

Abstract
In order to assess the frequency of mutations in the known Alzheimer's disease causative genes in Turkish dementia patients we screened amyloid precursor protein (APP), PSEN1 and PSEN2 for mutations in a cohort of 98 Turkish dementia families. Six families were found to carry PSEN1 mutations (p.H163R, p.P264L, and p.H214Y) or variants suggested to cause the disease (p.L134R, p.L262V, and p.A396T). In 4 other families, previously reported PSEN2 variants were identified (p.R62H, p.R71W, p.M174V (n = 2), and p.S130L). The phenotype of the carriers varied from rapid progressing Alzheimer's disease to frontotemporal dementia, with spasticity and seizures also observed. Here we report a frequency of 11.2% of mutations and variants in the known Alzheimer disease genes in the dementia cohort studied and 24% in the early onset subgroup of patients, suggesting that mutations in these genes are not uncommon in Turkey and are associated with various phenotypes. We thus believe that genetic analysis should become a standardized diagnostic implement, not only for the identification of the genetic disease, but also for appropriate genetic counseling.
Keywords: Alzheimer disease, Presenilin, Mutation, Turkey
1. Introduction
Alzheimer's disease (AD) is the most common cause of dementia and represents a major public health problem, especially in the face of the World's aging population. Approximately 25% of all AD is familial of which approximately 95% is late-onset (age >65 years) and 5% is early-onset (age <65 years) (Ballard et al., 2011). Turkey has a population of over 77 million, with a growth rate of 1.5% per year, according to the 2009 census (Turkish Statistical Institute, 2010). The average life expectancy is 73.2 years (71.1 years for men and 75.3 years for women). Characteristics for this population are large family units, high birth rates, and high rates of consanguineous marriages, making genetic diseases a common health problem. Over the last 2 decades, living standards and health services have improved (www.turkstat.gov.tr). This should increase survival, and thus the incidence of AD, which has been shown to be comparable with West European countries in recent studies (Gurvit et al., 2008). The genetic underpinnings of AD remain largely elusive despite successes in identifying mutations in 3 genes, presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP), causing mainly early-onset autosomal dominant, and rarely also sporadic forms of AD (Ballard et al., 2011). Additionally, some recent studies suggest mutations of APP to be related with autosomal recessive AD (Di Fede et al., 2009, Giaccone et al., 2010 and Tomiyama et al., 2008). In the last year, large genome-wide association studies identified CLU, PICALM, BIN1, ABCA7, EPHA1, CD33, CR1, and the MS4A gene cluster as new AD susceptibility loci (Chibnik et al., 2011, Hollingworth et al., 2011 and Naj et al., 2011). The reported risk effect of these new loci was lower when compared with the effect of the allelic variant in the apolipoprotein E gene, APOEε4, which is proven to be associated with increased risk for late-onset sporadic and familial AD. A daily update of the very rapidly increasing number of susceptibility genes and regions linked to AD can be taken from the AlzGene database web site (www.alzforum.org/res/com/gen/alzgene).
The current study is the first large molecular study of AD in Turkey. Our major objectives were to identify the frequency and nature of mutations in the 3 principal AD-associated genes and to search for new variants. The power of our study lies not only in the unique opportunity to study a large population with a high prevalence of consanguineous marriages, but also in the detailed characterization of patients by clinical and neuropsychological examination, as well as imaging.
2. Methods
2.1. Patients
A total of 106 patients from 98 families with a diagnosis of AD (n = 56), mild cognitive impairment (MCI; n = 31), frontotemporal dementia (FTD; n = 8), Lewy body disease (DLB; n = 4), Parkinson disease with dementia (PDD; n = 3), or atypical or mixed types of dementia (n = 4) participated in the study. All participants were recruited in the Behavioral Neurology and Movement Disorders Unit outpatient clinic in the Istanbul Faculty of Medicine of Istanbul University and underwent detailed clinical and neuropsychological examination and, if possible, cerebral magnetic resonance imaging (MRI) or positron emission tomography (PET) imaging. Diagnosis of dementia was based on the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer's disease (NINCDS/ADRDA) (McKhann et al., 1984), FTD was defined following the Lund and Manchester criteria (The Lund and Manchester Groups, 1994) and diagnosis of DLB was made on the consensus guidelines of the consortium on DLB (McKeith, 2006). Diagnostic procedures for PDD followed the recommendations from the movement disorder society task force (Dubois et al., 2007). All 31 MCI patients were defined as described by Frank and Petersen (2008). Sixteen of them had positive family history of different dementias; 6 patients were included either because of early-onset (<60 years, n = 3) or because of multiple consanguinity in the family (n = 3), and 9 patients were diagnosed with nonamnestic and multidomain MCI known to have a high risk of progression to dementia (Meyer et al., 2002). The study population comprised 61 females and 45 males with a mean age at time of examination (SD) of 69.5 (10.2) years (age range, 42–88) and age at onset of clinical signs 64.2 (10.4) years (age range, 33–84). The majority of families originated from Turkey with a few exceptions (Supplementary data).
Peripheral blood samples were collected and genomic DNA was extracted by standard procedures using the Qiagen DNA isolation maxi kit (Qiagen, Hilden, Germany). A neurologist took all blood samples and necessary clinical information after having obtained informed consent from the patients and their participating family members. The Ethics Committee of Istanbul Faculty of Medicine, Istanbul University approved the study.
2.2. Genetic analysis
The exonic regions of APP (exons 16 and 17), PSEN1 (exons 3–12), and PSEN2 (exons 3–12), as well as the flanking intronic sequences, were polymerase chain reaction (PCR) amplified using the respective primers and Roche FastStart PCR Master Mix polymerase (Roche Diagnostics, Corp., Indianapolis, IN, USA). The primers used to amplify PSEN1 and PSEN2 exons 3–12 were the ones described in Cruts et al. (1998). The primers used for the amplification of APP exon 16 were 5′-CTTCAGGCCTAGAAAGAAGT-3′ and 5′-GGATGAACCAGAGTTAATAGG-3′ and exon 17 5′-AACCTCATCCAAATGTCCCC-3′ and 5′-ATTCCCACTTGGAAACATGC-3′.
Each PCR product was sequenced using the same forward and reverse primers with Applied Biosystems BigDye terminator v3.1 sequencing chemistry and run on an ABI3730xl (Applied Biosystems, Carlsbad, CA, USA) genetic analyzer as per manufacturer's instructions. The sequences were analyzed with Sequencher software, version 4.2 (Genecodes, Ann Arbor, MI, USA).
