welch’s two-sample t-test (p < 0.05) to gain further insight into metabolite differences between groups. Fifty-four metabolites (12%) were present at significantly different levels in patients with BU, compared to controls (Table 2). They clustered into the hexose, fatty acid, lysolipid, steroid hormones, purine and heme metabolism, leading us to examine these pathways in greater detail.
Figure 1. Metabolic signature of BU.

PCA scatterplot of serum metabolites in patients with BU and controls. The most discriminating biochemicals (q-value ≤ 0.2) are shown, with their p-value and variation coefficient (Fold change) across groups (Blue: relatively increased; Yellow: relatively decreased in patients versus controls).
Table 2. Metabolic signature of BU in human patients.
| Metabolism | Biochemical Pathway | Metabolite | Fold change (Patient vs Ctrl) | p-value | q-value |
|---|---|---|---|---|---|
| Amino acid | Histidine | histidine | 0,85 | 0,002 | 0,090 |
| Lysine | N6-acetyllysine | 0,80 | 0,010 | 0,172 | |
| Phenylalanine and Tyrosine | p-cresol sulfate | 0,64 | 0,006 | 0,146 | |
| Cysteine and Methionine | S-methylcysteine | 0,67 | 0,049 | 0,388 | |
| Glutathione | 5-oxoproline | 0,84 | 0,002 | 0,090 | |
| Peptide | Dipeptide | glycylvaline | 2,03 | 0,000 | 0,023 |
| isoleucylglycine | 1,21 | 0,038 | 0,334 | ||
| phenylalanylleucine | 1,60 | 0,008 | 0,169 | ||
| Gamma-glutamyl amino acid | gamma-glutamylisoleucine | 0,77 | 0,046 | 0,386 | |
| gamma-glutamylmethionine | 0,81 | 0,025 | 0,295 | ||
| Fibrinogen cleavage peptide | ADSGEGDFXAEGGGVR | 1,47 | 0,029 | 0,316 | |
| DSGEGDFXAEGGGVR | 1,63 | 0,002 | 0,090 | ||
| Carbohydrate | Hexose | fructose | 1,50 | 0,031 | 0,316 |
| mannose | 1,51 | 0,000 | 0,023 | ||
| Glycolysis and Gluconeogenesis | glucose | 1,15 | 0,001 | 0,090 | |
| Nucleotide Sugar and Pentose | xylulose | 1,40 | 0,003 | 0,119 | |
| Energy | TCA cycle | citrate | 0,84 | 0,012 | 0,184 |
| malate | 0,67 | 0,001 | 0,090 | ||
| Lipid | Monohydroxy fatty acid | 4-hydroxybutyrate (GHB) | 0,72 | 0,048 | 0,388 |
| 2-hydroxyoctanoate | 0,74 | 0,028 | 0,316 | ||
| 3-hydroxyoctanoate | 0,74 | 0,011 | 0,181 | ||
| 2-hydroxystearate | 1,18 | 0,011 | 0,181 | ||
| 2-hydroxypalmitate | 1,21 | 0,006 | 0,146 | ||
| Beta-oxidation | palmitoylcarnitine | 0,66 | 0,012 | 0,181 | |
| oleoylcarnitine | 0,63 | 0,008 | 0,169 | ||
| Bile acid | glycodeoxycholate | 0,58 | 0,010 | 0,172 | |
| glycolithocholate sulfate | 0,33 | 0,000 | 0,023 | ||
| taurolithocholate 3-sulfate | 0,42 | 0,006 | 0,146 | ||
| Glycerolipid | choline | 0,91 | 0,035 | 0,334 | |
| Lysolipid | 1-oleoylglycerophosphoethanolamine | 0,71 | 0,009 | 0,169 | |
| 2-oleoylglycerophosphoethanolamine | 0,74 | 0,032 | 0,316 | ||
| 1-linoleoylglycerophosphoethanolamine | 0,73 | 0,028 | 0,316 | ||
| 1-palmitoylglycerophosphocholine | 0,88 | 0,042 | 0,367 | ||
| 2-palmitoylglycerophosphocholine | 0,73 | 0,024 | 0,284 | ||
| 1-oleoylglycerophosphocholine | 0,83 | 0,038 | 0,334 | ||
