

Abstract
Chronic obstructive pulmonary disease (COPD) affect millions of people worldwide and is known to be one of the leading causes of death. The highly sensitive airways protect themselves from irritants by cough and sneeze which propel endogenous and exogenous substances to minimize airway noxious effects. One noxious effect of these substances is activation of peripheral sensory nerve endings of nociceptor neurons innervating these airways lining thus transmitting dangerous signals from the environment to the central nervous system (CNS). Nociceptor neurons include transient receptor potential (TRP) ion channels, especially the vanilloid and ankyrin subfamilies, TRPV1/A1 which can be activated by noxious chemical challenges in models of airways disease. As oxidative stress may activate airways sensory neurons and contribute to COPD exacerbations we sought to review the role that TRP channel activation by oxidative signals may have on airway responses. It would be prudent to target the TRP channels with antagonists and lower systemic oxidative stress with agents that can modulate TRP expression and boost the endogenous levels of antioxidants for treatment and management of COPD.
Keywords: COPD, hyperresponsiveness, oxidative stress, ROS, transient receptor potential
Introduction
Chronic obstructive pulmonary disease (COPD) is a multi-factorial disorder affecting millions of people worldwide and is currently the fourth leading cause of death1. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines COPD as a disease state associated with progressive airflow obstruction that is not fully reversible2, and is closely tied to oxidative damage of cell. Increase in prevalence of COPD is not well understood in India as the important variables such as smoking, domestic smoke exposure, and outdoor pollution, socio-economic status and ethnicity are ill-defined3. Pena and co-workers4 reported prevalence ranging 5-18 per cent in seven regions of Spain. The 2005 Latin American Project5 for the Investigation of Obstructive Lung Disease (PLATINO) study reported prevalence ranging 8-20 per cent in five Latin American cities. Caballero and coworkers6 found prevalence of GOLD stage I COPD ranging 6-13 per cent in Colombia and the international Burden of Obstructive Lung Disease (BOLD) study identified prevalence in males ranging from 11 per cent in China to 24 per cent in South Africa7. Overall, the evidence suggests substantial geographical variations in COPD prevalence that remain largely unexplained7. COPD is not only confined to the lungs but is being recognized as a systemic disease with multisystem manifestations. In a sub-continent like India, there is a need to understand different phenotypes in relation to clinical presentation, spirometric parameters, exacerbations and finally prognosis3. In a multicentric study of Indian population on smoking and associated respiratory morbidities leading to the development of COPD/other respiratory disorders, it has been emphasized that benefits of quitting smoking need to be taken on a priority basis8,9. Apart from the exogenously produced oxidants, the body is also exposed to endogenously produced reactive oxygen species (ROS). These are quenched, to an extent by the antioxidant enzymes such as glutathione-S-transferase (GST) and superoxide dismutase (SOD) to maintain redox homeostasis. Oxidative stress generated due to cigarette smoke/environmental pollutants is known to irritate epithelial cells of the lungs and various cells related to immune response. This leads to an imbalance in redox homeostasis and causes deleterious damage to cell components (Fig. 1), resulting in COPD condtion10.
Fig. 1.
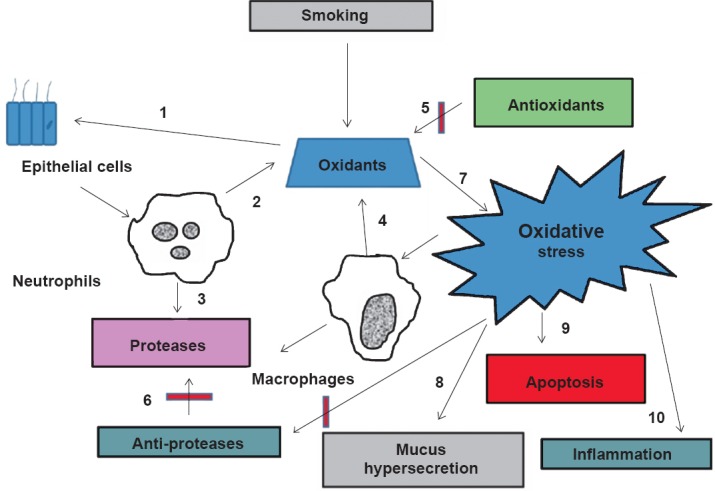
Cigarette smoke is a complex mixture of thousands of chemical compounds including free radicles and oxidants. Cigarette smoke activates alveolar and bronchial epithelial cells (1) to elicit inflammatory responses leading to the release of cytokines and recruitment of neutrophils. Endogenous oxidants (2) and proteases (3) are generated from neutrophils and other phagocytic cells (macrophages) (4) increasing the oxidative burden in the lung. The lung has an efficient antioxidant (5) and anti-protease systems (6). The balance between the oxidants and the anti- oxidants is deranged by the exogenously and endogenously produced oxidants by the cigarette smoke leading to oxidative stress (7). Oxidative stress also contributes to mucus hypersecretion (8), apoptosis (9) and inflammation (10).
Chemical airway exposures are detected by the olfactory, gustatory, and nociceptive sensory systems that initiate protective physiological and behavioural responses. Using physiological, imaging, and genetic approaches, the transient receptor potential (TRP) ion channels in sensory neurons were shown to respond to a wide range of noxious chemical stimuli, initiating pain, respiratory depression, cough, glandular secretions, and other protective responses. The reactive chemicals are detected by the peripheral sensory neurons by activating TRPV (vallinoid), TRPA1 (ankyrin), or ASIC (acid-sensitive ion channels). The activation of these channels induces neurogenic inflammatory and brain-mediated responses of the airways. These responses compromise breathing and can lead to disease states if these persist. The TRPA1, a TRP ion channel expressed in chemosensory C-fibers, is activated by almost all oxidizing and electrophilic chemicals, including chlorine, acrolein, tear gas agents, and isocyanates. Chemicals likely activate TRPA1 through covalent protein modification11.
Chemosensation to airways along with suitable reflex responses
Trigeminal chemosensory nerve endings in the nasal mucosa are in the first line of defense against noxious chemical challenges12. Calcitonin gene-related peptide (CGRP), tachykinins substance P (SP) and neurokinin A (NKA) released from chemically stimulated nerve endings, promote neurogenic inflammatory vasodilation and leakage, contributing to narrowing or obstruction of the nasal passages13,14. Vagal sensory nerves innervating the airways play a critical role in detection of the microenvironment in the airways. Oxidative stress and associated compounds activate unmyelinated bronchopulmonary C-fibers, initiating action potentials in these nerves that conduct centrally to evoke unpleasant sensations (e.g. urge to cough, dyspnoea, chest-tightness) and to stimulate/modulate reflexes (e.g. cough, bronchoconstriction, respiratory rate, inspiratory drive)15. Most of these factors are highly sensitive to intracellular calcium regulation in cells such as entry of nociceptors and other stimuli, highlighting the importance of Ca2+ transduction. The receptor-evoked Ca2+ signal causes Ca2+ release from internal stores, primarily the endoplasmic reticulum (ER), followed by activation of the store operated Ca2+ influx channels (SOCs) at the plasma membrane11. The two receptor stimulated SOCs, orai and transient receptor potential canonical (TRPCs) channels are gated by the ER Ca2+ sensor stromal interaction molecule 1 (STIM1)16. Also, the gating of peripheral terminal ion channels required for afferent nerve activation can occur through ionotropic and metabotropic mechanisms17. The ionotropic mechanism refers to an ion channel that has a self-contained activation/binding site for a specific stimulus (e.g. TRPV1 is directly gated by capsaicin). The metabotropic mechanism refers to the gating of certain ion channels downstream of second messenger systems (metabotropic), typically following the activation of G-protein-coupled receptors (GPCRs): for example, bradykinin, via the Gq-coupled B2 receptor can activate TRPV1 channels, inducing nerve depolarization and action potential discharge18,19,20. As COPD is characterized by epithelial cell damage, bronchoconstriction, lung parenchymal destruction and mucus hypersecretion, Ca2+ channels might impact COPD pathogenesis; because upon activation of TRP channel, these produce Ca2+ influx to trigger a variety of important physiological activities in the airways21.