The algorithm key to classify mutation's pathogenicity proposed by (Guerreiro et al., 2010) and PolyPhen-2 (Adzhubei et al., 2010) were used to determine the expected role of each single mutation.
2.2.1. Copy number variation analysis using Illumina single nucleotide polymorphism (SNP) BeadChips
All samples except 12 for which DNA was not available for this analysis, were assayed using either the Illumina Human610-Quad BeadChips (Illumina, Inc., San Diego, CA, USA) or the HumanOmniExpress Bead Chips as per manufacturer's instructions, using 200 ng of genomic DNA (Illumina, Inc., San Diego, CA, USA). All the samples analyzed had a genotype success rate of more than 99%. In order to examine theAPP locus for structural mutations we used the visualization tool Illumina Genome Viewer (IGV) within GenomeStudio version 2011.1 (Illumina, Inc.). Data were analyzed using the Human Genome Build 37 (www.ncbi.nlm.nih.gov/genome/assembly/12758/) and 2 metrics were visualized: B allele frequency and log R ratio for which, an R above 1 is indicative of an increase in copy number, and values below 1 suggest a deletion.
3. Results
A total of 98 families mainly with a Turkish ethnic background (n = 89) including 106 patients were recruited into the molecular part of the study. Fifty-three families had a positive family history of dementia, in 14 of these families first degree parental consanguinity was known and in 20 families parental consanguinity was suggested, however, due to a lack of information, the pedigree could not be completed. Forty-five patients had no family history of dementia, 7 among them had known and 20 highly suggested parental consanguinity. Additional cerebral diseases in the families were not taken into account even if idiopathic Parkinson's disease (n = 5) and cerebral vascular disorder (n = 22) were also frequently present. Four Turkish families, 1 family from Iran and 1 family with a mixed origin (Turkey/Bulgaria) were found to have PSEN1 gene mutations or variants (p.L134R, p.H163R, p.H214Y, p.L262V, p.P264L, p.A396T; average age at onset: 49.1 (4.5) years, age range, 41–53; Table 1, Fig. 1A). Four Turkish families with average age at onset of 63 (10.4) years (age range, 52–77; Table 1) were identified to carry PSEN2 known variants: p.R62H, and p. M174V (in 2 families), and the p.S130L variant (Fig. 1B). The index patient from a Macedonian family carried the p.R71W variant in PSEN2 (age at onset: 64 years). No mutations or any new nonsynonymous variants were found in the APP gene. Other previously reported SNPs and coding variants were identified including the PSEN1 E318G in 2 samples and the synonymous change G708G in APP. Three novel synonymous changes were also found: G78G; c.234C>T in PSEN1; P287P; c.861C>T in PSEN2 and L705L; c.2115C>T in APP. Common SNPs and synonymous variants were not studied any further. No APPcopy number mutations were found in the cohort studied.
Table 1.
Reported mutation and their prediction of pathogenicity according to the PSEN algorithm key and PolyPhen-2
| Mutation (protein; cDNA) |
Patient origin | Age at onset (y) |
Prediction of pathogenicity according to PolyPhen-2 |
Classification of pathogenicity according to the previously proposed algorithma |
Previous publication reporting these mutations |
|---|---|---|---|---|---|
| PSEN1: p.L134R; c.401T>G | Turkey | 53 | Probably damaging | Possible | — |
| PSEN1: p.H163R; c.488A>G | Turkey | 41 | Predicted to be benign | Definite | Boteva et al., 1996, Campion et al., 1995 and Campion et al., 1999; Gómez-Tortosa et al., 2010, Kamimura et al., 1998, Kamino et al., 1996, Lleó et al., 2002, Poduslo et al., 1996, Poorkaj et al., 1998, Rogaeva et al., 2001, Sherrington et al., 1995, Tanahashi et al., 1995, Tanahashi et al., 1996 and Zekanowski et al., 2003 |
| PSEN1: p.H214Y; c.640C>T | Iran | 51 | Possibly damaging | Definite | Raux et al. (2005) |
| PSEN1: p.L262V; c.784 T>G | Turkey | 51, 52 | Probably damaging | Probable | Forsell et al. (1997) |
| PSEN1: p.P264L; c.791C>T | Turkey | 51 (n= 2) | Probably damaging | Definite | Campion et al., 1995, Campion et al., 1999, Dumanchin et al., 2006, Jacquemont et al., 2002, Kwok et al., 1997, Martikainen et al., 2010, Poorkaj et al., 1998, Raux et al., 2005 and Wasco et al., 1995 |
| PSEN1: p.A396T: c.1186 G>A | Turkey/Bulgaria | 43 | Probably damaging | Possible | — |
| PSEN2: p.R62H; c.185G>A | Turkey | 63 | Predicted to be benign | Not pathogenic | Cruts et al., 1998, Gallo et al., 2010, Guerreiro et al., 2010 and Sleegers et al., 2004 |
| PSEN2: p.R71W; c.211C>T | Macedonia | 64 | Possibly damaging | Not pathogenic | Brouwers et al., 2008, Guerreiro et al., 2010 and Sleegers et al., 2004 |
| PSEN2: p.M174V; c.520A>Gb | Turkey | 52, 60 | Predicted to be benign | Probable | Guerreiro et al. (2010) |
| PSEN2: p.S130L; c.389C>T | Turkey | 77 | Possibly damaging | Probable | Li et al., 2006, Tedde et al., 2003 and Tomaino et al., 2007 |
This variant is found in 2 unrelated patients.
Fig. 1.
