

Abstract
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune connective tissue disease affecting predominantly females. To discover additional genetic risk variants for SLE on the X chromosome, we performed a follow-up study of our previously published genome-wide association study (GWAS) data set in this study.
Methods
Twelve single nucleotide polymorphisms (SNPs) within novel or unpublished loci with P-value < 1.00 × 10−02 were selected for genotype with a total of 2,442 cases and 2,798 controls(including 1,156 cases and 2,330 controls from central China, 1,012 cases and 335 controls from southern China and 274 cases and 133 controls from northern China) using Sequenom Massarry system. Associaton analyses were performed using logistic regression with sample region as a covariate through PLINK 1.07 software.
Results
Combined analysis in discovery and central validation dataset discovered a novel locus rs5914778 within LINC01420 associated with SLE at genome-wide significance (P = 1.00 × 10−08; odds ratio (OR) = 1.32). We also confirmed rs5914778 in the southern Chinese sample cohort (P = 5.31 × 10−05; OR = 1.51), and meta-analysis of the samples from the discovery, central and southern validations regions provided robust evidence for the association of rs5914778 (P = 5.26 × 10−12; OR = 1.35). However, this SNP did not show association with SLE in the northern sample (P = 0.33). Further analysis represent the association of northern was significantly heterogeneous compared to central and southern respectively.
Conclusions
Our study increases the number of established susceptibility loci for SLE in Han Chinese population and has further demonstrated the important role of X-linked genetic risk variants in the pathogenesis of SLE in Chinese Han population.
Electronic supplementary material
The online version of this article (doi:10.1186/s13075-015-0857-1) contains supplementary material, which is available to authorized users.
Keywords: Systemic lupus erythematosus, X chromosome, Genetics, Single nucleotide polymorphisms
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease that affects predominantly women aged 15–40 years, particularly women of child-bearing age. SLE has been estimated to affect 31–70 cases per 100,000 people in China [1] with a ratio of 9:1 between female and male patients. It is well known that both genetic and environmental factors contribute to disease susceptibility [2–6]. Numerous variants on autosomal loci have been found to be associated with SLE in multiple ethnic groups through candidate gene and genome-wide association studies (GWASs) [7]. Despite the advances in the genetic studies over recent years, the pathogenesis of SLE remains poorly understood.
Because of the huge gender difference in disease prevalence, involvement of the genetic variants on the X chromosome has long been suspected. In recent years, genetic variants of several genes on the X chromosome, such as MECP2, IRAK1, TLR7, and PRPS2, have been confirmed to be associated with SLE [8–11]. In particular, single nucleotide polymorphism (SNP) rs3853839 on the 3′ untranslated region (UTR) of TLR7 was shown to be associated with SLE, especially in Chinese and Japanese male subjects compared with females [10], and a fine mapping study by Kaufman et al. [11] in four different ancestral groups suggested that the nonsynonymous SNP rs1059702 (S196F) within IRAK1 might be a causal risk variant for SLE. More recently, we performed a meta-analysis of GWASs in Chinese Han populations and followed up the top findings in four additional Asian cohorts [12]. Besides confirming the previously reported associations within IRAK1-MECP2 (rs1059702) and L1CAM-MECP2 loci, we also identified a genetic variant (rs7062536) in PRPS2 on Xp22.3 as a novel susceptibility locus and novel independent associations within the NAA10 (rs2070028) and TMEM187 (rs17422) loci.
In this study, with the aim to discover additional X-linked genetic risk variants for SLE, we performed a follow-up study of our previously published GWAS dataset by improving the coverage of genetic variation through imputation and validating the top findings in an additional three independent Chinese Han sample collections [13]. We discovered a novel susceptibility locus LINC01420 on Xp11.21 associated with SLE.
Methods
Subjects
SLE cases and controls were all female and were recruited from multiple hospitals in three geographic regions of China (central, southern, and northern China). All subjects were of self-reported Chinese Han origin. Samples in the GWAS discovery stage (1017 SLE cases and 539 controls) were recruited from central China [13]. Samples in the replication studies were recruited from multiple regions in China, mainly from central (replication: 1156 cases and 2330 controls), southern (replication: 1012 cases and 335 controls), and northern (replication: 274 cases and 133 controls) China. All patients were diagnosed as cases by at least two experienced physicians using the American College of Rheumatology (ACR) criteria revised in 1997 [14]. Controls also were geographically and ethnically matched and clinically evaluated to be without SLE, autoimmune disorders, or family history of autoimmune diseases. Clinical information for all patients and controls was collected through a structured questionnaire. Written informed consent was acquired from all participants. This study was approved by the Institutional Ethical Committee of The First Affiliated Hospital of Anhui Medical University, China–Japan Friendship Hospital, Jiangmen Central Hospital, and The Third Affiliated Hospital of Sun Yat-Sen University, according to Declaration of Helsinki principles. The information for all subjects is summarized in Table 1.