| 1-linoleoylglycerophosphocholine | 0,74 | 0,005 | 0,146 | ||
| 2-linoleoylglycerophosphocholine | 0,70 | 0,005 | 0,146 | ||
| 1-dihomo-linoleoylglycerophosphocholine | 0,73 | 0,009 | 0,169 | ||
| Monoacylglycerol | 1-stearoylglycerol (1-monostearin) | 1,25 | 0,038 | 0,334 | |
| Sphingolipid | sphinganine | 0,55 | 0,001 | 0,090 | |
| Sterol/Steroid | cholesterol | 1,13 | 0,020 | 0,243 | |
| cortisol | 1,42 | 0,018 | 0,237 | ||
| cortisone | 1,26 | 0,007 | 0,157 | ||
| Nucleotide | Purine | xanthine | 1,27 | 0,004 | 0,146 |
| inosine | 2,01 | 0,004 | 0,146 | ||
| Cofactor | Heme | bilirubin (Z,Z) | 0,44 | 0,037 | 0,334 |
| bilirubin (E,E) | 0,28 | 0,014 | 0,204 | ||
| biliverdin | 0,66 | 0,019 | 0,243 | ||
| Vitamin | Riboflavin | riboflavin (Vitamin B2) | 1,82 | 0,048 | 0,388 |
| Tocopherol | gamma-CEHC | 0,67 | 0,017 | 0,236 | |
| Xenobiotic | Chemical | 4-methylcatechol sulfate | 0,74 | 0,016 | 0,221 |
| hexaethylene glycol | 1,20 | 0,030 | 0,316 | ||
| octaethylene glycol | 1,15 | 0,031 | 0,316 | ||
| pentaethylene glycol | 1,13 | 0,049 | 0,388 |
Hexoses
Compared to controls, patients with BU displayed elevated levels of all detected hexoses (glucose, fructose and mannose) (Fig. 2a). These sugars enter the cells via common membrane transporters of the solute carrier (SLC)-2 family. The 15–50% increase in serum hexoses may thus indicate defective uptake by SLC2 transporters, or increased hepatic gluconeogenesis. The PPP requires glucose for the generation of pentoses (Fig. 2a). We observed a relative accumulation of xylulose in patient serum (Table 2). Since serum levels of the xylulose precursor xylitol were unchanged, it suggested that generation of PPP intermediate xylulose-5-phosphate might be reduced. Finally, the TCA cycle intermediates citrate and malate were decreased in patients with BU, while alpha-ketoglutarate, succinate and fumarate were unchanged (Supplementary Figure S2 and Table 2). The TCA cycle is essential for the generation of ATP and precursors for various biosynthetic pathways. It requires equilibrated anaplerosis and cataplerosis (for entry and exit of TCA anions, respectively)27. In patients with BU, the imbalance between anaplerosis substrates (alpha-ketoglutarate) and cataplerosis substrates (citrate and malate) suggests that TCA cycle function may be impaired.
Figure 2. Increased serum hexoses in BU patients mirror decreased hexose concentrations in mycolactone-exposed cells.

(a) Differential serum levels of the detected hexoses in BU patients versus controls, shown as box and whiskers and in the context of energy-generating metabolic pathways. Biochemicals in bold red were relatively increased in patients versus controls. Those in bold black were detected at comparable levels. Those in grey were not detected. (b) Differential concentrations of detected hexoses in mycolactone- and vehicle-treated Jurkat T cells, shown as box and whiskers. *p < 0.05, ***p < 0.001.