The respiratory airways have a ramified network of peripheral sensory neurons expressing chemosensory receptors, including members of the TRP ion channel family. The TRP channels are expressed in all these tissues especially on lung epithelium and smooth muscles and are known to get activated by stimuli such as cigarette smoke, industrial pollutants, chlorine, aldehydes, and scents, which trigger the onset of such disease state. There exist 28 mammalian TRP channels which can be subdivided into seven main subfamilies on the basis of amino acid homology and are genetically conserved viz. the TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and the TRPA (ankyrin) groups, of which six have been reported in Homo sapiens22.
Each TRP channel subunit consists of six putative transmembrane spanning segments (TM 1–6), a pore-forming loop between TM 5 and TM 6, and intracellular located-NH2 and-COOH termini. Each of these channel subunits is either homo- or heterotetramers which results in the formation of cation-selective channels22 to allow selective molecules to pass through them as depicted in Fig. 2.
Fig. 2.
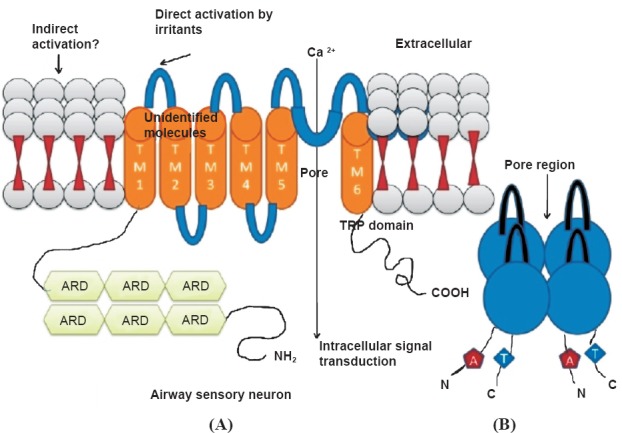
(A). A typical TRP channel containing six conserved transmembrane segments (TM1 to TM6) with a pore-forming reentrant loop between TM5 and TM6. TM1 carrying 6 ARDs (Ankyrin repeat domains); (B). the TRP pore structure formed of four subunits, each with six highly putative transmembrane helices. The pore contains a selectivity filter, which dictates through its stereochemical and electrostatic properties what kind of molecules are allowed through the pore.
Chronic obstructive pulmonary disease (COPD) and oxidative damage
Oxidative stress is the main reason behind COPD pathogenesis, as our lungs are being constantly bathed in oxidants. ROS is produced due to ageing as a result of various endogenously-generated oxidants and catalysis by inhaled toxic particulates. Overproduction of ROS (arising either from mitochondrial electron-transport chain or excessive stimulation of NADPH) results in oxidative stress, a deleterious process that can be an important mediator of damage to cell structures, including lipids and membranes, proteins, and DNA23. However, ROS is also required by the body during normal physiological functions for elimination of pathogens and other toxic metabolites produced in the body. Thus the body is constantly subjected to a redox control, which on imbalance results in disturbed homeostasis, resulting in degradation of normal tissues23.
Cigarette smoking has been a known factor responsible for development of COPD due to oxidant overload in the lower airways9. However, not all smokers develop COPD. The severity of COPD can be different among smokers with similar degree of exposure or no exposure. It is documented that lipid peroxidation occurs by the release of hydroxyl radicals by the cigarette smoke. These hydroxyl radicals react with unsaturated fatty acids of the membrane phospholipids to generate organic acid-free radicals which in turn react with oxygen to form lipid hydroperoxides. The latter act as free radicals, initiating an autocatalytic chain reaction and cause extensive membrane damage24. These are unstable compounds that tend to decompose rapidly to form secondary products which are responsible for deleterious effects of lipid peroxidation. These include alkanes, like ethane, pentane; and aldehydes, such as malondialdehyde. Among smokers, the thiobarbituric acid reactive substances (TBARS) levels were higher among those with COPD compared with those without COPD. These observations reflect increased lipid peroxidation because of oxidative stress due to smoking25.
Risk factors of COPD in non-smokers may include genetic factors (such as alfa-1-antitrypsin deficiency), long-standing asthma, exposure to exogenous ROS viz. outdoor air pollution (from traffic and other sources), environmental smoke exposure (ETS), biomass smoke, occupational exposure, diet, recurrent respiratory infection in early childhood, tuberculosis and several such factors. In Asian region, indoor/outdoor air pollution and poor socio-economic status may play important roles in the pathogenesis of non-smoking-related COPD25.
Alveolar macrophages of higher granular density are more prevalent in the lungs of smokers and leading to increased O2· − production26,27. The generation of ROS in epithelial lining fluid may be further enhanced by the presence of increased amounts of free ions in the airspaces in smokers28,29. An association between hydrogen peroxide (H2O2), superoxide anion, and hydroxyl radical is generate as byproducts of oxygen metabolism are released by peripheral blood neutrophils, resulting in bronchial hyper-responsiveness in patients with COPD30.
Cell-derived ROS: In a metabolically active cell, the chief ROS-generating enzymes are NADPH oxidase, haem peroxidases (myeloperoxidase, MPO) or eosinophil peroxidase (EPO)31. Reactive nitrogen species (RNS), superoxide anion and H2O2 are also generated by mitochondria32,33.
Environmental sources of ROS: Superoxide and nitric oxide generated from cigarette smoke34 and atmospheric ozone exposure cause lipid peroxidation and inflammation of airway epithelium35. Further, heavy metal ions can cause damage to cellular nuclear proteins and DNA36.
Role of TRP channels in chronic lung inflammation and onset of COPD
Exogenous pollutants act as lachrymatory agents, thus stimulating the corneal and airway passage nerves which are densely innervated by peripheral sensory nerve fibers (PSNF). These work by irritating mucous membranes that trigger the lachrymator reflex, ocular pain and blepharospasm after exposure to such noxious chemical stimulus37. Most TRP channels are located on plasma membranes (except nuclear membrane and mitochondrial membrane), where these have an essential role in the influx and/or transcellular transport of Ca2+, Mg2+ and trace metal ions38. The mechanism of involvement of various TRP channels in various disease states is still not clear, however, the channels involved in patients with respiratory distress is shown in Table I.