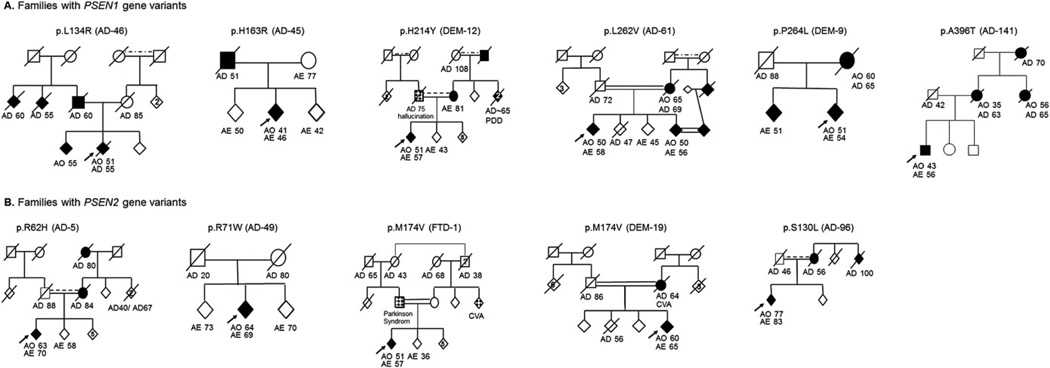
Families with PSEN gene variants. Diamond symbols indicate parkinsonism. Abbreviations: AE, age at examination (years); AO, age at onset (years); AD, age at death (years); CVA, cerebrovascular accident; PDD, Parkinson's disease with dementia.
3.1. Clinical details of the PSEN mutation carriers
3.1.1. PSEN1-p.L134R (family AD-46)
This novel variant in PSEN1 was identified in a male patient without any notable medical history, with the age of onset at 53 years. The first symptom experienced by the patient at age of 51 years was a very rapid progressive impairment of memory. After 18 months, his mood became labile, his communication abilities were considerably reduced and he was disoriented to time and space. A clear frontal syndrome associated with visual hallucination was noted. Neurological examination revealed next to his cognitive problems pyramidal signs such as a bilateral positive Babinski reflex and a mild spasticity in the lower limbs. Cerebral MRI examination revealed a marked cerebro-cerebellar atrophy as well as a mild microangiopathy. Symptoms progressed rapidly, at age of 55 years he was bedridden and after only 4 years from the onset of disease, the patient died.
3.1.2. PSEN1-p.H163R (family AD-45)
This Turkish female patient started memory problems at age of 41 years. Symptoms developed in a fairly short time and after only 2 years she was no longer able to look after herself, her attention was reported to be fluctuating, she permanently repeated short sentences and she suffered from urinary incontinence. One year later, marked visual hallucination next to a rigid-bradykinetic syndrome was noticed, and only a few months later the patient stopped speaking. Currently, at age of 49 years the patient is bedridden. A cerebral MRI was reported to show significant cerebral atrophy. 99mTc-HMPAO (technetium 99m-hexamethylpropylenamine oxime) single-photon emission computed tomography (SPECT) revealed marked brain hypoperfusion involving the right temporoparietal, left frontal, and also occipital lobes, far beyond the regions of brain volume loss. The father, who had died at age of 51 years, also suffered from memory problems.
3.1.3. PSEN1-p.H214Y (family DEM-12)
This Iranian patient was first observed presenting behavioral symptoms such as apathy and depression at age of 51 years. In the following months he developed disorientation and progressive impairment of memory, nonfluent aphasia with prominent word finding deficits and agrammatism. At clinical examination some myoclonic jerks were noted. Imaging studies at age of 52 years revealed global cortical and marked frontotemporal atrophy associated with white matter lesions. Research of protein 14-3-3 was negative. Five years after onset the patient is severely demented with some visual hallucination, falls, and stereotypic behavior. Family history was positive: the mother, who is still alive at age of 81 years, is reported to suffer from frontotemporal dementia, 1 aunt and 2 of her children are known to be affected by PDD, and 1 uncle apparently died because of dementia and the grandfather was known to be affected with AD.
3.1.4. PSEN1-p.L262V (family AD-61)
This PSEN1 variant was found in 2 siblings diagnosed with AD and frontotemporal dementia. Both patients had age at onset at approximately 50 years. The first symptoms of the older illiterate sibling included depression, inappropriate behavior, and memory problems. There was no notable medical history except frequent migraine attacks. Brain MRI at age of 57 years revealed a very large cisterna magna and globalized atrophy. Examination at age of 58 years showed marked depression (Geriatic Depression Score 23/30), important frontal symptoms including increased and disinhibited speech, perseveration, and temporospatial disorientation. The second sibling had a 5-year history of abnormal mental status including disorientation, aphasia, and memory deficits. Cerebral MRI examination 4 years after onset showed a marked frontotemporal atrophy, Mini Mental State Examination (MMSE) was found at 9/30. At age of 56 years the patient was mostly apathetic with no fluent speaking. The family came from a village in the Corum province in Turkey's central Black Sea Region, bordering on the Central Anatolia Region. The mother as well as the mother's sister was reported to have suffered from dementia, both of them died at age of approximately 70 years after a short and rapidly worsening disease. In this family there was a high number of consanguineous marriages and the second patient married his cousin who was also reported to have started to suffer from memory problems since a few months, but who refused further investigation. Together, they have 5 children between 29 and 37 years old for which no further details are known.
3.1.5. PSEN1-p.P264L (family DEM-9)
The initial symptom at age of 51 years encountered by this female patient from Turkey was memory impairment when she was examined for the first time in our clinic at age of 52 years. Additional symptoms including petulance, language difficulty, memory, and visual-spatial impairments were noticed. Her cerebral MRI showed next to a global atrophy a marked frontotemporal degeneration. At age of 54 years the patient presented reduced speaking, ideomotor apraxia, impairments in copying, but also some perseveration and hallucinations were noticed. The mother was known to have suffered from dementia with an onset at age of 60 years. The brother carrying the same mutation had no particular medical history and had received no treatment at age of 51 years when examined in the context of the study. He had a university degree and worked as an engineer. No complains about memory problems had been noted, though examination by Addenbrook's Cognitive Examination-Revised (ACE-R) was not normal (86/100, cutoff <88 gives 94% sensitivity and 89% specificity for dementia). In particular, his test results in memory (17/26) and language performances (22/26) were decreased compared to healthy subjects (17/26, cut off: 18/26 and 22/26, cut off: 24/26, respectively).
3.1.6. PSEN1-p.A396T (family AD-141)
This 56-year-old male with 1 parent originating from Bulgaria and the other from Turkey showed since the age of 43 years progressive indifferent and fractious behavior affecting daily living activities and intermittent visual hallucinations. Neuropsychological evaluation revealed a moderate overall mental decline (MMSE score 22/30 at age of 50 and 11/30 at age of 55 years) and cerebral MRI scan of the brain displayed generalized and marked atrophy of the temporal lobes. His mother and maternal aunt had died in their 60s and had developed symptoms of dementia at different ages, with a marked age gap of more than 15 years (35 and 56 years). His maternal grandmother also had dementia and died at approximately 70 years of age.