Table 1.
Summary of samples used in GWASs and replication studies
| Cases | Controls | ||||||
|---|---|---|---|---|---|---|---|
| Stage | Sample size | Mean (SD) age | Mean (SD) age of onset | Sex (female) | Sample size | Mean (SD) age | Sex (female) |
| GWAS | 1017 | 34.19 (11.47) | 29.84 (10.96) | 1017 | 539 | 34.31 (12.49) | 539 |
| Replication central | 1156 | 35.19 (12.16) | 30.70 (11.32) | 1156 | 2330 | 29.34 (11.20) | 2330 |
| Replication southern | 1012 | 33.01 (10.82) | 28.22 (10.03) | 1012 | 335 | 32.59 (13.21) | 335 |
| Replication northern | 274 | 35.64 (12.02) | 31.10 (11.79) | 274 | 133 | 29.48 (15.59) | 133 |
| Total | 3459 | 34.29 (11.60) | 29.76 (10.94) | 3459 | 3337 | 30.47 (11.98) | 3337 |
GWAS stage samples are from central China
GWAS genome-wide association study, SD standard deviation
Genotyping
The genotyping in the discovery stage for the central China cohort was conducted by Illumina 610-Quad Human Beadchip array (Illumina, Inc., San Diego, CA, USA). The genomic DNA was isolated from peripheral blood mononuclear cells (PBMCs) with standard procedures using Flexi Gene DNA kits (QIAGEN GmbH, Hilden, Germany) and was diluted to working concentrations of 50 ng/μl for genome-wide genotyping and 15–20 ng/μl for the validation study. The SNPs in the X chromosome for the validation stage were genotyped using the Sequenom MassArray iPlex Gold platform (Sequenom, Inc., San Diego, CA, USA).
Statistical analyses
Quality control criteria were applied to genotyped SNPs, and those with minor allele frequency (MAF) <5 % in cases and controls were excluded. SNPs with a genotype missing rate >10 % or Hardy–Weinberg equilibrium (HWE) P <3.14 × 10−6 in controls were also excluded. Association analysis was performed in PLINK v1.07 [15] using the logistic regression test. We selected 12 SNPs within novel or unpublished loci with P <1.00 × 10−2 for further validation in 2442 cases and 2798 controls (SNP missing rate <10 % and HWE for female controls with P >1.00 × 10−2).
To control the impact of population stratification in the validation and combined analysis, we matched cases and controls in terms of ethnic and geographic origins as independent validation samples for combined analysis. Fixed-effects meta-analysis of the four independent studies in the discovery GWAS and three validation cohorts (central, southern and northern) was performed using the inverse variants weighted effect size method in Metasoft version 2.0.0 [16].
We performed the combined analysis of the central region (both discovery and central validation) cohort, southern validation cohort, and northern validation cohort using fixed-effects meta-analysis. The I2 heterogeneity statistic shows the heterogeneity across studies, with I2 < 50 and Phet >0.05 considered insignificant (Table 2).
Table 2.
Validation of top SNP rs5914778 in southern and northern regions of China
| Central meta (discovery + central validation)a | Southern validationb | Northern validationc | Meta (discovery + central + southern validations)d | Meta (discovery + central + southern + northern validations)e | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Marker | Gene symbol | PVAL (FE) | OR (FE) | PVAL | OR | MAF (case) | MAF (control) | PVAL | OR | MAF (case) | MAF (control) | PVAL (FE) | I 2 (FE) | PVAL (Het) | OR (FE) | PVAL (FE) | I 2 (FE) | PVAL (Het) | OR (FE) |
| rs5914778 | LINC01420 | 1.00 × 10–8 | 1.32 | 5.31 × 10–5 | 1.51 | 0.352 | 0.265 | 3.28 × 10–1 | 0.85 | 0.252 | 0.284 | 5.26 × 10–12 | 0 | 4.62 × 10–1 | 1.35 | 1.22 × 10–10 | 65.3 | 3.44 × 10–2 | 1.31 |
aConsists of 2173 cases and 2869 controls
bConsists of 1012 cases and 335 controls
cConsists of 274 cases and 133 controls
dConsists of 3185 cases and 3204 controls
eConsists of 3459 cases and 3337 controls
FE fixed effects, Het heterogeneity, I 2 heterogeneity statistic, MAF minor allele frequency, OR odds ratio, PVAL P value, SNP single nucleotide polymorphism
Imputation
Imputation of the X chromosome SNPs was performed on the discovery dataset for female individuals using X chromosome nonpseudoautosomal region data from the 1000 Genomes project (phase 1 integrated version 3) as reference [17]. As part of the quality control, SNPs with accuracy score <0.8, missing rate >10 %, MAF <5 % in cases and controls, or HWE P <2.89 × 10−7 in controls were also excluded. Association was carried out by logistic regression test. The imputation results show that there is no substantial improvement of significant signals between imputed or genotyped SNPs (Fig. 1). No imputed SNPs show better P values that would warrant further validation on top of the genotyped SNPs. Therefore, we proceeded with the validation of the selected genotyped SNPs which resided in novel regions.