Table I.
Involvement of various TRP channels in patients with respiratory distress
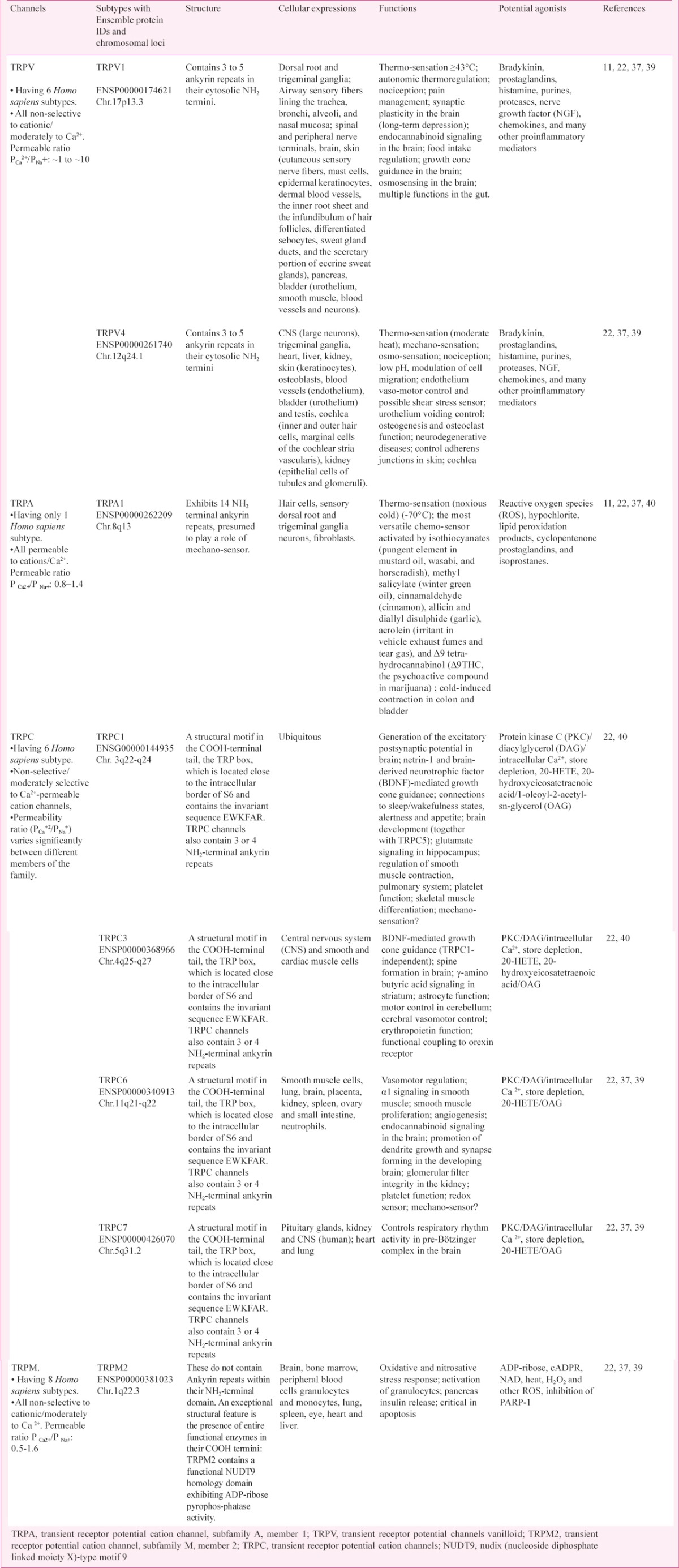
Other indications of the involvement of TRPs in several diseases come from correlations between the levels of channel expression and disease symptoms or from the mapping of TRP-encoding genes to susceptible chromosome regions41.
A study examining the role of TRPC6 on human alveolar macrophages and lung tissue macrophages reports an increase in the expression of TRPC6 mRNA in COPD patients as compared to other TRPs39. The pathophysiological role for non-neuronal TRPV1/TRPA1 is more apparent in the case of inflammation, infection and immunity. Though the effects of these TRP channels are non-neuronal, their impact is indirectly upon pain and/or neurogenic inflammation. While TRPV1 or TRPA1 activation causes airway neurogenic inflammation, non-neuronal TRPA1 produces an additional, prominent non-neurogenic inflammatory response, which may contribute to inflammatory airway diseases, offering a novel interpretation for the role of TRPA1 that could be a novel target for the treatment of inflammatory respiratory diseases40. TRP channels also play a role in the removal of foreign objects and irritants from lungs as Fig. 3 shows a simplified schematic diagram illustrating mechanisms and cell types with TRP expression in COPD progression. In lungs these are either found in sensory neurons (TRPA1 and TRPV1) and their activation results in altered vagal output and hence change respiratory pattern, blood flow and coughing or located in alveolar macrophages (TRPV2 and TRPV4) for initiation of an immune response42.
Fig. 3.
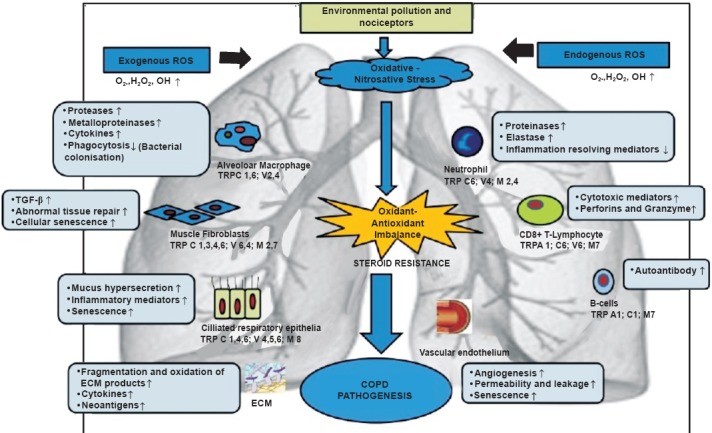
A simplified schematic diagram illustrating mechanisms and cell types with transient receptor potential (TRP) expression which are thought to be involved in the pathogenesis of COPD. Inhaled exogenous pollutants enter lungs causing oxidative/nitrative stress, resulting in activation of various cell types alongwith TRP expression. ROS reactive oxygen species; ↑ increase; ↓ decrease; OH∙ hydroxyl radical; H2O2 hydrogen peroxide; TGF-β, transforming growth factor- β; ECM, extra cellular matrix; TRPCs, transient receptor potential canonical.
TRPM4 and TRPM5, the Ca2+- activated non-selective cation channels represent a molecular candidate for a large number of functionally similar Ca2+-activated cation channels found in native cell types (phagocytic naïve cells) as a typical fingerprint, its lack of permeability for Ca2+ in the TRP superfamily. However, Ca2+ is a major regulator of their activity since both channels are activated by a rise in internal Ca2+ 43. The function of TRPM4 has been suggested to result in inappropriate release of cytokines triggering immunological hyperresponsiveness, proinflammatory conditions, or allergy22.
