

Abstract
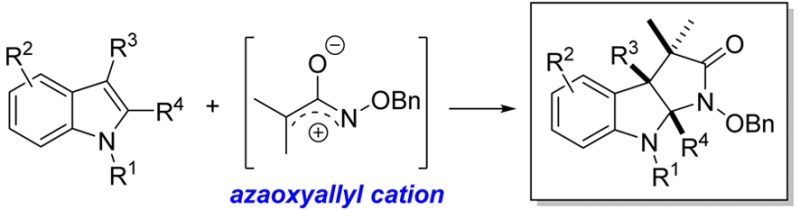
Herein, we report the first examples of the synthesis of pyrroloindolines by means of (3 + 2) dearomative annulation reactions between 3-substituted indoles and highly reactive azaoxyallyl cations. Computational studies using density functional theory (DFT) (B3LYP-D3/6-311G**++) support a stepwise reaction pathway in which initial C–C bond formation takes place at C3 of indole, followed by ring closure to give the observed products. Insights gleaned from these calculations indicate that the solvent, either TFE or HFIP, can stabilize the transition state through H-bonding interactions with oxygen of the azaoxyallyl cation and other relevant intermediates, thereby increasing the rates of these reactions.
Pyrroloindolines are important structural motifs found in numerous families of biorelevant alkaloid natural products. As a result, considerable resources have been expended to develop new and improved methods for their syntheses. By far, the most common way to prepare pyrroloindolines is through biomimetic pathways starting with either tryptophan, tryptamine, or derivatives thereof.1−20 Such a strategy involves the formation of bonds a and b (Scheme 1a) by alkylation at C3 with various electrophiles to generate an iminium intermediate, followed by ring closure to give the desired products. The alternative retrosynthetic disconnection at bonds b and c (Scheme 1b) would require 3-substituted indole 1 and 1,3-dipole 2 as synthons. In the forward direction, this approach constitutes a dearomative indole (3 + 2) annulation reaction and has been a relatively underexplored option for preparing pyrroloindolines as compared to the biomimetic strategy described above. Notable recent examples include Reisman’s use of 2-amidoacrylates,21−23 Davies’ reports of Rh-catalyzed annulation reactions,24,25 and annulations with aziridines by Chai.26
Scheme 1. Synthesis of Pyrroloindolines.

Consistent with our continuing interest in indole annulation27 and alkylation reactions,28 we recently disclosed a new method for carrying out formal (3 + 2) cycloadditions between 3-substituted indoles and oxyallyl cations 5 (Scheme 1c).29 We were, therefore, intrigued by Jeffrey and co-workers’ report in which they described the (4 + 3) cycloaddition reaction between azaoxyallyl cations and dienes such as furan and cyclopentadiene,30−32 later expanded to include diaminations.33,34 We postulated that azaoxyallyl cations 9 may also react with 3-substituted indoles in a fashion analogous to oxyallyl cations (Scheme 1d).29 In this manuscript and in the previous paper of this issue,38 the first examples of dearomative (3 + 2) annulation reactions between indoles and azaoxyallyl cations is described as a novel means to prepare pyrroloindolines.
We initiated our studies by examining the reaction between N-benzyl-3-methylindole (11) and bromohaloamide 8 under conditions we originally developed for the (3 + 2) annulation reaction of oxyallyl cations.29 Gratifyingly, this led to the formation of the desired product 12 in 94% isolated yield (Table 1, entry 1). Under these conditions (first reported by Föhlisch and co-workers for generating oxyallyl cations),35 formation of the azaoxyallyl cation likely occurs by hydrogen-bond-mediated tautomerization followed by unimolecular ionization of bromide. Despite this success, we later discovered that the use of TFE (2,2,2-trifluoroethanol) leads to competitive decomposition of haloamide 8 by solvolysis. By switching to the more sterically hindered solvent hexafluoroisopropanol (HFIP), we were able to minimize these solvolysis byproducts while maintaining high yields of the desired pyrroloindolines (entry 8).
Table 1. Optimization Reactions.