Fig. 1.
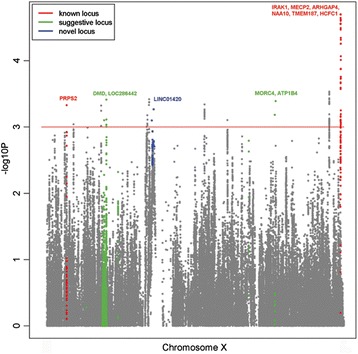
Manhattan plot of the X chromosome association analysis on SLE. Manhattan plot of association results (−log10(P value)) are depicted with regards to the physical location of SNPs and include both imputed and genotyped association results. Positions and genes were based on National Center for Biotechnology Information build 37 (http://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.25/). The color of gene labeling corresponds to the gene type of known loci, suggestive loci, and novel loci. Known loci are defined as loci published previously, whereby novel loci are defined as loci in the current study validated at genome-wide significance (P <5 × 10−8)
Results
X chromosome discovery and first-stage study
We conducted X chromosome association tests of SLE in the GWAS dataset which consists of 1017 cases and 539 controls, after stringent quality control filtering (see Statistical analyses). The discovery analysis revealed strong evidence of association for all previously identified susceptibility loci on the X chromosome and suggested additional novel risk loci (Additional file 1: Table S1).
To further investigate the observed associations, we imputed the genotypes of additional SNPs that were not genotyped using IMPUTE (v2.0) (Oxf, Oxford, Oxon, UK). After stringent quality control filtering (imputation), no imputed SNPs show better P values that would warrant further validation on top of the genotyped SNPs. Therefore, to validate the findings from the discovery analysis, we selected the top SNPs from 14 independent new loci with suggestive association with SLE (P <10−2) for a follow-up analysis in an additional 1156 cases and 2330 controls of Chinese Han descent from central China. Of the 12 successfully genotyped SNPs, two showed association at P <0.05 in the validation samples and six showed consistent effects between the discovery and validation samples. The meta-analysis results for the 12 SNPs in the combined discovery and central validation dataset totaling 2173 cases and 2869 controls, using fixed-effects and random-effects models, are presented in Table 3. The combined analysis discovered a novel locus rs5914778 within LINC01420 associated with SLE disease at genome-wide significance (P = 1.00 × 10−8; odds ratio (OR) = 1.32).
Table 3.
Validation of selected SNPs on the X chromosome in the central region
| Discovery set (central)a | Central validationb | Meta (discovery + central validation)c | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Marker | Minor allele | Position (Hg19) | Annotation | Gene symbol | PVAL | OR | MAF case | MAF control | PVAL | OR | MAF case | MAF control | PVAL (FE) | PVAL (RE) | OR (FE) | OR (RE) | I 2 | PVAL (Het) |
| rs5914778 | A | 56758231 | Intronic | LINC01420 | 3.59 × 10–3 | 1.29 | 0.286 | 0.238 | 7.15 × 10–7 | 1.33 | 0.296 | 0.24 | 1.00 × 10–8 | 1.00 × 10–8 | 1.32 | 1.32 | 0 | 7.58 × 10–1 |
| rs1527803 | A | 16421982 | 184 kb 3′ of CTPS2 | NA | 6.75 × 10–2 | 0.87 | 0.368 | 0.402 | 1.90 × 10–2 | 1.13 | 0.403 | 0.374 | 3.63 × 10–1 | 9.82 × 10–1 | 1.04 | 1.00 | 87.5 | 4.70 × 10–3 |