A potential mechanism to explain chronic cough in conditions where there is repeated or severe irritant-induced airway epithelial injury (e.g. RADS) is through persistent TRPV1 channel activation (e.g. TRPV1pathy) with accumulation of inflammatory mediators, tachykinins, and the release of neurotrophins leading to persistent cough and airway inflammation. The significance of this observation is that, if proven, it may provide new therapeutic approaches for the treatment of chronic cough44.
TRPA1 is activated by tear gas agents, chlorine, reactive oxygen species, and noxious constituents of smoke and smog, initiating irritation and airway reflex responses. Together with TRPV1, the capsaicin receptor, TRPA1 may contribute to chemical hypersensitivity, chronic cough, and airway inflammation in asthma, COPD, and reactive airway dysfunction syndrome45. TRPA1 agonist activity of a given endogenous compound depends on its site of origin, reactivity, membrane permeability and reach, tissue antioxidant levels, and many other factors. TRPA1 is likely to be activated or sensitized by a diverse cocktail of simultaneously present oxidants and electrophiles than by a single predominant agonist. This idea relates to the concept of the “inflammatory soup” describing the diverse mix of chemical and biological mediators promoting sensory neuronal sensitization and activation during tissue injury and inflammation46. It appears that endogenous oxidants and electrophiles need to be added to this particular mix45.
TRPA1 and TRPV1: an overprotective mechanism?: TRPA1 is expressed in vagal sensory nerves and in the sensory nerve fibers originating from the dorsal root ganglion (DRG), thus innervating the airways. TRPA1 receptors are co-expressed with TRPV1 in a subpopulation of primary afferent somatosensory neurons innervating mouse airways and containing the neuropeptides SP, NKA and CGRP47. TRPA1 in the airway nerve endings is activated by pungent plant constituents such as allicin (garlic), cinnamaldehyde (cinnamon), and isothiocyanates (horseradish)48,49,50 as well as from several volatile irritants and air pollutants such as formaldehyde51 and acrolein48. Various endogenous byproducts derived from peroxidation of membrane phospholipids, such as 4-hydroxy-2-nonenal (4-HNE) and 4-oxononeal (4-ONE) have been described to activate TRPA1 and, as a consequence, produce pain and neurogenic inflammation52. Given the pathophysiological relevance of the endogenous activators of TRPA1, a role for this receptor in mediating pulmonary inflammatory processes characterized by oxidative stress can be predicted53.
Among TRPA1 activators are some of the most harmful environmental/industrial irritants which, when inhaled, may cause a number of adverse reactions in the lung/airways, collectively known as RADS (reactive airways dysfunction syndrome)54. Therefore, TRPA1 can be considered either a chemoreceptor for environmental irritants or a mediator of neurogenic inflammation responses elicited by endogenous harmful stimuli in the airways.
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) activators of TRP channels: Pulmonary oxidant burden can be increased by infiltrating eosinophils, neutrophils and macrophages into alveolar space thereby generating ROS burden such as oxygen radicals (O2·–), hydrogen peroxide (H2O2) and hypochlorite. On imbalance between oxidants and antioxidants (oxidative stress), ROS produced in excess generate highly reactive nitrogen species (RNS), like peroxynitrite (ONOO-) and nitrogen dioxide (NO2), by reacting with nitric oxide (NO) which is overproduced in inflamed tissues, thus causing nitrative stress which is associated with various airway diseasess55.
A typical target of ROS/RNS signaling is Ca2+ channels (TRPA1/V1) which mediate both long-term as well as acute cellular responses to oxidative stress. RNS similar to ROS directly attack unsaturated fatty acids (e.g. oleic acid) of membrane proteins, by adding NO2 groups (nitration) to the organic acids56, thereby generating highly reactive electrophilic compounds such as nitro-oleic acid57. RNS have the ability to directly activate TRPA1 by oxidation of key cysteine residues within the N-terminal sequence of the channel58. Similarly, ROS is also known to activate TRPA1 by oxidative modification of the key cysteines of the channel. TRPA1 is not only sensitive to electrophiles, but is also activated by oxidizing agents entering the airways.
Oxidative stress a well known indicator of acute and chronic airway inflammation, produces ROS during oxidant exposures and through catalysis by inhaled toxic particulates thus results in inflammation caused by infiltrating macrophages and neutrophils. Similar to the oxidant gas chlorine, ROS such as hydrogen peroxide excite airway sensory nerve fibers, resulting in respiratory depression59.
There are several RNS derived from NO viz. peroxynitrite (ONOO-) with high biological activity, formed from NO and O2- One such product is 3-nitrotyrosine (3-NT), which can be used as a marker of ONOO- formation in vivo. Lipid peroxidation is an autocatalytic pathway that causes oxidative damage to cell membranes and results in the release of reactive lipid aldehydes. These cytotoxic metabolites such as 4-HNE diffuse from the site of production and react with cellular macromolecules. These in turn activate TRPA1and elicit hypersensitive reactions. Thus, ROS signals may alter behaviour of TRP channels60.
Antagonists/drugs modulating TRP expression
Intense efforts have been carried out to design both competitive and non-competitive TRPV1 antagonists. Thus, drugs to modify TRP channels may be useful targets in asthma, COPD and other airway diseases. Antagonists that bind to the agonist binding site, and lock the channel in the closed, non-conductive state are known as competitive antagonists61. On the other hand, antagonists that interact with additional binding sites on the receptor structure preventing receptor opening by the agonist or in other words, blocking its aqueous pore fall under the category of non-competitive antagonists. Non-competitive antagonists acting as open channel blockers are therapeutically attractive because of their recognition of over-activated TRPV1 channels, which can reduce the potential of unwanted side effects, preventing receptor opening by the agonist or blocking its aqueous pore60. Despite the structural heterogeneity of TRPV1 antagonists, a general model for their binding interaction with TRPV1 antagonists has been proposed62. In brief, the unifying structural feature of TRPV1 antagonists is the presence of a central hydrogen-bond acceptor/donor motif flanked by a lipophilic side chain on one side and a more polar aromatic group that incorporates a hydrogen-bond acceptor on the other. A hydrogen-bonding motif is present in most known TRPV1 antagonist structures63. In the classic antagonists, the central core can act as an H-bonding donor and acceptor, whereas in some non-classic antagonists, it can only act as an H-bonding acceptor. Inflammatory response to cigarette smoke is mediated entirely by neuronal TRPA1. Therefore, TRPV1 and TRPA1 antagonists may represent potential antitussive and anti-inflammatory therapeutics for respiratory airway diseases, as illustrated by Vriens and co-workers62 (Table II).
Table II.
Antagonists representing potential antitussive and anti-inflammatory therapeutics for various respiratory airway diseases

Although the development of antagonists has been slow, but there is increasing interest in TRPA1 being used as a therapeutic target. One of these is the non-selective cation channel blocker ruthenium red, which, although described to be a potent blocker, is not selective and blocks several of the TRP channels, including TRPV1. Identification of toxicity issues has prevented the development of this compound. Interestingly, (±) camphor and related compounds have also been reported to be weak TRPA1 antagonists64,65. Potential therapeutic utility of TRPV1 antagonists in somatic pain, migraine, respiratory disease, bladder and gut related pain has been suggested66,67.