To determine if some of these effects could result from the action of mycolactone, we profiled the metabolome of Jurkat T cells exposed for 16 h to 20 nM of the purified factor, or vehicle as control. In accordance with previous studies19, this treatment decreased the production of membrane receptor CD62L without altering the cell viability (Supplementary Figure S1). Total cell metabolites were extracted and analyzed similar to serum samples, leading to the identification and relative quantification of 292 metabolites. Among them, 59 differed significantly between experimental groups (Table 3). Notably, glucose, galactose and mannose were relatively less concentrated in mycolactone-exposed T cells (Fig. 2b), arguing for a defect in cellular uptake by membrane transporters. Intracellular levels of glucose-1-phosphate and mannose-6-phosphate were downregulated in mycolactone-treated T cells (Table 3), indicative of altered glycolysis. Mycolactone also triggered the intracellular accumulation of acetylcarnitine, propionylcarnitine and butyrylcarnitine (Table 3). On the contrary, serum levels of palmitoylcarnitine and oleoylcarnitine were decreased in patients with BU (Table 2). Because they facilitate the transport of fatty acids across mitochondrial membranes, a rate-limiting step in fatty acid oxidation (FAO), circulating acylcarnitines are clinically-used biomarkers of FAO disorders28. At the cellular level, the accumulation of acylcarnitines correlates with reduced oxidation of glucose and insulin resistance29. The increased levels of serum hexoses in patients with BU may thus be due, at least partially, to mycolactone-induced defects in hexose uptake and FAO. With the exception of mannose, none of the above-described alterations were observed in patients with TB30.
Table 3. Metabolic signature of mycolactone in human T cells.
| Metabolism | Biochemical Pathway | Metabolite | Fold change (Myco vs Ctrl) | p-value | q-value |
|---|---|---|---|---|---|
| Amino acid | Glycine, Serine and Threonine | betaine | 1,38 | 0,003 | 0,028 |
| beta-alanine | 1,41 | 0,038 | 0,109 | ||
| Glutamate | glutamate | 0,77 | 0,011 | 0,065 | |
| pyroglutamine | 1,31 | 0,023 | 0,089 | ||
| gamma-aminobutyrate (GABA) | 1,43 | 0,014 | 0,066 | ||
| Histidine | histidine | 0,78 | 0,017 | 0,072 | |
| Lysine | lysine | 0,46 | 0,008 | 0,053 | |
| 2-aminoadipate | 0,53 | 0,000 | 0,005 | ||
| Phenylalanine and Tyrosine | phenylalanine | 0,79 | 0,026 | 0,089 | |
| tyrosine | 0,71 | 0,002 | 0,024 | ||
| Tryptophan | tryptophan | 0,77 | 0,025 | 0,089 | |
| Valine, Leucine and Isoleucine | leucine | 0,80 | 0,031 | 0,100 | |
| valine | 0,81 | 0,049 | 0,128 | ||
| Cysteine and Methionine | taurine | 1,34 | 0,027 | 0,089 | |
| methionine | 0,76 | 0,023 | 0,089 | ||
| 2-hydroxybutyrate (AHB) | 1,25 | 0,047 | 0,124 | ||
| Urea cycle, Arginine and Proline | dimethylarginine (SDMA + ADMA) | 0,65 | 0,010 | 0,063 | |
| N-acetylornithine | 0,79 | 0,024 | 0,089 | ||
| argininosuccinate | 0,43 | 0,009 | 0,056 | ||
| Creatine | creatine | 1,44 | 0,001 | 0,018 | |
| Polyamine | putrescine | 1,84 | 0,001 | 0,015 | |
| Peptide | Dipeptide | glycylproline | 0,68 | 0,036 | 0,106 |
| glycylleucine | 0,69 | 0,039 | 0,109 | ||
| glycylthreonine | 0,70 | 0,003 | 0,031 | ||
| prolylglycine | 0,58 | 0,004 | 0,034 | ||
| prolylalanine | 0,56 | 0,004 | 0,034 | ||
| prolylglutamine | 0,58 | 0,005 | 0,039 | ||
| cysteinylglycine | 0,58 | 0,031 | 0,100 | ||
| prolylglutamate | 0,62 | 0,013 | 0,065 | ||
| phenylalanylaspartate | 0,61 | 0,008 | 0,053 | ||
| Gamma-glutamyl amino acid | gamma-glutamylvaline | 1,77 | 0,000 | 0,004 | |
| gamma-glutamylleucine | 1,58 | 0,001 | 0,018 | ||
| gamma-glutamylisoleucine | 1,40 | 0,012 | 0,065 | ||
| gamma-glutamylmethionine | 0,57 | 0,006 | 0,047 | ||