| entry | base | solvent | concn (M) | temp (°C) | time (h) | yielda (%) |
|---|---|---|---|---|---|---|
| 1 | Na2CO3 | TFE | 0.25 | rt | 4 | 94 |
| 2 | Na2CO3 | TFE | 0.25 | 0 | 6 | 61 |
| 3b | Na2CO3 | TFE | 0.25 | rt | 6 | 91 |
| 4 | Na2CO3 | THF | 0.25 | rt | 18 | no rxn |
| 5 | NEt3 | HFIP | 0.25 | rt | 16 | 42 |
| 6 | K2CO3 | HFIP | 0.25 | rt | 8 | 40 |
| 7 | Cs2CO3 | HFIP | 0.25 | rt | 12 | 46 |
| 8 | Na2CO3 | HFIP | 0.25 | rt | 4 | 89 |
| 9b | Na2CO3 | HFIP | 0.25 | rt | 18 | 66 |
| 10 | Na2CO3 | HFIP | 0.50 | rt | 6 | 39 |
Isolated yield.
2 equiv of indole, 1 equiv of haloamide.
With optimized reaction conditions in hand, we sought to delineate the scope of the reaction. Alkylation on the indole nitrogen could be either methyl or benzyl. The reaction was compatible with a wide range of functional groups at the C3 position of indole. These include esters, free alcohols, esters, nitriles, phthalimide, isopropyl, and methyl. We also demonstrated that substitution at C2 (entry 6), electron-rich groups at C4/6 (entry 17), and electronegative groups at C5 are well tolerated (entry 15). For the electron-rich substrate, we observed significant amounts of byproducts resulting from electrophilic aromatic substitution with the azaoxyallyl cation at C7 (entry 17). We were able to overcome this problem by reversing the stoichiometry of the indole and haloamide starting materials. N–O bond cleavage could be achieved using Mo(CO)6 to give 14 (eq 1).
 |
1 |
When indole 15 was reacted with monomethyl-substituted α-haloamide 16a, the expected product 17a was generated in 54% yield as an inseparable mixture of diastereomers with respect to the stereocenter bearing the methyl group (Scheme 2). It was also possible to prepare unsubstituted pyrroloindolines such as 18. We accomplished this by carrying out the annulation reaction of 15 with dichlorohaloamide 16b to initially give 17b in a 1:1 diastereomeric ratio with respect to the stereocenter bearing the chloride. Protodehalogenation using the Zn/Cu couple provided the desired compound 18.
Scheme 2. Synthesis of Monosubstituted and Unsubstituted Pyrroloindolines.

Several of the yields in Table 2 (entries 1, 7, 11, 12, 13, 15) and for compound 17 are slightly different from the corresponding reactions reported in the paper preceding this one.38 These differences are likely attributable to a combination of higher reaction concentration (i.e., 1 M versus 0.25 M) and, in some instances, longer reaction times (entries 1, 11, 15) and higher temperatures (50 °C versus room temperature for compound 17) that were utilized by Jeffrey.
Table 2. Scope of Dearomative Indole (3 + 2) Reactiona.


Indole (1 equiv), α-haloamide (2 equiv), Na2CO3 (4 equiv) in HFIP, 0.25 M.
Isolated yield.
Significant decomposition products observed.
The mechanism of this reaction was examined using Density Functional Theory (DFT) using the dispersion corrected B3LYP-D3 functional and the 6-311G**++ basis set; this method and basis set have been demonstrated to be competent in predicting the energetics of reactions of oxyallyl cations with indoles.29 The model starting materials used were indole 15 and azaoxyallyl cation 9-Me. An implicit solvent correction for 2,2,2-trifluoroethanol (TFE) was used. Full details of all calculations are provided in the Supporting Information.
Attempts to locate a transition state for a concerted reaction were unsuccessful, but energy surfaces for various stepwise processes were found. Formation of the expected product 12 could occur in a stepwise fashion by initial construction of the C–C bond at C3 of indole 15 to give intermediate 19a as shown in Figure 1. Alternatively, formation of a C–N bond at C2 of the indole could occur first to give intermediate 19b. The energy profiles for these two reactions are illustrated in Figure 2.
Figure 1.
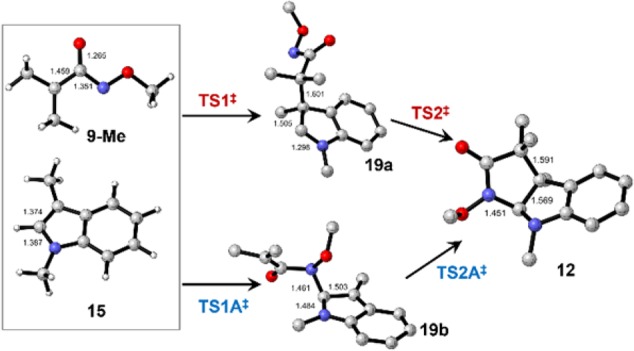
Calculated structures of starting materials, product and intermediates for the stepwise reaction of 15 with 9-Me to give 12. Except for 9-Me and 15, H atoms are not shown.