3.1.7. PSEN2-p.R62H (family AD-5)
We identified 1 more patient from Trabzon, city on the Black Sea coast of northeastern Turkey, with autosomal-dominant family history of AD carrying the PSEN2 p.R62H variant. The patient, who underwent a hemicolectomy because of colon cancer at the age of 55 years, started to suffer from memory problems including particularly to remember recent events at age 63 years. At age 70 years MMSE was found to be 22/30 and ACE-R was 65/100. Cerebral MRI showed mild leukoariosis and cortical atrophy.
3.1.8. PSEN2-p.R71W (family AD-49)
This 69-year-old female patient is reported to have started typical AD symptoms associated with postural hand tremor from age 64 years. There was no known family history of dementia, but her mother had died of tuberculosis at age 20 years and there was no more information about any additional family members from the mother's side of the family.
3.1.9. PSEN2-p.M174V (families DEM-19 and FTD-1)
This variant was found in 2 unrelated Turkish patients, both with parental first degree consanguinity and frontotemporal dementia phenotype. The first patient (FTD-1) started to suffer from rapid onset progressive nonfluent aphasia with agrammatism and speech apraxia at age of 52. At age of 54, when examined for the first time in our outpatient clinic he could only communicate by phonemes, also suffered from gait disturbances linked to a rigid-bradykinetic syndrome and postural instability and had dystonia in the right leg. Cerebral MRI examination showed next to peri- and supraventricular gliotic lesions bilateral marked anterior temporal and cortical atrophy. The parents were cousins and the father was reported to suffer from a parkinson syndrome as well. Additionally, 1 sister of the mother had had a cerebral vascular accident and the maternal grandfather was reported to have died at age of 38 years because of a neurological disease. The mutation was also found in 1 of the patient's sisters: at age of 36 years her clinical examination was normal.
The second patient (DEM-19) who started to show aggressiveness and personality changes followed by progressive speech and attention deficit at age of 60 years was examined at age of 63 for the first time in our clinic. At clinical examination a marked frontal syndrome, some agrammatisms and prominent executive function deficits like prolonged Stroop interference and perseverations in Luria drawings were noted. ACE-R was 75/100 and MMSE was 30/30. Cerebral MRI showed right predominant frontal atrophy. Two years later, at age of 65 years clinical symptoms worsened and clear, predominantly right fronto-parieto-temporal hypometabolism was found at F-18 fluoro-deoxy-glucose (FDG)-PET. In addition, the patient was treated for arterial hypertension and noninsulin-dependent diabetes. His mother was known to have suffered from memory problems; she died at age of 64 years of a cerebral vascular accident.
3.1.10. PSEN2-p.S130L (family AD-96)
This patient originating from the Artvin province in Turkey, on the Black Sea coast in the northeastern corner of the country, started to show depression, progressive memory impairment, and episodes of spatial disorientation at the age of 77. His mother and 1 maternal uncle were reported to have had memory problems. Diagnosis of AD was made and at age of 82 years at first examination in our clinic the patient was mildly bradykinetic; MMSE was 17/30. Cerebral MRI showed cortical and hippocampal atrophy and a mild hydrocephalus. At 1-year follow-up, some worsening of cognitive impairment was noted with MMSE at 12/30 but the physical examination did not change.
4. Discussion
This is the first large-scale study to evaluate genetics and phenotypes of AD and other dementia in a Turkish population. Overall we found a PSEN mutation prevalence of 11.2% in this population of different types of dementia. Focusing only on early-onset (EO) familial dementia (<65 years, n = 50) the prevalence of PSENgene mutations and disease related variants is clearly increased (24%).
The pathogenicity of each PSEN1 and PSEN2 variants found in this study as well as their relation to clinical findings and pedigrees will be discussed next.
4.1. PSEN1-p.L134R
This novel variant in PSEN1 is suggested to be associated with an early-onset and rapid progression of cognitive decline. At least 4 members of our family AD-46 had also died because of dementia with short but fulminate evolution, 1 uncle died even after 2 years from disease onset. For this, autosomal dominant pattern of inheritance of the disease in this family can be suggested. Families carrying the very close p.N135S mutation were also reported, with autopsy-confirmed AD patients presenting spastic paraparesis (Finckh et al., 2005 and Rudzinski et al., 2008). This is a novel variant that was found in 1 patient with a positive family history. We were unable to prove segregation of the variant within this family because no other DNA samples from affected members of the family were available for genetic testing. This variant occurs in a residue ofPSEN1 in the hydrophilic loop I (HL-I) that is conserved in PSEN2. According to the algorithm key proposed by Guerreiro et al. to classify PSENs variants' pathogenicity (Guerreiro et al., 2010), this variant is now classified as possibly pathogenic, pending on the report of additional cases with the same mutation. In silico evaluation of pathogenicity using PolyPhen-2 p.L134R is defined as a probably damaging variant (Table 1).
4.2. PSEN1-p.H163R
To date, 15 families with AD patients carrying the p.H163R mutation have been reported including also AD phenotypes associated with seizures or myoclonus (www.molgen.ua.ac.be/ADMutations). Age of onset of our patient (family AD-45) is comparable to the previous findings (41 vs. 45.6 years), but the clinical picture is not associated with any myoclonus or seizures but with hallucination. The pathogenic nature of the mutation has not only been proven by segregation within the families, Nagasaka and colleagues (Nagasaka et al., 2005) could also show that mutation carriers differed significantly in their gene expression compared to their wild type siblings. Interestingly, Kaneko et al. (Kaneko et al., 2007) showed on N2a mouse neuroblastoma and C6 rat glioblastoma cell lines, that the levels of phosphorylated α-synuclein do not increase in neuronal and non-neuronal cells expressing the wild type PSEN1 or PSEN1 p.H163R mutant compared to cells expressing PSEN1-p.T440 mutant. The author suggested that the mechanism by which the PSEN1-p.T440 mutation caused DLB is by enhancing the phosphorylation of α-synuclein in the brain. Following the clinical diagnosis criteria of DLB as prescribed by McKeith (2006) our patient is diagnosed as DLB. As he carries the p.H163R mutation it does not seem probable that the mechanism leading to DLB in p.T440 or p.H163R is by phosphorylation of α-synuclein in the brain. The p.H163R variant is considered as definitely pathogenic. It is interesting to note that PolyPhen-2 predicts this mutation to be benign, illustrating how in silico predictions can be misleading and how specific models for the known AD genes can be more accurate.