| rs5963635 | A | 39166975 | None | LOC286442 | 1.40 × 10–3 | 1.34 | 0.247 | 0.197 | 2.17 × 10–1 | 1.08 | 0.230 | 0.217 | 5.17 × 10–3 | 1.08 × 10–1 | 1.15 | 1.19 | 74.1 | 4.94 × 10–2 |
| rs1860995 | G | 119502122 | Intronic | ATP1B4 | 7.48 × 10–4 | 1.30 | 0.417 | 0.355 | 6.74 × 10–1 | 1.02 | 0.395 | 0.389 | 1.69 × 10–2 | 2.87 × 10–1 | 1.10 | 1.11 | 81.4 | 4.33 × 10–2 |
| rs2536576 | G | 147417249 | 165 kb 5′ of AFF2 | NA | 5.73 × 10–3 | 1.34 | 0.172 | 0.134 | 9.43 × 10–1 | 1.01 | 0.152 | 0.152 | 1.12 × 10–1 | 3.36 × 10–1 | 1.10 | 1.15 | 80.2 | 2.45 × 10–2 |
| rs17326228 | G | 106239318 | Intronic | MORC4 | 2.56 × 10–4 | 1.33 | 0.438 | 0.370 | 3.01 × 10–1 | 0.95 | 0.418 | 0.432 | 3.82 × 10–1 | 6.14 × 10–1 | 1.02 | 1.05 | 88.5 | 1.75 × 10–3 |
| rs17329976 | G | 142130195 | 8.1 KB 3′ of SPANXN4 | NA | 4.28 × 10–2 | 0.83 | 0.179 | 0.209 | 1.58 × 10–1 | 0.91 | 0.160 | 0.174 | 2.23 × 10–2 | 3.62 × 10–2 | 0.85 | 0.83 | 51.6 | 1.62 × 10–1 |
| rs5936901 | G | 67975499 | 30 kb 3′ of STARD8 | NA | 4.45 × 10–4 | 1.33 | 0.371 | 0.308 | 5.53 × 10–1 | 0.97 | 0.342 | 0.350 | 1.28 × 10–1 | 4.50 × 10–1 | 1.07 | 1.13 | 90.3 | 1.30 × 10–3 |
| rs10218247 | A | 83476018 | 33 kb 5′ of RPS6KA6 | NA | 3.67 × 10–3 | 0.78 | 0.241 | 0.289 | 5.47 × 10–1 | 0.96 | 0.248 | 0.255 | 2.85 × 10–2 | 3.06 × 10–1 | 0.91 | 0.86 | 82.4 | 1.24 × 10–2 |
| rs5970959 | G | 22987793 | 30 kb 5′ of DDX53 | DDX53 | 1.42 × 10–2 | 1.25 | 0.254 | 0.215 | 1.57 × 10–1 | 0.92 | 0.251 | 0.267 | 3.76 × 10–1 | 6.41 × 10–1 | 1.03 | 1.05 | 86.7 | 2.06 × 10–3 |
| rs6631753 | A | 33199451 | Intronic | DMD | 6.94 × 10–3 | 0.81 | 0.376 | 0.426 | 5.09 × 10–1 | 1.04 | 0.374 | 0.366 | 7.12 × 10–2 | 4.69 × 10–1 | 0.93 | 0.91 | 89.2 | 4.35 × 10–3 |
| rs2411864 | G | 16377763 | 206 kb 3′ of GRPR | NA | 5.04 × 10–3 | 0.76 | 0.154 | 0.194 | 4.81 × 10–1 | 1.05 | 0.165 | 0.159 | 2.98 × 10–1 | 5.16 × 10–1 | 0.94 | 0.90 | 86.2 | 7.10 × 10–3 |
aFrom Han et al. [13] with 1017 cases and 539 controls from central China
bConsists of 1156 cases and 2330 controls
cConsists of 2173 cases and 2869 controls
FE fixed effects, Het heterogeneity, I 2 heterogeneity statistic, MAF minor allele frequency, NA not available, OR odds ratio, PVAL P value, RE random effects, SNP single nucleotide polymorphism
Further replication of selected SNPs and the heterogeneity test
We performed further replication analysis of rs5914778 in two additional independent samples of Chinese Han descent from the southern and northern regions of China. The replication in the southern Chinese sample cohort, consisting of a total of 1012 cases and 335 controls, provided strong supporting evidence for the association of rs5914778 with SLE (P = 5.31 × 10−5; OR = 1.51). The meta-analysis of the samples from the central and southern regions, totaling 3185 cases and 3204 controls, provided robust evidence for the association of rs5914778 (P = 5.26 × 10−12; OR = 1.35). In addition, the strength of the association is very consistent without any evidence of heterogeneity (Phet = 0.46, I2 = 0) (Table 2).