| gamma-glutamylglutamine | 0,62 | 0,000 | 0,007 | ||
| gamma-glutamylthreonine | 2,10 | 0,000 | 0,000 | ||
| Carbohydrate | Aminosugar | Isobar: UDP-acetylglucosamine, UDP-acetylgalactosamine | 1,45 | 0,017 | 0,072 |
| Hexose | galactose | 0,54 | 0,012 | 0,065 | |
| 6′-sialyllactose | 2,30 | 0,000 | 0,005 | ||
| mannose | 0,39 | 0,014 | 0,066 | ||
| mannose-6-phosphate | 0,53 | 0,012 | 0,065 | ||
| sorbitol | 1,30 | 0,042 | 0,114 | ||
| Glycolysis | glucose 1-phosphate | 0,58 | 0,033 | 0,100 | |
| glucose | 0,46 | 0,023 | 0,089 | ||
| Lipid | Beta-oxidation | propionylcarnitine | 1,80 | 0,001 | 0,018 |
| butyrylcarnitine | 2,84 | 0,002 | 0,021 | ||
| Carnitine | acetylcarnitine | 1,34 | 0,013 | 0,065 | |
| Glycerolipid | choline phosphate | 1,27 | 0,026 | 0,089 | |
| Lysolipid | 1-myristoylglycerophosphocholine | 0,60 | 0,033 | 0,100 | |
| 1-pentadecanoylglycerophosphocholine | 0,64 | 0,038 | 0,109 | ||
| Neurotransmitter | acetylcholine | 9,40 | 0,000 | 0,000 | |
| Sterol/Steroid | lathosterol | 1,66 | 0,025 | 0,089 | |
| Nucleotide | Purine | xanthine | 0,72 | 0,002 | 0,024 |
| hypoxanthine | 0,66 | 0,020 | 0,084 | ||
| inosine | 0,60 | 0,015 | 0,070 | ||
| inosine 5’-monophosphate (IMP) | 0,68 | 0,042 | 0,114 | ||
| N1-methyladenosine | 0,70 | 0,033 | 0,100 | ||
| Cofactor and Vitamin | Biotin | biotin | 1,94 | 0,000 | 0,000 |
| Pantothenate and CoA | pantothenate | 1,51 | 0,000 | 0,004 |
Steroid hormones
Glucocorticoids assist in the regulation of glucose homeostasis through the stimulation of hepatic gluconeogenesis and downregulation of glucose transport systems. There was a relative augmentation in serum cholesterol and downstream glucocorticoid hormones cortisol in patients with BU (Fig. 3a). Cortisone, a conversion product of cortisol with weaker glucocorticoid activity, was also increased whereas other steroidal hormones were not significantly impacted. To validate these findings with an independent and quantitative approach, an additional cohort of patients and controls was assayed for serum cortisol (Cohort 2, Table 1). In agreement with our metabolomics data, the mean cortisol level was higher in patients with BU, compared to controls (Fig. 3b). Although variable, cortisol levels trended higher in patients with more severe lesions (Fig. 3b). No relationship could be demonstrated between serum cortisol and paradoxical reaction, or the clinical form of lesions (nodule, plaque, oedema or ulcer). Together with the data in Fig. 2, these observations suggest that glucocorticoid hormones may be induced in patients with progressive ulcers, in order to raise blood sugars. Since corticosteroids inhibit wound healing, increased circulation in patients may delay their clinical response to antibiotic treatment.
Figure 3. Increased serum glucocorticoids in BU patients.

(a) Differential serum levels of glucocorticoid hormones and cholesterol in BU patients and controls. (b) Absolute concentrations of serum cortisol in BU patients versus controls (left), and patients with different ulcer category (right). Data are presented as box and whiskers. *p < 0.05, **p < 0.01.
Purine catabolites
Patients with BU displayed elevated serum levels of the purine catabolites inosine and xanthine (Supplementary Figure S3a and Table 2). These metabolites were not augmented in patients with active TB30, suggesting that they do not reflect a general response to mycobacterial infection. Interestingly, opposite variations were observed in T cells exposed to mycolactone (Supplementary Figure S3b and Table 2), whereas adenine and guanine, and the pyrimidine catabolite uracil remained unchanged. Although the underlying molecular mechanism is unclear, increased serum inosine and xanthine may thus constitute specific traits of BU.