4.3. PSEN1-p.H214Y
The p.H214Y mutation was previously described in 1 family with 5 affected patients (Raux et al., 2005), but with a significantly earlier onset compared with our patient (37–45 years vs. 51 years, family DEM-12) and, in the same residue, the mutation p.H214D has also been previously identified (Guerreiro et al., 2010) associated with atypical dementia including bradykinesia and action tremor. In comparison with this, our patient showed progressive nonfluent aphasia (PNFA) phenotype associated by myoclonia and also hallucinations. The p.H214Y mutation is classified as definitely pathogenic using the PSEN algorithm key, and the underlying pathomechanism is suggested to have a broad effect as the phenotype is very variable even within the same family.
4.4. PSEN1-p.L262V
This is a novel variant leading to an amino acid substitution (leucine to valine) localized in exon 8 where the p.L262F mutation was previously described in 1 family with 3 affected mutation carriers (Forsell et al., 1997). Age at onset (approximately 50 years) and clinical findings (decreased short time memory and decreased word finding ability) are comparable with our family (AD-61). Based not only on our study, but also on these previous findings, the PSEN1-p.L262V variant is classified as probably pathogenic.
4.5. PSEN1-p.P264L
This mutation was previously found in 11 families (Dumanchin et al., 2006; Martikainen et al., 2010; Raux et al., 2005). The diversity of phenotypic expression associated with this point mutation is surprising and includes: spastic paraparesis, DLB, atypical dementia, and lobar hemorrhage. This variant is localized in exon 8 of the PSEN1 gene; it has been suggested that exon 8 exclusion leads to cotton wool plaques/spastic paraparesis (CWP/SP) (Kwok et al., 1997), but in a study conducted by Dumanchin and colleagues (Dumanchin et al., 2006), no effect on exon splicing nor an increase of β-amyloid (Aβ) 42/40 ratio on cell experiments for the p.P264L variant could be determined. Nevertheless, cosegregation of this mutation with AD has been established in several families (Campion et al., 1999) and for this, p.P264L is predicted to be definitely pathogenic in according with the PSEN algorithm key.
4.6. PSEN1-p.A396T
This 56-year-old patient (family AD-141) suffering from AD was found to carry the new p.A396T variant located within exon 11, in a highly conserved region. To assess if p.A396T is a common variant in the Turkish population we have sequenced exon 11 of PSEN1 in 75 Turkish healthy controls. This variant was not found in any of the control samples studied, indicating this is not a common polymorphism specific of the Turkish population. However, in absence of familial segregation and any other reports of this same variant in other AD patients, this novel variant is classified as possibly pathogenic.
4.7. PSEN2-p.R62H
The pathogenicity of PSEN2 p.R62H variant is questionable: functional analysis of this variant could not prove any effect on neurodegeneration (Walker et al., 2005) and different authors reported this variant either without familial segregation (Sleegers et al., 2004) or in healthy subjects (Guerreiro et al., 2010) or even as a relatively common polymorphism (Cruts et al., 1998). This variant is probably not pathogenic.
4.8. PSEN2-p.R71W
The pathogenic nature of this variant is unclear: previous findings are similar to our patient's presentation (family AD-49): to date 3 families have been reported to have late-onset AD carriers, but in none of these the variant segregated with the disease (Brouwers et al., 2008, Guerreiro et al., 2010 and Sleegers et al., 2004). Additionally, the p.R71W variation was also detected in 1 healthy control (Sleegers et al., 2004). Similarly to p.R62H, this variant is classified as possibly damaging by PolyPhen-2, but following the PSEN algorithm key it is probably not pathogenic. Further studies in larger numbers of cases and controls are needed in order to determine if these 2 variants (p.R62H and p.R71W) may act as risk factors and influence the course of the disease or the age at onset of carriers.
4.9. PSEN2-p.M174V
This variant has been previously described in 1 patient diagnosed with AD and was classified as possibly pathogenic (Guerreiro et al., 2010). Additionally, we can report 2 more independent cases but with frontotemporal dementia phenotype (family DEM-19 and FTD-1). According to the PSEN algorithm key, this variant should now be considered as probably pathogenic as at least 3 independent cases are known and this variant has not been found in healthy controls until now. Nonetheless, no segregation data are available for any of these families and PolyPhen-2 predicts this variant to be benign.
4.10. PSEN2-p.S130L
The p.S130L has been previously described in 3 different families (Li et al., 2006, Tedde et al., 2003 and Tomaino et al., 2007). Segregation of the mutant allele with disease could be not be demonstrated in any of the families including the presented case (family AD-96), Moreover, no significant effect on either Aβ42 levels, Aβ40 levels, or the Aβ 42/40 ratio could be found (Walker et al., 2005). Nevertheless, we cannot classify this variant as benign, even if others and we have failed to identify it in healthy controls (Li et al., 2006 and Tomaino et al., 2007). Additionally, it is interesting to note, that all variant carriers had late onset AD (72, 75, 77, and 81 years) and slow progressing cognitive impairment. This variant's pathogenicity is classified as probable.
With this background, we could also show that 2 subgroups differ as to the age at which the first clinical symptoms become apparent: early-onset dementia (<65 years) which is highly susceptible to be related to 1 of the 3 identified AD genes and late-onset (>65 years) dementia. Nevertheless, the impact and function of the different mutations and variants in the 3 genes known to harbor AD causative mutations on the different phenotypes still need to be elucidated. Guerreiro et al. (2010) proposed an algorithm key to classify mutation pathogenicity, which turned out to be a very helpful instrument for the interpretation of mutation interpretation. This interpretation takes into account findings from functional studies and is specific to PSENvariants (although it can also be used with some modifications for APP variants). Interestingly, by comparing PolyPhen-2 and the PSEN algorithm key, classification of only 2 out of 10 variants was similar (PSEN1p.L262V and PSEN2 p.R62H) and interpretation of at least 2 variants were contradictory (PSEN1 p.H163R,PSEN1 p.H214Y). Our data show that larger and detailed studies on the prevalence, function and associate phenotype of PSEN variants are necessary to develop more precise tools to predict the pathogenicity of a variant in order to allow the neurogeneticist to perform adequate genetic counseling. However, the PSENalgorithm key applied during genetic counseling might be beneficial for both the counselor who can decide more appropriately about the value of molecular diagnostic results and the related potential risk of additional family members in developing AD, and for the patient who will receive more detailed information. The recently proposed guidelines for genetic counseling and testing for AD by the American College of Medical Genetics (Goldman et al., 2011) is lacking in advice on how to recognize and how to handle the different pathogenic impact of the variants. The PSEN algorithm key will be helpful for the former. This is the first molecular study in a large Turkish cohort including patients with dementia. The prevalence rate of PSENmutations with regard to the age of onset in our population is comparable with those previously described. Nevertheless further studies are required and a standardized diagnostic procedure needs to be implemented in Turkey. The genetic architecture of AD and related dementia is far from being completely understood, but conducting more detailed analysis also using different techniques such as genome-wide genotyping analyses, exome sequencing or genetic linkage analyses in the Turkish population might be particularly successful due to the high rate of consanguinity as gene frequency and genetic structure are changed by consanguineous marriages.