Intriguingly, this SNP did not show association with SLE in the northern sample with a total of 274 cases and 133 controls (P = 0.33, OR = 0.85), and the SNP actually showed an opposite effect in the northern sample as compared with the central and southern samples (Table 2). This could be because of the very small sample size of the northern replication cohort. Further studies are needed to confirm the heterogeneity of this association between the northern and central/southern Chinese populations.
Lastly, we performed a joint analysis for all of the discovery, central, southern, and northern validation samples totaling 3459 cases and 3337 controls, using a fixed-effects meta-analysis. The association at rs5914778 (LINC01420) on Xp11.21 surpassed the genome-wide significance (P = 1.22 × 10−10; OR = 1.31), but a moderate heterogeneity of association was observed within the samples (Phet = 0.034, I2 = 65.3) (Table 2 and Fig. 2).
Fig. 2.
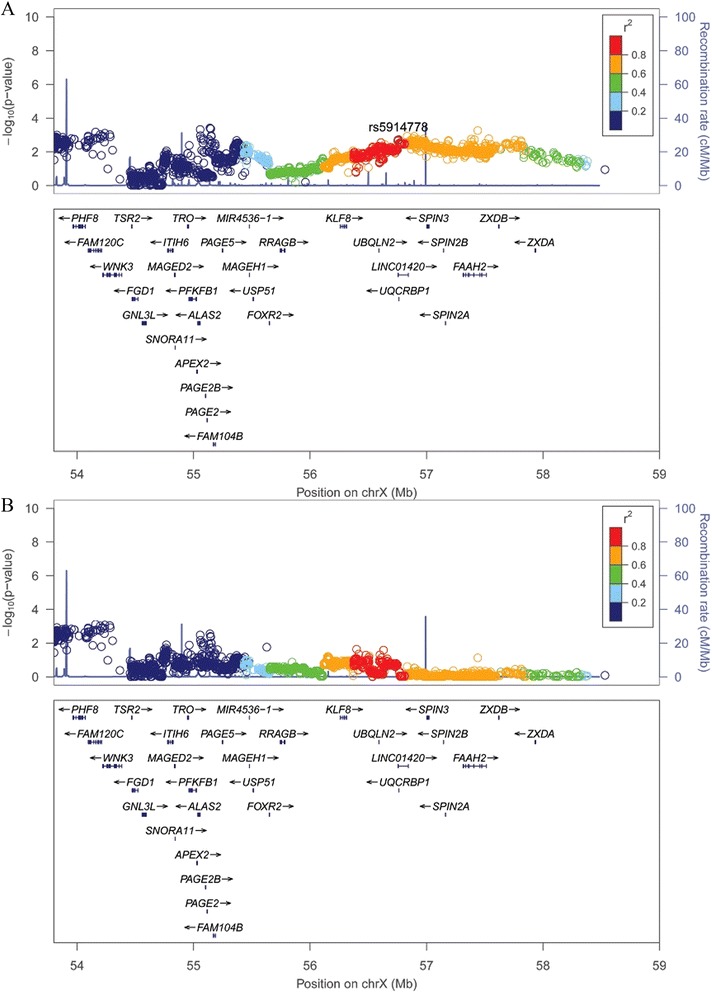
a Regional association plots of new loci (Xp11.21, LINC01420). The association results (−log10(P value)) of SNPs from the discovery analysis were shown against their map positions (NCBI build 37). Validated SNP rs5914778 is labeled purple. b Regional association plots after conditioning on rs5914778. The association results (−log10(P value)) of SNPs from the discovery analysis after conditioning on rs5914778. All map coordinates are based on NCBI build 37. chrX X chromosome, NCBI National Center for Biotechnology Information
Discussion
Through the discovery and validation analyses in two independent female samples from the central region of China, we have discovered a novel SLE susceptibility locus at rs5914778 (LINC01420) on Xp11.21 at the genome-wide significance. Further replication analysis in the independent sample of southern Chinese confirmed the association with strong evidence. The analysis of the independent sample of northern Chinese failed to replicate the association, but the sample size of the northern cohort is very small.