In 2007, GlaxoSmithKline disclosed its Phase 1 results obtained with its selective and potent TRPV1 antagonist, SB-705498. In the first part of the study, single doses of SB- 705498 ranging from 2 to 200 mg did not display efficacy in the capsaicin-evoked flare test68. However, in the second part of the study, a single oral dose of 400 mg SB-705498 substantially reduced pain from cutaneous capsaicin challenge (0.075% capsaicin cream applied to the forearm) compared to placebo. Importantly, SB-705498 did not show any serious adverse effects in the study. Topical formulations of SB-705498 have been recently evaluated in two Phase 2 clinical trials in chronic cough and non allergic rhinitis patients69. Another Phase 1 trial was started in 2011 with topical formulation in pruritus70.
Gene controlling exacerbation of COPD
An interaction between gene and various environmental factors may be responsible for distinct aberrant pathophysiological processes/pathways. Like many chronic complex diseases, it has been difficult to unravel the genetic predisposition and pathogenetic mechanisms for COPD. Pulmonary function is influenced by heredity71,72. There is also familial aggregation of COPD, indicating probable heritability of risk factors for the disease72. It is still obscure how genetic factors contribute to the development and progression of COPD. Exogenous pollutants have pleotrophic effects on human bronchial epithelium as they coordinate recruitment of pivotal inflammatory cells in several pathologies, including chronic COPD73.
Case control studies suggested associations between COPD and polymorphisms of the alpha1-antitrypsin, tumour necrosis factor alpha (TNFα), and surfactant protein B genes74,75,76. In a study on Central Indian population, genetic alteration has been found to be one of the late effects of industrial pollutants, thereby increasing the susceptibility to COPD. This study showed microsatellite instability (MSI) to be weakly associated with smoking, age, and exposure to exogenous toxins, which are instrumental in the rise of COPD cases77.
Candidate genes involved in established pathogenetic pathways have been investigated for their association with COPD (oxidative stress, protease-anti-protease imbalance, chemokines, cytokines, and extracellular matrix breakdown and repair). It has been documented that the importance of genetic factors in the development of COPD especially in the young, is attributed to alpha-1-antitrypsin (A1AT) deficiency, a protein required for inhibition of neutrophil elastase, proteinase 3 (PR3), cathepsin G, kallikreins, matriptase, caspase-3 and ADAM-173,78. A1AT encoded by the SERPINA1 gene, is a member of the serpine protease inhibitor super family (SERPIN), and is mainly produced by hepatocytes. Some candidate genes have been identified, such as ADRB2, CHRNA5, CSF3, EPHX1, GSTO2, HMOX1, MMP12, SERPINA1, SERPINE2, SFTPB, SMOC2, TGFB1, TNF, IL1RN/IL1B, IL4R, IL6, IL8, IL10, INF-γ, ADAM33, MMP1, SOD3, GSTP1, GSTT1 and GSTM179,80,81,82,83,84.
Studies on TRPV4P19S, a human genetic polymorphism previously identified as a COPD susceptibility locus21, displayed an increase in MMP-1 (matrix-metalloproteinase) activation via increased Ca2+ influx, providing a mechanistic link between human airway epithelium signaling, airway disease, and air pollution. The TRPV4 is expressed in ciliated bronchial epithelial cells where it is believed to play a pivotal role in regulating ciliary movements85. This function of TRPV4 is one possible mechanism that may explain the genetic association between multiple TRPV4 SNPs (single nucleotide polymorphismsims) and COPD. The COPD-associated TRPV4P19S SNP located in the coding region (C144T) in exon 2, at the N-terminal of the ankyrin repeats, results in change in charge from non-polar to polar causing less conductivity, and thus displays gain-of-function characteristics in human airway epithelial cells, where it increases Ca2+ influx and secretion of matrix metalloprotease-1 in response to diesel exhaust21,86,87. The increased intracellular Ca2+ may compromise ciliary movements, leading to accumulation of harmful particles in the lungs. If clinically safe and effective inhalable TRPV4 antagonists are synthesized, these can be given preventively to at risk patients (e.g. heavy smokers) who carry COPD-associated TRPV4 mutations.
Overall this suggests a clinical benefit of inhibiting TRPV4 function in the treatment of altered lung function. Additional benefit is suggested in inhibiting TRPV4 function in pulmonary-based pathologies presenting with symptoms including lung oedema/congestion, infection, inflammation, pulmonary remodelling and/or altered airway reactivity. A genetic link between TRPV4 and COPD has been identified21 suggesting potential efficacy for TRPV4 modulation in treatment of COPD with or without coincident emphysema. Expression of few TRP channels is enhanced in airway disease. Thus making them promising targets for the treatment of chronic cough88.
Further studies are being carried on COPD patients by focusing primarily on genes involved in protease-anti-protease and oxidant-antioxidant pathways. However, given the diverse pathways (such as inflammation, innate immunity, cell death, matrix repair mechanisms and lung development) involved in COPD pathogenesis it is likely that other genes contribute as well78.
Conclusions & perspectives
TRP channels are emerging as vital cellular switches that allow us to sense our environment. Their multifunctional role as cellular sensors is important in understanding human pathophysiology and consequently disease development as well as progression. These can be potential drugs to curb the rampant progress of COPD especially in a sub-continent like India. Further understanding of the effects and roles of TRP channels/ROS in basic cellular functions as transduction of Ca2+ ions or amplification of pro-inflammatory and immunological responses, signaling pathways, activation of transcription factors, chromatin remodelling and gene expression will provide important information regarding basic pathological processes contributing to chronic lung diseases such as COPD. Identification of genes that predispose to the development of chronic lung diseases may identify novel therapeutic targets such as the genotypic/phenotypic disposition of the population at large.
In summary, the oxidative stress is associated with the pathogenesis of various chronic lung diseases. Modulation of selected TRP channels may have beneficial effects at targeting key features of these respiratory diseases including airways inflammation, airways hyper-reactivity, mucus secretion and cough. Blockers of TRP channels may offer a useful strategy for curbing the exaggerated chemosensory responses accompanying these conditions, thereby reducing sensory irritation and, potentially, prevent adverse long-term health effects elicited by neurogenic inflammatory mechanisms.
Acknowledgment
Authors thank Dr Manoj Pandey, Director, Bhopal Memorial Hospital and Research Centre, Bhopal for technical suggestions.
Footnotes
Conflicts of Interest: None.
References
- 1.Decramer M, Janssen W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379:1341–51. doi: 10.1016/S0140-6736(11)60968-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Global initiative for chronic obstructive lung disease. [accessed on October 8, 2012]. Available from: http://www.goldcopd.org/uploads/users/files/GOLD_Report_2011_Feb21.pdf .