Supplementary Material
Acknowledgements
The authors thank all the patients and their families. Burcu Atasu helped with DNA management and Ms. Aysun Dulger and Canan Barikan provided administrative help.
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services, project number Z01 AG000950-06. Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study.
Footnotes
Disclosure statement
The study was approved by the Ethics Committee of Istanbul Faculty of Medicine, Istanbul University, and informed consent obtained from the patients and their participating family members.
The authors disclose no conflicts of interest.
References
- 1.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 3.Boteva K, Vitek M, Mitsuda H, de Silva H, Xu PT, Small G, Gilbert JR. Mutation analysis of presenillin 1 gene in Alzheimer's disease. Lancet. 1996;347:130–131. doi: 10.1016/s0140-6736(96)90261-5. [DOI] [PubMed] [Google Scholar]
- 4.Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer's disease: an update. Ann. Med. 2008;40:562–583. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- 5.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campion D, Flaman JM, Brice A, Hannequin D, Dubois B, Martin C, Moreau V, Charbonnier F, Didierjean O, Tardieu S, et al. Mutations of the presenilin I gene in families with early-onset Alzheimer's disease. Hum. Mol. Genet. 1995;4:2373–2377. doi: 10.1093/hmg/4.12.2373. [DOI] [PubMed] [Google Scholar]
- 7.Chibnik LB, Shulman JM, Leurgans SE, Schneider JA, Wilson RS, Tran D, Aubin C, Buchman AS, Heward CB, Myers AJ, Hardy JA, Huentelman MJ, Corneveaux JJ, Reiman EM, Evans DA, Bennett DA, De Jager PL. CR1 is associated with amyloid plaque burden and age-related cognitive decline. Ann. Neurol. 2011;69:560–569. doi: 10.1002/ana.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruts M, van Duijn CM, Backhovens H, Van den Broeck M, Wehnert A, Serneels S, Sherrington R, Hutton M, Hardy J, St George-Hyslop PH, Hofman A, Van Broeckhoven C. Estimation of the genetic contribution of presenilin-1 and –2 mutations in a population-based study of presenile Alzheimer disease. Hum. Mol. Genet. 1998;7:43–51. doi: 10.1093/hmg/7.1.43. [DOI] [PubMed] [Google Scholar]
- 9.Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, Merlin M, Giovagnoli AR, Prioni S, Erbetta A, Falcone C, Gobbi M, Colombo L, Bastone A, Beeg M, Manzoni C, Francescucci B, Spagnoli A, Cantù L, Del Favero E, Levy E, Salmona M, Tagliavini F. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubois B, Burn D, Goetz C, Aarsland D, Brown RG, Broe GA, Dickson D, Duyckaerts C, Cummings J, Gauthier S, Korczyn A, Lees A, Levy R, Litvan I, Mizuno Y, McKeith IG, Olanow CW, Poewe W, Sampaio C, Tolosa E, Emre M. Diagnostic procedures for Parkinson's disease dementia: recommendations from the movement disorder society task force. Mov. Disord. 2007;22:2314–2324. doi: 10.1002/mds.21844. [DOI] [PubMed] [Google Scholar]
- 11.Dumanchin C, Tournier I, Martin C, Didic M, Belliard S, Carlander B, Rouhart F, Duyckaerts C, Pellissier JF, Latouche JB, Hannequin D, Frebourg T, Tosi M, Campion D. Biological effects of four PSEN1 gene mutations causing Alzheimer disease with spastic paraparesis and cotton wool plaques. Hum. Mutat. 2006;27:1063. doi: 10.1002/humu.9458. [DOI] [PubMed] [Google Scholar]
- 12.Finckh U, Kuschel C, Anagnostouli M, Patsouris E, Pantes GV, Gatzonis S, Kapaki E, Davaki P, Lamszus K, Stavrou D, Gal A. Novel mutations and repeated findings of mutations in familial Alzheimer disease. Neurogenetics. 2005;6:85–89. doi: 10.1007/s10048-005-0211-x. [DOI] [PubMed] [Google Scholar]
- 13.Forsell C, Froelich S, Axelman K, Vestling M, Cowburn RF, Lilius L, Johnston JA, Engvall B, Johansson K, Dahlkild A, Ingelson M, St George-Hyslop PH, Lannfelt L. A novel pathogenic mutation (Leu262Phe) found in the presenilin 1 gene in early-onset Alzheimer's disease. Neurosci. Lett. 1997;234:3–6. doi: 10.1016/s0304-3940(97)00603-4. [DOI] [PubMed] [Google Scholar]
- 14.Frank AR, Petersen RC. Alzheimer disease—Mild cognitive impairment. In: Duyckaerts C, Litvan I, editors. Handbook of Clinical Neurology--Dementias, Volume 89, third series. Elsevier; New York: 2008. pp. 217–221. [DOI] [PubMed] [Google Scholar]
- 15.Gallo M, Tomaino C, Puccio G, Frangipane F, Curciom SA, Bernardi L, Geracitano S, Anfossi M, Mirabelli M, Colao R, Vasso F, Smirne N, Maletta RG, Bruni AC. Novel MAPT Val75Ala mutation and PSEN2 Arg62Hys in two siblings with frontotemporal dementia. Neurol. Sci. 2010;31:65–70. doi: 10.1007/s10072-009-0132-9. [DOI] [PubMed] [Google Scholar]
- 16.Giaccone G, Morbin M, Moda F, Botta M, Mazzoleni G, Uggetti A, Catania M, Moro ML, Redaelli V, Spagnoli A, Rossi RS, Salmona M, Di Fede G, Tagliavini F. Neuropathology of the recessive A673V APP mutation: Alzheimer disease with distinctive features. Acta Neuropathol. 2010;120:803–812. doi: 10.1007/s00401-010-0747-1. [DOI] [PubMed] [Google Scholar]