rs5914778 is located within a long intronic region between the first and second exons of LINC01420. LINC01420 is a long noncoding RNA with enhancers marked by histone modifications in human umbilical vein endothelial cells (HUVEC) and HSMM based on HaploReg annotation [18] (Additional file 2: Table S2). LINC01420 was found to have sex-specific DNAse I hypersensitivity patterns which showed H3K4me3 histone enrichment and strong expression in females only [19]. According to the regulatory annotation information from the ENCODE project [20], this SNP is within a DNase I hypersensitive site that was detected in the lymphoblastoid cell line. LINC01420 may maintain the X inactivation which avoids X-linked gene overexpression through dosage compensation in females [21]. Long noncoding RNAs have been shown to be associated with many complex diseases such as psoriasis, breast cancer, gastric cancer, colorectal cancer, osteosarcoma, adrenocortical cancer, and cardiovascular diseases in recent years [22–28]. Some noncoding RNAs also play a role in the pathogenesis and progression of hepatocellular carcinoma, and may act as therapeutic targets for hepatocellular carcinoma [29]. In order to reveal whether there are expression difference of LINC01420 between females and males, we performed gene expression analyses using the gene expression data from CD4+ T cells and monocytes from 461 healthy individuals [30] and the gene expression data from PBMCs of 82 controls [31] in GEO datasets. However, we did not obtain the gene expression result of LINC01420, indicating that LINC01420 might express too low to be detected in blood cells from healthy individuals. Hence, more work will be needed to elucidate the biological mechanism through which LINC01420 influences SLE pathogenesis.
We also observed another SNP, rs5913992, in perfect linkage disequilibrium with our top SNP rs5914778 (R2 = 1) that was predicted to be functional by Regulome DB (LSJU, Stanford, CA, USA) with a score of 2b (likely to affect binding of motifs, transcription factors, and enhancer histone marks) in this locus [32]. rs5913992 is also within the region of the binding sites of six overlapping transcription factors (transcription factor binding sites)—RELA, CTCF, CEBPB, RAD21, ZNF143, and SMC3—that were detected by ChIP-Seq analysis in lymphoblastoid, epithelial, endothelial, breast cancer, and myeloid leukemia cell lines (Fig. 3). The prediction by Regulome DB indicates that this SNP overlaps a potential consensus EWSR1-FLI1 binding motif within the binding sites of the six transcription factor binding sites (Additional file 3: Table S3).
Fig. 3.
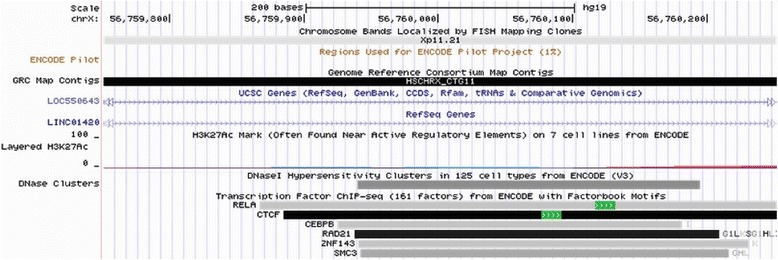
Transcription factor binding sites of rs5913992 SNP (ENCODE). The transcription track from the UCSC (University of California, Santa Cruz) genome browser showed regions where transcription factors responsible for modulating gene transcription bind to DNA as assayed by ChIP-seq
We observed the same risk effect at rs5914778 in the central and southern validation results, while the opposite effect was observed in the northern validation results. The association of the northern cohort was significantly heterogeneous compared with the central and southern cohorts respectively (Phet = 0.034, I2 = 65.3). Several previous studies have demonstrated differences in disease risk between northern and southern Chinese, and further studies in more northern Chinese samples will be needed to confirm the genetic heterogeneity of this susceptibility locus among the central, southern, and northern Chinese populations [33–35].
Conclusions
We performed a three-stage X chromosome association analysis of SLE in the Chinese Han population and discovered a novel susceptibility locus on Xp11.21. Although further studies will be required to understand how the locus influences the etiology of SLE, the discovery of this novel locus has further expanded the role of the X chromosome in the development of SLE in the Chinese Han population.
Acknowledgements
This study was approved by the respective institutional Ethical Committee at each institution and according to Declaration of Helsinki principles. Patient consent was also obtained.
The authors thank all study participants and all of the volunteers who have willingly participated in this study. This study was supported by grants from the National Key Basic Research Program of China (2014CB541901, 2012CB722404, 2011CB512103), the International (Regional) Cooperation and Exchanges major project (81320108016), the National Natural Science Foundation of China (81573033, 81171505, 81402590, 81371722), the Research Project of the Chinese Ministry of Education (213018A), the Program for New Century Excellent Talents in University (NCET-12-0600), and the Natural Science Fund of Anhui province (1408085MKL27).