Figure 2.
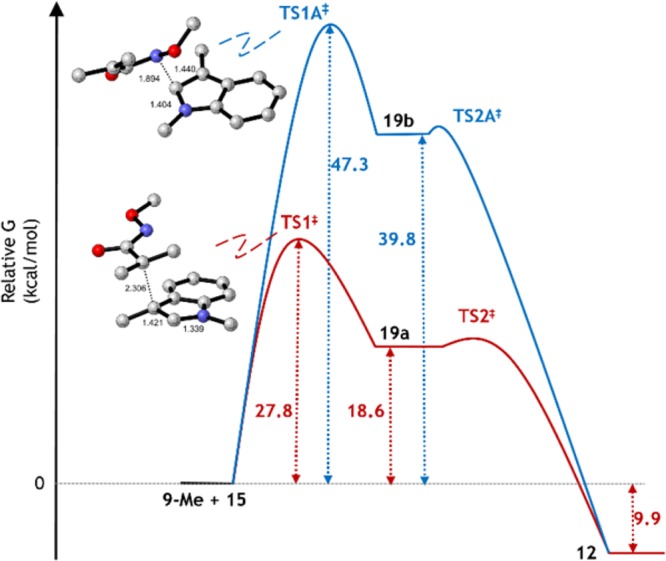
Free energy profiles for the stepwise formation of product 12 via intermediates 19a (red) and 19b (blue) with calculated structures of the transition states for the rate limiting steps. H atoms are not shown.
The lowest energy stepwise pathway is via intermediate 19a, which is over 21 kcal/mol more stable than its regioisomer 19b. Moreover, transition state TS1‡ which leads to 19a is almost 20 kcal/mol lower in energy than TS1A‡ leading to 19b. Formation of product 12 is downhill by almost 10 kcal/mol with respect to the starting materials. There are other conformers of intermediate 19a with essentially equal energies, none of which shows any interaction of N with C2 of the indole; likewise conformers essentially isoenergetic with TS1‡ were also located. We were unable to find TS2‡, suggesting that it is a very low energy process that evolves smoothly during rotation of the newly formed C–C bond of intermediate 19a.
Our calculations also reveal that the observed regiochemistry of addition to give 12 is kinetic in origin. The calculated intermediate 20a en route to the formation of 21 (the regioisomer of 12) is shown in Figure 3. Product 21 is 1.8 kcal/mol more stable than 12, and intermediate 20a is also 1.8 kcal/mol more stable than the corresponding intermediate 19a. However, the transition state leading to 20a lies 2.9 kcal/mol higher in energy than TS1‡ (leads to 19a). From these data, we conclude that the preference for the formation of product 12 over 21 is governed by kinetic control.
Figure 3.
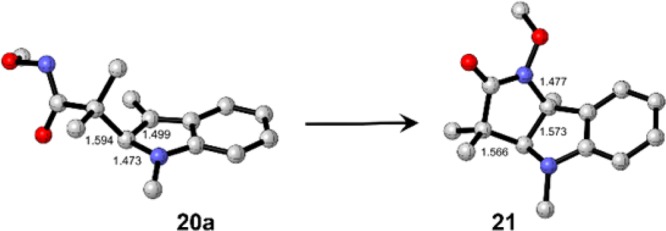
Calculated structures of starting materials, product, and intermediates for stepwise reaction of 15 with 9-Me to give 21 (the regioisomer of 12). H atoms are not shown.
The strong hydrogen-bonding solvents TFE and HFIP also play a significant role in this reaction. As an extreme limiting case, the effect of a very strong H-bonding solvent was modeled by calculating the energetics of the reaction between a fully O-protonated azaoxyallyl cation 22 and indole 15 with an implicit TFE solvent model. Key calculated structures are shown in Figure 4. O-Protonation dramatically lowers the activation barrier of the first step TS1‡(H+) (located 16.4 kcal/mol above reactants) and also strongly stabilizes the intermediate of the corresponding protonated intermediate 19a(H+) which lies only 1.9 kcal/mol higher than the reactants. The energy of regioisomeric intermediate 20a(H+) becomes 3.4 kcal/mol higher than that of 19a(H+) with the transition state for formation of 20a(H+) lying 4.3 kcal/mol above TS1‡(H+), thereby increasingly favoring the observed regioselectivity.