- 3.Vijayan VK. Chronic obstructive pulmonary disease. Indian J Med Res. 2013;127:251–69. [PMC free article] [PubMed] [Google Scholar]
- 4.Pena VSM, Miravitlles MM, Gabriel RM, Jimenez-Ruiz CA, Villasante C, Masa JF, et al. Geographic variations in prevalence and underdiagnosis of COPD: Results of the IBERPOC Multicentre Epidemiological Study. Chest. 2000;118:981–9. doi: 10.1378/chest.118.4.981. [DOI] [PubMed] [Google Scholar]
- 5.Menezes AM, Perez-Padilla R, Jardim JR, Muiño A, Lopez MV, Valdivia G, et al. Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study): a prevalence study. Lancet. 2005;366:1875–81. doi: 10.1016/S0140-6736(05)67632-5. [DOI] [PubMed] [Google Scholar]
- 6.Caballero A, Torres-Duque CA, Jaramillo C, Bolívar F, Sanabria F, Osorio P, et al. Prevalence of COPD in five Columbian cities situated at low, medium, and high altitude (PREPOCOL Study) Chest. 2008;133:343–9. doi: 10.1378/chest.07-1361. [DOI] [PubMed] [Google Scholar]
- 7.Buist AS, McBurnie MA, Vollmer WM, Gillespie S, Burney P, Mannino DM, et al. International variation in the prevalence of COPD (The BOLD Study): a population-based prevalence study. Lancet. 2007;370:741–9. doi: 10.1016/S0140-6736(07)61377-4. [DOI] [PubMed] [Google Scholar]
- 8.Jindal SK, Aggarwal AN, Chaudhry K, Chhabra SK, D'souza GA, Gupta D, et al. Tobacco smoking in India: Prevalence, quit-rates and respiratory morbidity. Indian J Chest Dis Allied Sci. 2006;48:37–42. [PubMed] [Google Scholar]
- 9.Kumar R, Vijayan VK. Smoking cessation programs and other preventive strategies for chronic obstructive pulmonary disease. J Assoc Physicians India. 2012;60:53–6. [PubMed] [Google Scholar]
- 10.Mach WJ, Thimmesch AR, Pierce JT, Pierce JD. Consequences of hyperoxia and the toxicity of oxygen in the lung. Nurs Res Pract. 2011 doi: 10.1155/2011/260482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bessac BF, Jordt SE. Sensory detection and responses to toxic gases Mechanisms, health effects, and countermeasures. Proc Am Tho Soc. 2010;7:269–77. doi: 10.1513/pats.201001-004SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baraniuk JN, Kim D. Nasonasal reflexes, the nasal cycle, and sneeze. Curr Allergy Asthma Rep. 2007;7:105–11. doi: 10.1007/s11882-007-0007-1. [DOI] [PubMed] [Google Scholar]
- 13.Baraniuk JN, Lundgren JD, Goff J, Mullol J, Castellino S, Merida M, et al. Calcitonin gene-related peptide in human nasal mucosa. Am J Physiol Lung Cell Mol Physiol. 1990;258:L81–8. doi: 10.1152/ajplung.1990.258.2.L81. [DOI] [PubMed] [Google Scholar]
- 14.Petersson G, Malm L, Ekman R, Hakanson R. Capsaicin evokes secretion of nasal fluid and depletes substance P and calcitonin gene-related peptide from the nasal mucosa in the rat. Br J Pharmacol. 1989;98:930–6. doi: 10.1111/j.1476-5381.1989.tb14623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor-Clark TE, Undem BJ. Sensing pulmonary oxidative stress by lung vagal afferents. Respir Physiol Neurobiol. 2011;178:406–13. doi: 10.1016/j.resp.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KP, Choi S, Hong JH, Ahuja M, Graham S, Ma R, et al. Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1) J Biol Chem. 2014;289:6372–82. doi: 10.1074/jbc.M113.546556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor-Clark T, Undem BJ. Transduction mechanisms in airway sensory nerves. J Appl Physiol. 2006;101:950–9. doi: 10.1152/japplphysiol.00222.2006. [DOI] [PubMed] [Google Scholar]
- 18.Carr MJ, Kollarik M, Meeker SN, Undem BJ. A role for TRPV1 in bradykinin-induced excitation of vagal airway afferent nerve terminals. J Pharmacol Exp Ther. 2003;304:1275–9. doi: 10.1124/jpet.102.043422. [DOI] [PubMed] [Google Scholar]
- 19.Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns (4, 5) P2-mediated inhibition. Nature. 2001;411:957–62. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- 20.Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, et al. Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA. 2002;99:10150–5. doi: 10.1073/pnas.152002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu G, Gulsvik A, Bakke P, Ghatta S, Anderson W, David AL, et al. ICGN Investigators. Association of TRPV4 gene polymorphisms with chronic obstructive pulmonary disease. Hum Mol Genet. 2009;18:2053–62. doi: 10.1093/hmg/ddp111. [DOI] [PubMed] [Google Scholar]
- 22.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 23.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Rahman I, Morrison D, Donaldson K, MacNee W. Oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med. 1996;154:1055–60. doi: 10.1164/ajrccm.154.4.8887607. [DOI] [PubMed] [Google Scholar]
- 25.Premanand R, Kumar S, Mohan A. Study of thiobarbituric reactive substances and total reduced glutathione as indices of oxidative stress in chronic smokers with and without chronic obstructive pulmonary disease. Indian J Chest Dis Allied Sci. 2007;49:9–12. [PubMed] [Google Scholar]
- 26.Drath DB, Larnovsky ML, Huber GL. The effects of experimental exposure to tobacco smoke on the oxidative metabolism of alveolar macrophages. J Reticul Soc. 1970;25:597–604. [PubMed] [Google Scholar]
- 27.Schaberg T, Klein U, Rau M, Eller J, Lode H. Subpopulation of alveolar macrophages in smokers and nonsmokers: relation to the expression of CD11/CD18 molecules and superoxide anion production. Am J Respir Crit Care Med. 1995;151:1551–8. doi: 10.1164/ajrccm.151.5.7735614. [DOI] [PubMed] [Google Scholar]
- 28.Mateos F, Brock JF, Perez-Arellano JL. Iron metabolism in the lower respiratory tract. Thorax. 1998;53:594–600. doi: 10.1136/thx.53.7.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapenna D, Gioia SD, Mezzetti A, Ciofani G, Consoli A, Marzio L, et al. Cigarette smoke, ferritin, and lipid peroxidation. Am J Respir Crit Care Med. 1995;151:431–5. doi: 10.1164/ajrccm.151.2.7842202. [DOI] [PubMed] [Google Scholar]
- 30.Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28:219–42. doi: 10.1183/09031936.06.00053805. [DOI] [PubMed] [Google Scholar]
- 31.Hayashi Y, Yawa S, Nishimura M, Fukuyama N, Ichikawa H, Ohtake S, et al. Peroxynitrite, a product between nitric oxide and superoxide anion, plays a cytotoxic role in the development of post-bypass systemic inflammatory response. Eur J Cardiothorac Surg. 2004;26:276–80. doi: 10.1016/j.ejcts.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 32.Churc DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–26. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, Sont JK, et al. Ozone-induced inflammation assessed in sputum and bronchial lavage fluid from asthmatics: a new non invasive tool in epidemiologic studies on air pollution and asthma. Free Radic Biol Med. 1999;27:1448–54. doi: 10.1016/s0891-5849(99)00191-4. [DOI] [PubMed] [Google Scholar]