Gamma-glutamyl amino acids
Among the metabolites discriminating patients from controls was the gamma-glutamyl amino acid degradation product 5-oxoproline (Fig. 1), which serum level was relatively lower in patients. Interestingly, patients also displayed reduced levels of gamma-glutamylisoleucine and gamma-glutamylmethionine (Table 2). Gamma-glutamyl amino acids result from the transfer of the gamma-glutamyl moiety of glutathione to acceptor amino acids by the liver enzyme gamma-glutamyl transferase (GGT). We reported previously that BU patients display normal serum GGT23. Since isoleucine and methionine were unchanged in patients versus controls in the present study, we can speculate that downregulation of their gamma-glutamyl derivatives is due to limited glutathione availability. Extracellular glutathione results from synthesis, consumption and extrusion by producing cells31. Binding of glutamate to cysteine is the first and rate-limiting step in the biosynthesis of this tripeptide. In Jurkat cells, mycolactone had mixed effects on the intracellular levels of gamma-glutamyl amino acids. It did not alter significantly the intracellular levels of cysteine, glutamate nor glutathione (reduced and oxidized forms). Although extracellular glutathione measurements would be required to confirm it, these data suggest that BU-associated alterations in serum gamma-glutamyl amino acids, and potentially glutathione and redox homeostasis, are independent of mycolactone.
Bile acids
Bile acids are synthesized from cholesterol by 7-alpha-hydroxylase (CYP7A1) in the liver (Fig. 4). Bile acids facilitate cholesterol elimination, intestinal absorption and excretion of lipids and lipid-soluble molecules. They are also important signaling molecules regulating energy homeostasis, inflammation and liver regeneration32. Compared to controls, patients with BU displayed significantly higher levels of cholesterol (Fig. 2a) and normal levels of 7-alpha-hydroxycholesterol and cholate, suggesting that precursors of bile acid synthesis are not limiting. Yet glycodeoxycholate, glycolithocholate sulfate and taurolithocholate 3-sulfate were significantly downregulated in patients with BU (Fig. 4). No variation in bile acids was reported in patients with TB30. The reduced levels of bile acids in BU patients may be indicative of decreased synthesis, increased intestinal absorption or urinary excretion. Since bile acid synthesis requires contribution from the microbial community, they may also reflect changes in the intestinal flora.
Figure 4. Decreased bile acids levels in the serum of BU patients.

Differential serum levels of detected bile acids in patients and controls, shown as box and whiskers and in the context of their metabolic pathways. Biochemicals in bold red were relatively increased in patients versus controls. Those in bold green were relatively decreased. Those in grey were not detected. **p < 0.01, ***p < 0.001.
Heme products
Heme, the most common porphyrin found in the human body, complexes with cellular proteins to form hemoglobin, myoglobin and cytochromes. Heme is synthesized from glycine and succinyl-CoA and can be oxidized into bilirubin and vasodilator carbon dioxide. In patients with BU, heme levels trended higher compared to controls. Conversely, the heme catabolic products biliverdin, bilirubin ZZ and EE were diminished in these subjects (Fig. 5 and Table 2). No such variation was detected in patients with TB, possibly because M. tuberculosis possesses its own heme-degrading enzyme MhuD, producing an unusual tetrapyrole called mycobilin33. The M. ulcerans MhuD gene orthologue (MUL_4167) is a predicted pseudogene due to the introduction of premature stop codon34. Consistent with this prediction, no metabolite with a mass corresponding to mycobilin was detected in the serum of patients with BU.
Figure 5. Decreased levels of heme catabolic products in the serum of BU patients.

Differential serum levels of biliverdin and bilirubin in patients and controls, shown as box and whiskers and in the context of the heme metabolic pathway. Biochemicals in bold green were relatively decreased in patients versus controls. Those in bold black were detected at comparable levels. Those in grey were not detected. *p < 0.05.