- 17.Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, Strecker MN, Roberts JS, Burke W, Mayeux R, Bird T. American College of Medical Genetics and the National Society of Genetic Counselors. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet. Med. 2011;13:597–605. doi: 10.1097/GIM.0b013e31821d69b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gómez-Tortosa E, Barquero S, Barón M, Gil-Neciga E, Castellanos F, Zurdo M, Manzano S, Muñoz DG, Jiménez-Huete A, Rábano A, Sainz MJ, Guerrero R, Gobernado I, Pérez-Pérez J, Jiménez-Escrig A. Clinical-genetic correlations in familial Alzheimer's disease caused by presenilin 1 mutations. J. Alzheimers Dis. 2010;19:873–884. doi: 10.3233/JAD-2010-1292. [DOI] [PubMed] [Google Scholar]
- 19.Guerreiro RJ, Baquero M, Blesa R, Boada M, Brás JM, Bullido MJ, Calado A, Crook R, Ferreira C, Frank A, Gómez-Isla T, Hernández I, Lleó A, Machado A, Martínez-Lage P, Masdeu J, Molina-Porcel L, Molinuevo JL, Pastor P, Pérez-Tur J, Relvas R, Oliveira CR, Ribeiro MH, Rogaeva E, Sa A, Samaranch L, Sánchez-Valle R, Santana I, Tàrraga L, Valdivieso F, Singleton A, Hardy J, Clarimón J. Genetic screening of Alzheimer's disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging. 2010;31:725–731. doi: 10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurvit H, Emre M, Tinaz S, Bilgic B, Hanagasi H, Sahin H, Gurol E, Kvaloy JT, Harmanci H. The prevalence of dementia in an urban Turkish population. Am. J. Alzheimers Dis. Other Demen. 2008;23:67–76. doi: 10.1177/1533317507310570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ER, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Rüther E, Schürmann B, Heun R, Kölsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Gallacher J, Hüll M, Rujescu D, Giegling I, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Pankratz VS, Sando SB, Aasly JO, Barcikowska M, Wszolek ZK, Dickson DW, Graff-Radford NR, Petersen RC, van Duijn CM, Breteler MM, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, Berr C, Campion D, Epelbaum J, Dartigues JF, Tzourio C, Alpérovitch A, Lathrop M, Feulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snaedal J, Björnsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia A, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossù P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O'Donovan M, Amouyel P, Williams J Alzheimer's Disease Neuroimaging Initiative, CHARGE Consortium, EADI1 consortium. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacquemont ML, Campion D, Hahn V, Tallaksen C, Frebourg T, Brice A, Durr A. Spastic paraparesis and atypical dementia caused by PSEN1 mutation (P264L), responsible for Alzheimer's disease. J. Med. Genet. 2002;39:E2. doi: 10.1136/jmg.39.2.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamimura K, Tanahashi H, Yamanaka H, Takahashi K, Asada T, Tabira T. Familial Alzheimer's disease genes in Japanese. J. Neurol. Sci. 1998;160:76–81. doi: 10.1016/s0022-510x(98)00219-6. [DOI] [PubMed] [Google Scholar]
- 4.Kamino K, Sato S, Sakaki Y, Yoshiiwa A, Nishiwaki Y, Takeda M, Tanabe H, Nishimura T, Ii K, St. George-Hyslop PH, Miki T, Ogihara T. Three different mutations of presenilin 1 gene in early-onset Alzheimer's disease families. Neurosci. Lett. 1996;208:195–198. doi: 10.1016/0304-3940(96)12587-8. [DOI] [PubMed] [Google Scholar]
- 5.Kaneko H, Kakita A, Kasuga K, Nozaki H, Ishikawa A, Miyashita A, Kuwano R, Ito G, Iwatsubo T, Takahashi H, Nishizawa M, Onodera O, Sisodia SS, Ikeuchi T. Enhanced accumulation of phosphorylated alpha-synuclein and elevated beta-amyloid 42/40 ratio caused by expression of the presenilin-1 deltaT440 mutant associated with familial Lewy body disease and variant Alzheimer's disease. J. Neurosci. 2007;27:13092–13097. doi: 10.1523/JNEUROSCI.4244-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwok JB, Taddei K, Hallupp M, Fisher C, Brooks WS, Broe GA, Hardy J, Fulham MJ, Nicholson GA, Stell R, St. George Hyslop PH, Fraser PE, Kakulas B, Clarnette R, Relkin N, Gandy SE, Schofield PR, Martins RN. Two novel (M233T and R278T) presenilin-1 mutations in early-onset Alzheimer's disease pedigrees and preliminary evidence for association of presenilin-1 mutations with a novel phenotype. Neuroreport. 1997;8:1537–1542. doi: 10.1097/00001756-199704140-00043. [DOI] [PubMed] [Google Scholar]
- 7.Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Partain J, Nixon RR, Allen CN, Irwin RP, Jakobs PM, Litt M, Hershberger RE. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006;79:1030–1039. doi: 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lleó A, Blesa R, Queralt R, Ezquerra M, Molinuevo JL, Peña-Casanova J, Rojo A, Oliva R. Frequency of mutations in the presenilin and amyloid precursor protein genes in early-onset Alzheimer disease in Spain. Arch. Neurol. 2002;59:1759–1763. doi: 10.1001/archneur.59.11.1759. [DOI] [PubMed] [Google Scholar]
- 9.Martikainen P, Pikkarainen M, Pöntynen K, Hiltunen M, Lehtovirta M, Tuisku S, Soininen H, Alafuzoff I. Brain pathology in three subjects from the same pedigree with presenilin-1 (PSEN1) P264L mutation. Neuropathol. Appl. Neurobiol. 2010;36:41–54. doi: 10.1111/j.1365-2990.2009.01046.x. [DOI] [PubMed] [Google Scholar]
- 10.McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J. Alzheimers Dis. 2006;9:417–423. doi: 10.3233/jad-2006-9s347. [DOI] [PubMed] [Google Scholar]