Figure 4.
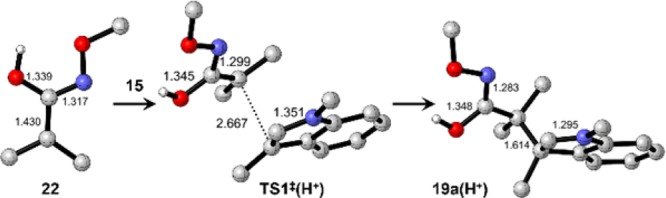
Calculated structures of protonated oxyallyl cation 22, the protonated intermediate 19a(H+), and the protonated transition state TS1‡(H+) for its reaction with indole 15. H atoms are not shown except for the OH.
These calculations suggest that, when strong hydrogen bond-donor solvents are used, the actual energy of the transition state should be intermediate between that of TS1‡ and TS1‡(H+). This should accelerate the rates of these reactions and also increase their regioselectivity. We and others have seen similar effects of protonation on the reaction energetics calculated for the corresponding reactions between oxyallyl cations and 15.29,36 Moreover, MacMillan and co-workers have empirically observed a similar phenomenon.37 Collectively, these results offer the intriguing possibility that annulation reactions of azaoxyallyl or oxyallyl cations can be catalyzed with strong H-bond donor catalysts in aprotic solvents.
In summary, we have disclosed the first report of preparing pyrroloindolines by means of dearomative (3 + 2) annulation reactions between indoles and azaoxyallyl cations. DFT calculations indicate a stepwise reaction mechanism and that the hydrogen bond donor ability of the solvent plays an important role in the stabilization of transition states and intermediates.
Acknowledgments
J.W. acknowledges the generous support of the NIH NIGMS (1R01GM111638). M.C.D. was also supported, in part, by a GAANN fellowship provided by the United States Department of Education. We thank Prof. Christopher S. Jeffrey (University of Nevada, Reno) and his students for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b10221.
Author Present Address
Department of Chemistry, Dartmouth College, 6128 Burke Laboratories, Hanover, New Hampshire 03755, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Marsden S. P.; Depew K. M.; Danishefsky S. J. J. Am. Chem. Soc. 1994, 1162411143. 10.1021/ja00103a034. [DOI] [Google Scholar]
- Kamenecka T. M.; Danishefsky S. J. Angew. Chem., Int. Ed. 1998, 37212995.. [DOI] [PubMed] [Google Scholar]
- Crich D.; Huang X. J. Org. Chem. 1999, 64197218. 10.1021/jo991093+. [DOI] [PubMed] [Google Scholar]
- Tan G. H.; Zhu X.; Ganesan A. Org. Lett. 2003, 5101801. 10.1021/ol034516+. [DOI] [PubMed] [Google Scholar]
- Austin J. F.; Kim S.-G.; Sinz C. J.; Xiao W.-J.; MacMillan D. W. Proc. Natl. Acad. Sci. U. S. A. 2004, 101155482. 10.1073/pnas.0308177101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M.; Futamata M.; Mukai R.; Tamaru Y. J. Am. Chem. Soc. 2005, 127134592. 10.1021/ja0501161. [DOI] [PubMed] [Google Scholar]
- López-Alvarado P.; Caballero E.; Avendaño C.; Menéndez J. C. Org. Lett. 2006, 8194303. 10.1021/ol061631m. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Quancard J. J. Am. Chem. Soc. 2006, 128196314. 10.1021/ja0608139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movassaghi M.; Schmidt M. A. Angew. Chem., Int. Ed. 2007, 46203725. 10.1002/anie.200700705. [DOI] [PubMed] [Google Scholar]
- Movassaghi M.; Schmidt M. A.; Ashenhurst J. A. Angew. Chem., Int. Ed. 2008, 4781485. 10.1002/anie.200704960. [DOI] [PubMed] [Google Scholar]
- Newhouse T.; Baran P. S. J. Am. Chem. Soc. 2008, 1303310886. 10.1021/ja8042307. [DOI] [PubMed] [Google Scholar]
- Shangguan N.; Hehre W. J.; Ohlinger W. S.; Beavers M. P.; Joullié M. M. J. Am. Chem. Soc. 2008, 130196281. 10.1021/ja800067q. [DOI] [PubMed] [Google Scholar]