- 34.Nightingale JA, Rogers DF, Barnes PJ. Effect of inhaled ozone on exhaled nitric oxide, pulmonary function, and induced sputum in normaland asthmatic subjects. Thorax. 1999;54:1061–9. doi: 10.1136/thx.54.12.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Comhair SA, Thomassen M, Erzurum SC. Differential induction of extracellular glutathione peroxidase and nitric oxide synthase 2 in airways of healthy individuals exposed to 100% O(2) or cigarette smoke. Am J Respir Cell Mol Biol. 2000;23:350–4. doi: 10.1165/ajrcmb.23.3.4076. [DOI] [PubMed] [Google Scholar]
- 36.Lima ES, Abdalla DSP. Peroxidação lipídica: mecanismos e avaliação em amostras biológicas”. Brazilian J Pharmaceutical Sci. 2001;37:293–303. [Google Scholar]
- 37.Ferguson JS, Alarie Y. Long term pulmonary impairment following a single exposure to methyl isocyanate. Toxicol Appl Pharmacol. 1991;107:253–68. doi: 10.1016/0041-008x(91)90207-u. [DOI] [PubMed] [Google Scholar]
- 38.Alarie Y, Ferguson JS, Stock MF, Weyel DA, Schaper M. Sensory and pulmonary irritation of methyl isocyanate in mice and pulmonary irritation and possible cyanide like effects of methyl isocyanate in guinea pigs. Environ Health Perspect. 1987;72:159–67. doi: 10.1289/ehp.8772159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finney-Hayward TK, Popa MO, Bahra P, Li S, Poll CT, Gosling M, et al. Expression of transient receptor potential C6 channels in human lung macrophages. Am J Respir Cell Mol Biol. 2009;43:296–304. doi: 10.1165/rcmb.2008-0373OC. [DOI] [PubMed] [Google Scholar]
- 40.Nassini R, Pedretti P, Moretto N, Fusi C, Carnini C, Facchinetti F, et al. Transient receptor potential ankyrin 1 channel localized to non-neuronal airway cells promotes non-neurogenic inflammation. PLoS One. 2012;7:e42454. doi: 10.1371/journal.pone.0042454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nilius B, Voets T, Peters J. TRP channels in disease. Sci STKE. 2005;295:re8. doi: 10.1126/stke.2952005re8. [DOI] [PubMed] [Google Scholar]
- 42.Earley S, Gonzales AL, Crnich R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+- activated K+ channels. Circ Res. 2009;104:987–94. doi: 10.1161/CIRCRESAHA.108.189530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brooks SM. Irritant-induced chronic cough: irritant-induced TRPpathy. Lung. 2008;186:S88–93. doi: 10.1007/s00408-007-9068-0. [DOI] [PubMed] [Google Scholar]
- 44.Bessac BF, Jordt SE. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology. 2008;23:360–70. doi: 10.1152/physiol.00026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer RA, Davis KD, Cohen RH, Treede RD, Campbell JN. Mechanically insensitive afferents (MIAs) in cutaneous nerves of monkey. Brain Res. 1991;561:252–61. doi: 10.1016/0006-8993(91)91601-v. [DOI] [PubMed] [Google Scholar]
- 46.Nassenstein C, Kwong K, Taylor-Clark T, Kollarik M, Macglashan DM, Braun A, et al. Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. J Physiol. 2008;586:1595–604. doi: 10.1113/jphysiol.2007.148379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, et al. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell. 2006;124:1269–82. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 48.Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, et al. Mustard oils and cannabinoids excite sensory nerve fibers through the TRP channel ANKTM1. Nature. 2004;427:260–5. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 49.Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and Bradykinin. Neuron. 2004;41:849–57. doi: 10.1016/s0896-6273(04)00150-3. [DOI] [PubMed] [Google Scholar]
- 50.McNamara CR, Mandel-Brehm J, Bautista DM, Siemens J, Deranian KL, Zhao M, et al. TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci USA. 2007;104:13525–30. doi: 10.1073/pnas.0705924104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B, et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci USA. 2007;104:13519–24. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor-Clark TE, Undem BJ, Macglashan DW, Jr, Ghatta S, Carr MJ, McAlexander MA. Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1) Mol Pharmacol. 2008;73:274–81. doi: 10.1124/mol.107.040832. [DOI] [PubMed] [Google Scholar]
- 53.Shakeri MS, Dick FD, Ayres JG. Which agents cause reactive airways dysfunction syndrome (RADS)?. A systematic review. Occup Med (Lond) 2008;58:205–11. doi: 10.1093/occmed/kqn013. [DOI] [PubMed] [Google Scholar]
- 54.Facchinetti F, Patacchini R. The rising role of TRPA1 in asthma. Open Drug Discov J. 2010;2:71–80. [Google Scholar]
- 55.Jain K, Siddam A, Marathi A, Roy U, Falck JR, Balazy M. The mechanism of oleic acid nitration by ∙NO2. Free Radic Biol Med. 2008;45:269–83. doi: 10.1016/j.freeradbiomed.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 56.Trostchansky A, Rubbo H. Nitrated fatty acids, mechanisms of formation, chemical characterization, and biological properties. Free Radic Biol Med. 2008;44:1887–96. doi: 10.1016/j.freeradbiomed.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 57.Andersson DA, Gentry C, Moss S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci. 2008;28:2485–94. doi: 10.1523/JNEUROSCI.5369-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J Clin Invest. 2008;118:1899–910. doi: 10.1172/JCI34192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walker LM, York JL, Imam SZ, Ali SF, Muldrew KL, Mayeux PR. Oxidative stress and reactive nitrogen species generation during renal ischemia. Toxicol Sci. 2001;63:143–8. doi: 10.1093/toxsci/63.1.143. [DOI] [PubMed] [Google Scholar]
- 60.Messeguer A, Planells-Cases R, Ferrer-Montiel A. Physiology and pharmacology of the vanilloid receptor. Curr Neuropharmacol. 2006;4:1–15. doi: 10.2174/157015906775202995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357–72. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- 62.Vriens J, Appendino G, Nilius B. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol. 2009;75:1262–79. doi: 10.1124/mol.109.055624. [DOI] [PubMed] [Google Scholar]
- 63.Nassini R, Materazzi S, De Siena G, De Cesaris F, Geppetti P. Transient receptor potential channels as novel drug targets in respiratory diseases. Curr Opin Investig Drugs. 2010;11:535–42. [PubMed] [Google Scholar]