Abbreviations
- ACR
American College of Rheumatology
- GWAS
Genome-wide association study
- HUVEC
Human umbilical vein endothelial cells
- HWE
Hardy–Weinberg equilibrium
- MAF
Minor allele frequency
- OR
Odds ratio
- PBMC
Peripheral blood mononuclear cell
- SLE
Systemic lupus erythematosus
- SNP
Single nucleotide polymorphism
- UTR
Untranslated region
Additional files
Presenting association results for the X chromosome SLE GWAS cohort. (DOC 241 kb)
Presenting HaploReg annotation for rs5914778 (Query SNP: rs5914778 and variants with r 2 ≧0.8). (DOC 46 kb)
Presenting the motifs predicted to be affected by rs5913992 SNP (Regulome DB). (DOC 171 kb)
Footnotes
Zhengwei Zhu, Zhuoyuan Liang, Herty Liany, Chao Yang and Leilei Wen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JJL shared senior author. XJZ, YCu, WLY, and JJL conceived of this study, obtained financial support, participated in the study design, and revised the manuscript. ZWZ, ZYL, HL, CY, and LLW were responsible for sample selection, genotyping, and project management, and drafted the manuscript. ZML, YJShe, YL, LY, YYC, YCh, LL, LLY, YJShi, CBS, HYT, LDS, and SY conducted sample selection, undertook recruitment, collected clinic data, managed recruitment, obtained biological samples, and helped to revise the manuscript. JZ, BL, YTD, and YZ were responsible for sample management and DNA extraction, and helped to draft the manuscript. HL, YJShe, XDZ, XYY, and XBZ conducted data management, undertook related data handling and calculation, and revised the manuscript. FSZ and JZ performed genotyping analysis and helped to draft the manuscript. HL, YJShe, XDZ, XYY, XBZ, HYT, and J-XB performed data processing and statistical analysis, and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Zhengwei Zhu, Email: 631183574@qq.com.
Zhuoyuan Liang, Email: liangzy2r@163.com.
Herty Liany, Email: lianyh@gis.a-star.edu.sg.
Chao Yang, Email: xotong@163.com.
Leilei Wen, Email: wenleilei_2012@163.com.
Zhiming Lin, Email: lzm-zj99@163.com.
Yujun Sheng, Email: ahmusyj@163.com.
Yan Lin, Email: lara862@163.com.
Lei Ye, Email: yelei8778@sina.cn.
Yuyan Cheng, Email: chengyil567@sina.com.
Yan Chang, Email: 458210265@qq.com.
Lu Liu, Email: 501335910@qq.com.
Lulu Yang, Email: 892319580@qq.com.
Yinjuan Shi, Email: 345016785@qq.com.
Changbing Shen, Email: cambridge2008@126.com.
Fusheng Zhou, Email: biozhoufs@163.com.
Xiaodong Zheng, Email: zhengxiaodong.1234@163.com.
Jun Zhu, Email: 805866340@qq.com.
Bo Liang, Email: anyiliangbo@126.com.
Yantao Ding, Email: ding2008yantao@163.com.
Yi Zhou, Email: zhcym198228@163.com.
Xianyong Yin, Email: xianyongyin@gmail.com.
Huayang Tang, Email: tanghuayang@hotmail.com.
Xianbo Zuo, Email: zuoxianbo@gmail.com.
Liangdan Sun, Email: ahmusld@163.com.
Jin-Xin Bei, Email: beijx@gis.a-star.edu.sg.
Jianjun Liu, Email: liuj3@gis.a-star.edu.sg.
Sen Yang, Email: yangsen@medmail.com.cn.
Wanling Yang, Phone: (852) 283-15077, Email: yangwl@hkucc.hku.hk.
Yong Cui, Phone: +86-010-84205147, Email: wuhucuiyong@vip.163.com.
Xuejun Zhang, Phone: +86-551-65161002, Email: ayzxj@vip.sina.com.