- Shen L.; Zhang M.; Wu Y.; Qin Y. Angew. Chem., Int. Ed. 2008, 47193618. 10.1002/anie.200800566. [DOI] [PubMed] [Google Scholar]
- Jones S. B.; Simmons B.; MacMillan D. W. C. J. Am. Chem. Soc. 2009, 1313813606. 10.1021/ja906472m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Movassaghi M. J. Am. Chem. Soc. 2011, 1333814940. 10.1021/ja206743v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell R. H.; Woodward R. L.; Willis M. C. Angew. Chem., Int. Ed. 2011, 50399116. 10.1002/anie.201103864. [DOI] [PubMed] [Google Scholar]
- Boyer N.; Movassaghi M. Chem. Sci. 2012, 361798. 10.1039/c2sc20270k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q.; Liu C.; Liang X.-W.; You S.-L. Org. Lett. 2012, 14174588. 10.1021/ol302043s. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Antilla J. C. Angew. Chem., Int. Ed. 2012, 514711778. 10.1002/anie.201203553. [DOI] [PubMed] [Google Scholar]
- Zhu S.; MacMillan D. W. C. J. Am. Chem. Soc. 2012, 1342610815. 10.1021/ja305100g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repka L. M.; Ni J.; Reisman S. E. J. Am. Chem. Soc. 2010, 1324114418. 10.1021/ja107328g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J.; Wang H.; Reisman S. E. Tetrahedron 2013, 6927–285622. 10.1016/j.tet.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Reisman S. E. Angew. Chem., Int. Ed. 2014, 53246206. 10.1002/anie.201402571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Y.; Davies H. M. J. Am. Chem. Soc. 2010, 1322440. 10.1021/ja9078094. [DOI] [PubMed] [Google Scholar]
- Spangler J. E.; Davies H. M. L. J. Am. Chem. Soc. 2013, 135186802. 10.1021/ja4025337. [DOI] [PubMed] [Google Scholar]
- Chai Z.; Zhu Y.-M.; Yang P.-J.; Wang S.; Wang S.; Liu Z.; Yang G. J. Am. Chem. Soc. 2015, 1373210088. 10.1021/jacs.5b05820. [DOI] [PubMed] [Google Scholar]
- Han X.; Li H.; Hughes R. P.; Wu J. Angew. Chem., Int. Ed. 2012, 514110390. 10.1002/anie.201205238. [DOI] [PubMed] [Google Scholar]
- Han X.; Wu J. Angew. Chem., Int. Ed. 2013, 52174637. 10.1002/anie.201209810. [DOI] [PubMed] [Google Scholar]
- Li H.; Hughes R. P.; Wu J. J. Am. Chem. Soc. 2014, 136176288. 10.1021/ja412435b. [DOI] [PubMed] [Google Scholar]
- Jeffrey C. S.; Barnes K. L.; Eickhoff J. A.; Carson C. R. J. Am. Chem. Soc. 2011, 133207688. 10.1021/ja201901d. [DOI] [PubMed] [Google Scholar]
- Acharya A.; Eickhoff J. A.; Jeffrey C. S. Synthesis 2013, 45, 1825. 10.1055/s-0033-1338883. [DOI] [Google Scholar]
- Barnes K. L.; Koster A. K.; Jeffrey C. S. Tetrahedron Lett. 2014, 55344690. 10.1016/j.tetlet.2014.06.050. [DOI] [Google Scholar]
- Jeffrey C. S.; Anumandla D.; Carson C. R. Org. Lett. 2012, 14225764. 10.1021/ol302771z. [DOI] [PubMed] [Google Scholar]
- Anumandla D.; Littlefield R.; Jeffrey C. S. Org. Lett. 2014, 16195112. 10.1021/ol502460j. [DOI] [PubMed] [Google Scholar]
- Föhlisch B.; Gehrlach E.; Herter R. Angew. Chem., Int. Ed. Engl. 1982, 212137. 10.1002/anie.198201371. [DOI] [Google Scholar]
- Harmata M.; Huang C.; Rooshenas P.; Schreiner P. R. Angew. Chem., Int. Ed. 2008, 47458696. 10.1002/anie.200803487. [DOI] [PubMed] [Google Scholar]
- Vander Wal M. N.; Dilger A. K.; MacMillan D. W. C. Chem. Sci. 2013, 483075. 10.1039/c3sc51266e. [DOI] [Google Scholar]
- Jeffrey C. S.; Anumandla D.; Acharya A.. J. Am. Chem. Soc., DOI: 10.1021/jacs.5b10184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.