Riboflavin
Riboflavin (vitamin B2) was recently reported to be upregulated in mosquitoes exposed to live M. ulcerans, compared to untreated mosquitoes or mosquitoes exposed to dead bacteria35. Interestingly in the present work, patients with BU also displayed increased levels of riboflavin (Table 2). Inspection of the M. ulcerans genome predicts an intact riboflavin anabolic pathway. M. ulcerans is predicted to possess intact inosine-5′-monophosphate dehydrogenases (e.g. MUL_0901) and GMP synthase (MUL_0913) and the subsequent enzymes to convert these molecules to GTP and enter the riboflavin biosynthesis pathway. Increases in riboflavin are also consistent with the increased levels of the purine metabolism intermediates, inosine and xanthine (see above). The enhanced detection of riboflavin in infected hosts may thus reflect either bacterial growth, or the host response to infection. In any case, the observation that riboflavin levels are associated with M. ulcerans infection in both humans and mosquitoes suggest that it could potentially serve as a pathogen-specific correlate of infection.
Fibrinogen cleavage peptides
Upon vascular injury, soluble fibrinogen is cleaved into insoluble fibrin, which is the main component of blood clots. Fibrinogen A-α cleavage peptides ADSGEGDFXAEGGGVR and DSGEGDFXAEGGGVR were elevated in patients with BU (Supplementary Figure S4 and Table 2), likely reflecting vascular remodeling in lesions. Comparable augmentations were seen in patients with active TB and diabetes30,36, indicating that this process is not specific to BU.
Lysolipids
Phospholipids (also called glycerophospholipids) are the main lipid constituents of cell membranes. They are a highly diverse family of compounds containing diacylglycerol, a phosphate head group and organic molecules like ethanolamine or choline. Lysolipids and fatty acids are the natural products of their hydrolysis by phospholipases. Compared to controls, patients with BU displayed lower serum levels of choline and all detected lysophosphatidylcholine (LysoPC) compounds (Fig. 6a and Table 2). Lysophosphatidylethanolamines (LysoPE) were comparably impacted. No such variations were reported in patients infected with M. tuberculosis30, suggesting that they are specific to infection with M. ulcerans. In line with this hypothesis, several LysoPC compounds were decreased in mosquitoes exposed to live but not killed preparations of the bacteria35. Together with our observations in human patients, these data indicate that M. ulcerans interaction with its host may alter phospholipid turnover in biomembranes. In T cells exposed to mycolactone, two LysoPC species were decreased compared to controls (Fig. 6b and Table 3), suggesting that mycolactone may contribute to these changes.
Figure 6. Decreased serum lysolipids in BU patients.

(a) Differential levels of choline and representative lysolipids in patients and controls, shown as box and whiskers and in the context of their metabolic pathway. Biochemicals in bold green were relatively decreased in patients versus controls. Those in bold black were detected at comparable levels. Those in grey were not detected. (b) Differential levels of the detected lysolipids in mycolactone- and vehicle-treated Jurkat T cells, presented as box and whiskers *p < 0.05, **p < 0.01.
Conclusion
Here, we report the metabolomic profiles of serum samples of patients infected with M. ulcerans, and mycolactone-exposed cells. Figure 7 summarizes our principal findings, and highlights which metabolites/pathways were modulated in both BU patients and mycolactone-exposed cells. Among them were hexoses, purine products and lysolipids, suggesting that mycolactone released by bacteria interferes with blood cell production of biochemical energy, membrane lipid turnover and degradation of nucleic acids. Interestingly, patients with BU also displayed distinctive downregulation of bile acids and heme products, and upregulation of riboflavin in serum. Intermediates of these metabolic pathways may have potential as biomarkers of BU progression, and inspire new avenues for therapeutic interventions.
Figure 7. Metabolic alterations in patients with BU and mycolactone-exposed T cells partially overlap.