- 11.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;77:333. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 12.Meyer J, Xu G, Thornby J, Chowdhury M, Quach M. Longitudinal analysis of abnormal domains comprising mild cognitive impairment (MCI) during aging. J. Neurol. Sci. 2002;201:19–25. doi: 10.1016/s0022-510x(02)00159-4. [DOI] [PubMed] [Google Scholar]
- 13.Nagasaka Y, Dillner K, Ebise H, Teramoto R, Nakagawa H, Lilius L, Axelman K, Forsell C, Ito A, Winblad B, Kimura T, Graff C. A unique gene expression signature discriminates familial Alzheimer's disease mutation carriers from their wild-type siblings. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14854–14859. doi: 10.1073/pnas.0504178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St. George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat. Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poduslo SE, Herring K, Neal M. A presenilin 1 mutation in an early onset Alzheimer's family: no association with presenilin 2. Neuroreport. 1996;7:2018–2020. doi: 10.1097/00001756-199608120-00033. [DOI] [PubMed] [Google Scholar]
- 16.Poorkaj P, Sharma V, Anderson L, Nemens E, Alonso ME, Orr H, White J, Heston L, Bird TD, Schellenberg GD. Missense mutations in the chromosome 14 familial Alzheimer's disease presenilin 1 gene. Hum. Mutat. 1998;11:216–221. doi: 10.1002/(SICI)1098-1004(1998)11:3<216::AID-HUMU6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 17.Raux G, Guyant-Maréchal L, Martin C, Bou J, Penet C, Brice A, Hannequin D, Frebourg T, Campion D. Molecular diagnosis of autosomal dominant early onset Alzheimer's disease: an update. J. Med. Genet. 2005;42:793–795. doi: 10.1136/jmg.2005.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogaeva EA, Fafel KC, Song YQ, Medeiros H, Sato C, Liang Y, Richard E, Rogaev EI, Frommelt P, Sadovnick AD, Meschino W, Rockwood K, Boss MA, Mayeux R, St. George-Hyslop P. Screening for PS1 mutations in a referral-based series of AD cases:21 novel mutations. Neurology. 2001;57:621–625. doi: 10.1212/wnl.57.4.621. [DOI] [PubMed] [Google Scholar]
- 19.Rudzinski LA, Fletcher RM, Dickson DW, Crook R, Hutton ML, Adamson J, Graff-Radford NR. Early onset familial Alzheimer Disease with spastic paraparesis, dysarthria, and seizures and N135S mutation in PSEN1. Alzheimer Dis. Assoc. Disord. 2008;22:299–307. doi: 10.1097/WAD.0b013e3181732399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 1.Sleegers K, Roks G, Theuns J, Aulchenko YS, Rademakers R, Cruts M, van Gool WA, Van Broeckhoven C, Heutink P, Oostra BA, van Swieten JC, van Duijn CM. Familial clustering and genetic risk for dementia in a genetically isolated Dutch population. Brain. 2004;127:1641–1649. doi: 10.1093/brain/awh179. [DOI] [PubMed] [Google Scholar]
- 2.Tanahashi H, Kawakatsu S, Kaneko M, Yamanaka H, Takahashi K, Tabira T. Sequence analysis of presenilin-1 gene mutation in Japanese Alzheimer's disease patients. Neurosci. Lett. 1996;218:139–141. doi: 10.1016/s0304-3940(96)13138-4. [DOI] [PubMed] [Google Scholar]
- 3.Tanahashi H, Mitsunaga Y, Takahashi K, Tasaki H, Watanabe S, Tabira T. Missense mutation of S182 gene in Japanese familial Alzheimer's disease. Lancet. 1995;346:440. doi: 10.1016/s0140-6736(95)92810-3. [DOI] [PubMed] [Google Scholar]
- 4.Tedde A, Nacmias B, Ciantelli M, Forleo P, Cellini E, Bagnoli S, Piccini C, Caffarra P, Ghidoni E, Paganini M, Bracco L, Sorbi S. Identification of new presenilin gene mutations in early-onset familial Alzheimer disease. Arch. Neurol. 2003;60:1541–1544. doi: 10.1001/archneur.60.11.1541. [DOI] [PubMed] [Google Scholar]
- 5.The Lund and Manchester Groups. Clinical and neuropathological criteria for frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tomaino C, Bernardi L, Anfossi M, Costanzo A, Ferrise F, Gallo M, Geracitano S, Maletta R, Curcio SA, Mirabelli M, Colao R, Frangipane F, Puccio G, Calignano C, Muraca MG, Paonessa A, Smirne N, Leotta A, Bruni AC. Presenilin 2 Ser130Leu mutation in a case of late-onset “sporadic” Alzheimer's disease. J. Neurol. 2007;254:391–393. doi: 10.1007/s00415-006-0373-y. [DOI] [PubMed] [Google Scholar]
- 7.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann. Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 8.Turkish Statistical Institute. 2009 Census, Population statistics access in 2009. 2010 www.turkstat.gov.tr.
- 9.Walker ES, Martinez M, Brunkan AL, Goate A. Presenilin 2 familial Alzheimer's disease mutations result in partial loss of function and dramatic changes in Abeta 42/40 ratios. J. Neurochem. 2005;92:294–301. doi: 10.1111/j.1471-4159.2004.02858.x. [DOI] [PubMed] [Google Scholar]
- 10.Wasco W, Pettingell WP, Jondro PD, Schmidt SD, Gurubhagavatula S, Rodes L, DiBlasi T, Romano DM, Guenette SY, Kovacs DM, Growdon JH, Tanzi RE. Familial Alzheimer's chromosome 14 mutations. Nat. Med. 1995;1:848. doi: 10.1038/nm0995-848a. [DOI] [PubMed] [Google Scholar]
- 11.Zekanowski C, Styczyńska M, Pepłońska B, Gabryelewicz T, Religa D, Ilkowski J, Kijanowska-Haładyna B, Kotapka-Minc S, Mikkelsen S, Pfeffer A, Barczak A, Łuczywek E, Wasiak B, Chodakowska-Zebrowska M, Gustaw K, Łaczkowski J, Sobów T, Kuźnicki J, Barcikowska M. Mutations in presenilin 1, presenilin 2 and amyloid precursor protein genes in patients with early-onset Alzheimer's disease in Poland. Exp. Neurol. 2003;184:991–996. doi: 10.1016/S0014-4886(03)00384-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.