References
- 1.Zeng QY, Chen R, Darmawan J, Xiao ZY, Chen SB, Wigley R, et al. Rheumatic diseases in China. Arthritis Res Ther. 2008;10:R17. doi: 10.1186/ar2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet. 2009;10:285–90. doi: 10.1038/nrg2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel DR, Richardson BC. Epigenetic mechanisms in lupus. Curr Opin Rheumatol. 2010;22:478–82. doi: 10.1097/BOR.0b013e32833ae915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaiser R, Criswell LA. Genetics research in systemic lupus erythematosus for clinicians: methodology, progress, and controversies. Curr Opin Rheumatol. 2010;22:119–25. doi: 10.1097/BOR.0b013e3283361943. [DOI] [PubMed] [Google Scholar]
- 5.Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009;10:373–9. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng Y, Tsao BP. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat Rev Rheumatol. 2010;6:683–92. doi: 10.1038/nrrheum.2010.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun. 2013;41:25–33. doi: 10.1016/j.jaut.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, et al. Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2009;106:6256–61. doi: 10.1073/pnas.0901181106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawalha AH, Webb R, Han S, Kelly JA, Kaufman KM, Kimberly RP, et al. Common variants within MECP2 confer risk of systemic lupus erythematosus. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2010;107:15838–43. doi: 10.1073/pnas.1001337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis. 2013;72:437–44. doi: 10.1136/annrheumdis-2012-201851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Zhang J, Yang J, Wang Y, Zhang L, Zuo X, et al. Meta-analysis of GWAS on two Chinese populations followed by replication identifies novel genetic variants on the X chromosome associated with systemic lupus erythematosus. Hum Mol Genet. 2014;24:274–80. doi: 10.1093/hmg/ddu429. [DOI] [PubMed] [Google Scholar]
- 13.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–7. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 14.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 15.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han B, Eskin E. Interpreting meta-analyses of genome-wide association studies. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–4. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joo JE, Novakovic B, Cruickshank M, Doyle LW, Craig JM, Saffery R. Human active X-specific DNA methylation events showing stability across time and tissues. Eur J Hum Genet. 2014;22:1376–81. doi: 10.1038/ejhg.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57-74. [DOI] [PMC free article] [PubMed]
- 21.Brooks WH, Renaudineau Y. Epigenetics and autoimmune diseases: The X chromosome-nucleolus nexus. Front Genet. 2015;6:22. doi: 10.3389/fgene.2015.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsoi LC, Iyer MK, Stuart PE, Swindell WR, Gudjonsson JE, Tejasvi T, et al. Analysis of long non-coding RNAs highlights tissue-specific expression patterns and epigenetic profiles in normal and psoriatic skin. Genome Biol. 2015;16:24. doi: 10.1186/s13059-014-0570-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y, Li J, Liu Y, Ding J, Fan Y, Tian Y, et al. The long noncoding RNA SPRY4-IT1 increases the proliferation of human breast cancer cells by upregulating ZNF703 expression. Mol Cancer. 2015;14:51. doi: 10.1186/s12943-015-0318-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan W, Liu L, Wei J, Ge Y, Zhang J, Chen H, et al. A functional lncRNA HOTAIR genetic variant contributes to gastric cancer susceptibility. Mol Carcinog. 2015 doi: 10.1002/mc.22261. [DOI] [PubMed] [Google Scholar]
- 25.Chu H, Xia L, Qiu X, Gu D, Zhu L, Jin J, et al. Genetic variants in noncoding PIWI-interacting RNA and colorectal cancer risk. Cancer. 2015;121:2044–52. doi: 10.1002/cncr.29314. [DOI] [PubMed] [Google Scholar]
- 26.Wang B, Su Y, Yang Q, Lv D, Zhang W, Tang K, et al. Overexpression of long non-coding RNA HOTAIR promotes tumor growth and metastasis in human osteosarcoma. Mol Cells. 2015;38:432–40. doi: 10.14348/molcells.2015.2327. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Glover AR, Zhao JT, Ip JC, Lee JC, Robinson BG, Gill AJ, et al. Long noncoding RNA profiles of adrenocortical cancer can be used to predict recurrence. Endocr Relat Cancer. 2015;22:99–109. doi: 10.1530/ERC-14-0457. [DOI] [PubMed] [Google Scholar]
- 28.Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. 2015;116:737–50. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 29.George J, Patel T. Noncoding RNA as therapeutic targets for hepatocellular carcinoma. Semin Liver Dis. 2015;35:63–74. doi: 10.1055/s-0034-1397350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raj T, Rothamel K, Mostafavi S, Ye C, Lee MN, Replogle JM, et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science. 2014;344:519–23. doi: 10.1126/science.1249547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong SW, Collins CD, Shimizu-Motohashi Y, Holm IA, Campbell MG, Lee IH, et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao YG, Kong QP, Bandelt HJ, Kivisild T, Zhang YP. Phylogeographic differentiation of mitochondrial DNA in Han Chinese. Am J Hum Genet. 2002;70:635–51. doi: 10.1086/338999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao YG, Kong QP, Man XY, Bandelt HJ, Zhang YP. Reconstructing the evolutionary history of China: a caveat about inferences drawn from ancient DNA. Mol Biol Evol. 2003;20:214–9. doi: 10.1093/molbev/msg026. [DOI] [PubMed] [Google Scholar]
- 35.Yao YG, Kong QP, Wang CY, Zhu CL, Zhang YP. Different matrilineal contributions to genetic structure of ethnic groups in the silk road region in china. Mol Biol Evol. 2004;21:2265–80. doi: 10.1093/molbev/msh238. [DOI] [PubMed] [Google Scholar]