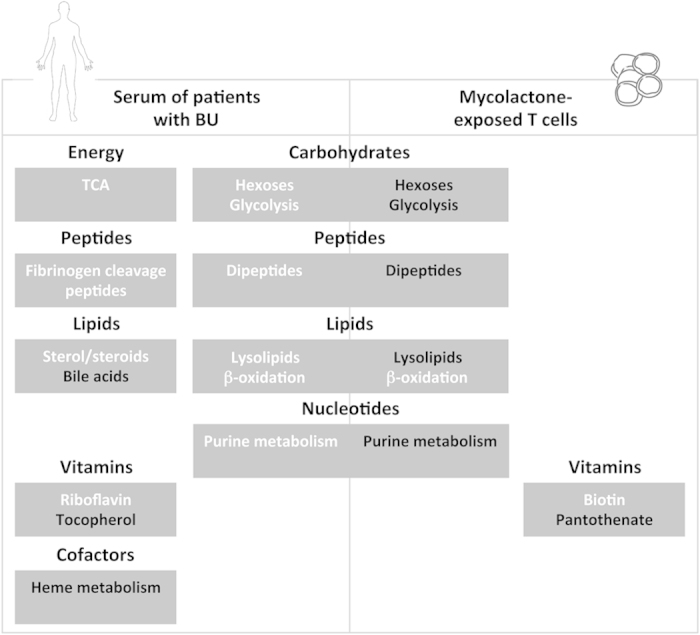
Metabolic alterations are grouped by biochemical pathway/biochemical structure, within boxes entitled with the corresponding metabolic pathway. In white: metabolites in this pathway were detected at higher levels in BU patients vs controls, or mycolactone-treated cells vs controls. In black: inversely.
Additional Information
How to cite this article: Niang, F. et al. Metabolomic profiles delineate mycolactone signature in Buruli ulcer disease. Sci. Rep. 5, 17693; doi: 10.1038/srep17693 (2015).
Supplementary Material
Acknowledgments
The authors wish to thank Magnus Fontes (Institut Pasteur) for help with PCA analysis. This work was supported by the Fondation de la Recherche Médicale (FRM 2012 DEQ20120323704), the Association Raoul Follereau and the Région Ile de France (dim130027).
Footnotes
Author Contributions C.D. and R.O.P. conceived the experiments; S.F., M.F., M.W.-J. and R.O.P. selected human participants and collected samples; L.G.-M. performed the Jurkat T cell studies; F.N., T.S. and C.D. analyzed the data; F.N. and C.D. prepared the figures and edited the text. All authors reviewed the manuscript.
References
- Yotsu R. R. et al. Revisiting Buruli ulcer. J Dermatol ; 10.1111/1346-8138.13049. (2015). [DOI] [PubMed] [Google Scholar]
- Huang G. K. & Johnson P. D. Epidemiology and management of Buruli ulcer. Expert Rev Anti Infe 12, 855–865 (2014). [DOI] [PubMed] [Google Scholar]
- Lavender C. J. et al. Risk of Buruli ulcer and detection of Mycobacterium ulcerans in mosquitoes in southeastern Australia. PLoS Negl Trop Dis 5, e1305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt R. W. et al. Ecology and transmission of Buruli ulcer disease: a systematic review. PLoS Negl Trop Dis 4, e911 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. S., Portaels F. & Meyers W. M. Buruli ulcer (Mycobacterium ulcerans infection). Trans R Soc Trop Med Hyg 102, 969–978 (2008). [DOI] [PubMed] [Google Scholar]
- Wansbrough-Jones M. & Phillips R. Buruli ulcer: emerging from obscurity. Lancet 367, 1849–1858 (2006). [DOI] [PubMed] [Google Scholar]
- Stinear T. et al. Identification and characterization of IS2404 and IS2606: two distinct repeated sequences for detection of Mycobacterium ulcerans by PCR. J Clin Microbiol 37, 1018–1023 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etuaful S. et al. Efficacy of the combination rifampin-streptomycin in preventing growth of Mycobacterium ulcerans in early lesions of Buruli ulcer in humans. Antimicrob Agents Chemother 49, 3182–3186 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfo F. S. et al. Clinical efficacy of combination of rifampin and streptomycin for treatment of Mycobacterium ulcerans disease. Antimicrob Agents Ch 54, 3678–3685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenin-Mace L., Oldenburg R., Chretien F. & Demangel C. Pathogenesis of skin ulcers: lessons from the Mycobacterium ulcerans and Leishmania spp. pathogens. Cell Mol Life Sci (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- George K. M., et al. Mycolactone: a polyketide toxin from Mycobacterium ulcerans required for virulence. Science 283, 854–857 (1999). [DOI] [PubMed] [Google Scholar]
- Stinear T. P. et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc Natl Acad Sci USA 101, 1345–1349 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H., Demangel C., Pidot S. J., Leadlay P. F. & Stinear T. Mycolactones: immunosuppressive and cytotoxic polyketides produced by aquatic mycobacteria. Nat Prod Rep 25, 447–454 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demangel C., Stinear T. P. & Cole S. T. Buruli ulcer: reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat Rev Microbiol 7, 50–60 (2009). [DOI] [PubMed] [Google Scholar]