- 64.Belvisi MG, Dubuis E, Birrell MA. Transient receptor potential A1 channels. Insights into cough and airway inflammatory disease. Chest. 2011;140:1040–7. doi: 10.1378/chest.10-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rami HK, Gunthorpe MJ. The therapeutic potential of TRPV1 (VR1) antagonists: Clinical answers await. Drug Disc Today: Ther Strategies. 2004;1:97–104. [Google Scholar]
- 66.Jia Y, McLeod RL, Hey JA. TRPV1 receptor: a target for the treatment of pain, cough, airway disease and urinary incontinence. Drug News Perspect. 2005;18:165–71. doi: 10.1358/dnp.2005.18.3.892761. [DOI] [PubMed] [Google Scholar]
- 67.Chizh BA, O’Donnell MB, Napolitano A, Wang J, Brooke AC, Aylott MC, et al. The effects of the TRPV1 antagonist SB-705498 on TRPV1 receptor-mediated activity and inflammatory hyperalgesia in humans. Pain. 2007;1-2:132–41. doi: 10.1016/j.pain.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 68.Trevisani M, Gatti R. TRPV1 Antagonists as analgesic agents. Open Pain J. 2013;6:108–18. [Google Scholar]
- 69.Bareille P, Murdoch R, Denyer J, Bentley J, Smart K, Yarnall K, et al. The effects of a TRPV1 antagonist, SB-705498, in the treatment of seasonal allergic rhinitis. Int J Clin Pharmacol Ther. 2013;51:576–84. doi: 10.5414/CP201890. [DOI] [PubMed] [Google Scholar]
- 70.Redline S, Tishler PV, Rosner B, Lewitter FI, Vandenburgh M, Weiss ST, et al. Genotypic and phenotypic similarities in pulmonary function among family members of adult monozygotic and dizygotic twins. Am J Epidemiol. 1989;129:827–36. doi: 10.1093/oxfordjournals.aje.a115197. [DOI] [PubMed] [Google Scholar]
- 71.Givelber RJ, Couropmitree NN, Gottlieb DJ, Evans JC, Levy D, Myers RH, et al. Segregation analysis of pulmonary function among families in the Framingham Study. Am J Respir Crit Care Med. 1998;157:1445–51. doi: 10.1164/ajrccm.157.5.9704021. [DOI] [PubMed] [Google Scholar]
- 72.Li J, Kanju P, Patterson M, Chew WL, Cho SH, Gilmour I, et al. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ Health Persp. 2011;119:784–93. doi: 10.1289/ehp.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sandford AJ, Chagani T, Weir TD, Connett JE, Anthonisen NR, Paré PD. Susceptibility genes for rapid decline of lung function in the Lung Health Study. Am J Respir Crit Care Med. 2001;163:469–73. doi: 10.1164/ajrccm.163.2.2006158. [DOI] [PubMed] [Google Scholar]
- 74.Keatings VM, Cave SJ, Henry MJ, Morgan K, O’Connor CM, FitzGerald MX, et al. A polymorphism in the tumor necrosis factor-alpha gene promoter region may predispose to a poor prognosis in COPD. Chest. 2000;118:971–5. doi: 10.1378/chest.118.4.971. [DOI] [PubMed] [Google Scholar]
- 75.Guo X, Lin H-M, Lin Z, Montaño M, Sansores R, Wang G, et al. Polymorphisms of surfactant protein gene A, B, D, and of SP-B-linked microsatellite markers in COPD of a Mexican population. Chest. 2000;117:249S–50S. doi: 10.1378/chest.117.5_suppl_1.249s-a. [DOI] [PubMed] [Google Scholar]
- 76.Bose P, Bathri R. Association of microsatellite instability and chronic obstructive pulmonary disorder in isocyanate-exposed population of Bhopal. Indian J Hum Genet. 2012;18:172–6. doi: 10.4103/0971-6866.100754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berndt A, Leme AS, Shapiro SD. Emerging genetics of COPD. EMBO Mol Med. 2012;4:1144–55. doi: 10.1002/emmm.201100627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Molfino NA. Genetic predisposition to accelerated decline of lung function in COPD. Int J Chron Obstruct Pulmon Dis. 2007;2:117–9. [PMC free article] [PubMed] [Google Scholar]
- 79.Bose P, Bathri R. Glutathione S-Transferase gene polymorphisms (GSTT1, GSTM1, GSTP1) as increased risk factors for asthma and COPD among Isocyanate exposed population of Bhopal, India. Res J Rec Sci. 2012;1:219–23. (ISC-2011) [Google Scholar]
- 80.He J, Shumansky K, Connett JE, Anthonisen NR, Pare´ PD, Sandford AJ. Association of genetic variations in the CSF2 and CSF3 genes with lung function in smoking-induced COPD. Eur Respir J. 2008;32:25–34. doi: 10.1183/09031936.00040307. [DOI] [PubMed] [Google Scholar]
- 81.Joos L, McIntyre L, Ruan J, Connett JE, Anthonisen NR, Weir TD, Pare PD, Sandford AJ, et al. Association of IL-1beta and IL-1 receptor antagonist haplotypes with rate of decline in lung function in smokers. Thorax. 2001;56:863–6. doi: 10.1136/thorax.56.11.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He J, Connett JE, Anthonisen NR, Sandford AJ. Polymorphisms in the IL13, IL13RA1, and IL4RA genes and rate of decline in lung function in smokers. Am J Respir Cell Mol Biol. 2003;28:379–85. doi: 10.1165/rcmb.4885. [DOI] [PubMed] [Google Scholar]
- 83.He JQ, Foreman MG, Shumansky K, Zhang X, Akhabir L, Sin DD, et al. Associations of IL6 polymorphisms with lung function decline and COPD. Thorax. 2009;64:698–704. doi: 10.1136/thx.2008.111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Van Diemen C, Postma D, Vonk J, Bruinenberg M, Schouten J, Boezen HM. A disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general population. Am J Respir Crit Care Med. 2005;172:329–33. doi: 10.1164/rccm.200411-1486OC. [DOI] [PubMed] [Google Scholar]
- 85.Kang SS. Rajnikant Mishra., editor. The mutation of transient receptor potential vanilloid 4 (TRPV4) cation channel in human diseases, mutagenesis. 2012. [accessed on August 21, 2015]. Available from: http://cdn.intechopen.com/pdfs/38250/InTech-The_mutation_of_transient_receptor_potential_vanilloid_4_trpv4_cation_channel_in_human_diseases.pdf .
- 86.Tian W, Fu Y, Garcia-Elias A, Fernandez-Fernandez JM, Vicente R, Kramer PL, et al. A loss-of-function nonsynonymous polymorphism in the osmoregulatory TRPV4 gene is associated with human hyponatremia. Proc Natl Acad Sci USA. 2009;106:14034–9. doi: 10.1073/pnas.0904084106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsushima H, Mori M. Antidipsogenic effects of a TRPV4 agonist, 4-alphaphorbol 12,13-didecanoate, injected into the cerebroventricle. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1736–41. doi: 10.1152/ajpregu.00043.2005. [DOI] [PubMed] [Google Scholar]
- 88.Bonvini SJ, Birrell MA, Smith JA, Belvisi MG. Targeting TRP channels for chronic cough: from bench to bedside. Naunyn-Schmiedeberg's Arch Pharmacol. 2015;388:401–20. doi: 10.1007/s00210-014-1082-1. [DOI] [PubMed] [Google Scholar]