- Hong H. et al. Mycolactone diffuses from Mycobacterium ulcerans-infected tissues and targets mononuclear cells in peripheral blood and lymphoid organs. PLoS Negl Trop Dis 2, e325 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfo F. S. et al. Mycolactone diffuses into the peripheral blood of buruli ulcer patients - implications for diagnosis and disease monitoring. PLoS Negl Trop Dis 5, e1237 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulkroun S. et al. Mycolactone suppresses T cell responsiveness by altering both early signaling and posttranslational events. J Immunol 184, 1436–1444 (2010). [DOI] [PubMed] [Google Scholar]
- Guenin-Mace L. et al. Shaping mycolactone for therapeutic use against inflammatory disorders. Sci Transl Med 7, 289ra285 (2015). [DOI] [PubMed] [Google Scholar]
- Guenin-Mace L. et al. Mycolactone impairs T cell homing by suppressing microRNA control of L-selectin expression. Proc Natl Acad Sci USA 108, 12833–12838 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds R. E., Lali F. V., Smallie T., Small P. L. & Foxwell B. M. Mycolactone inhibits monocyte cytokine production by a posttranscriptional mechanism. J Immunol 182, 2194–2202 (2009). [DOI] [PubMed] [Google Scholar]
- Hall B. & Simmonds R. Pleiotropic molecular effects of the Mycobacterium ulcerans virulence factor mycolactone underlying the cell death and immunosuppression seen in Buruli ulcer. Biochem Soc Trans 42, 177–183 (2014). [DOI] [PubMed] [Google Scholar]
- Hall B. S. et al. The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog 10, e1004061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R. O. et al. Combined inflammatory and metabolic defects reflected by reduced serum protein levels in patients with Buruli ulcer disease. PLoS Negl Trop Dis 8, e2786 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe J. A. M. et al. Development and application of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl Environ Microb 73, 4733–4740 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R. et al. Sensitivity of PCR targeting the IS2404 insertion sequence of Mycobacterium ulcerans in an assay using punch biopsy specimens for diagnosis of Buruli ulcer. J Clin Microbiol 43, 3650–3656 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T. & Kishi Y. Highly sensitive, operationally simple, cost/time effective detection of the mycolactones from the human pathogen Mycobacterium ulcerans. Chem Commun 46, 1410–1412 (2010). [DOI] [PubMed] [Google Scholar]
- Owen O. E., Kalhan S. C. & Hanson R. W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277, 30409–30412 (2002). [DOI] [PubMed] [Google Scholar]
- Rinaldo P., Cowan T. M. & Matern D. Acylcarnitine profile analysis. Genet Med 10, 151–156 (2008). [DOI] [PubMed] [Google Scholar]
- Schooneman M. G., Vaz F. M., Houten S. M. & Soeters M. R. Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes 62, 1–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner J. et al. Biomarkers of inflammation, immunosuppression and stress with active disease are revealed by metabolomic profiling of tuberculosis patients. Plos One 7(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak V. I. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 2012, 736837 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P., Cariou B., Lien F., Kuipers F. & Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89, 147–191 (2009). [DOI] [PubMed] [Google Scholar]
- Nambu S., Matsui T., Goulding C. W., Takahashi S. & Ikeda-Saito M. A new way to degrade heme: the Mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J Biol Chem 288, 10101–10109 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinear T. P. et al. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res 17, 192–200 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxmeier J. C. et al. Analysis of the metabolome of Anopheles gambiae mosquito after exposure to Mycobacterium ulcerans. Sci Rep 5, 9242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K. et al. Metabolic footprint of diabetes: a multiplatform metabolomics study in an epidemiological setting. Plos One 5, e13953 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.