

Abstract
The Abelson (ABL) tyrosine kinases were identified as drivers of leukemia in mice and humans. Emerging data has shown a role for the ABL family kinases, ABL1 and ABL2, in the progression of several solid tumors. This review will focus on recent reports of the involvement of the ABL kinases in tumor progression using mouse models as well as recent data generated from genomic and proteomic studies linking enhanced expression and hyper-activation of the ABL kinases to some human cancers. Preclinical studies on small molecule inhibitors of the ABL kinases suggest that their use may have beneficial effects for the treatment of selected solid tumors.
Keywords: ABL, cancer, leukemia, metastasis, metabolism, therapy resistance
An introduction to ABL kinases
Pioneering studies on the Abelson (ABL) tyrosine kinases opened the door to seminal discoveries of the molecular basis of cancer and the development of targeted therapies. Among these was the finding that structural alterations of the cellular Abl (c-Abl, Abl1) tyrosine kinase as a consequence of viral fusion (Gag-Abl) and chromosomal translocation (BCR-ABL1) events promoted leukemia in mice and humans, respectively [1]. Studies of the cell of origin of BCR-ABL-positive chronic myeloid leukemia (CML) demonstrated its presence in the hematopoietic stem cell (HSC). The recognition that small molecule tyrosine kinase inhibitors (TKIs) could effectively treat human CML opened the door to the era of targeted cancer therapies [2]. Subsequently, the emergence of resistance to ATP-competitive inhibitors of the BCR-ABL1 kinase led to the identification of diverse drug resistance mechanisms and provided a road-map for the development of alternative therapies in the treatment of leukemias and other malignancies. Recently, the ABL family kinases, ABL1 and ABL2 (also known as Abl-related gene, Arg) have been shown to play a role in the progression of several solid tumors through activation mechanisms distinct from those involved in the generation of ABL-induced leukemias. This review will focus on the involvement of ABL kinases in solid tumors, supported by evidence from genomic and proteomic studies as well as the use of mouse tumor models for breast, kidney, intestinal and colorectal cancer.
ABL structural domains and enzymatic regulation
The ABL family of protein tyrosine kinases, ABL1 and ABL2, function to link diverse stimuli to signaling pathways controlling cell growth, survival, invasion, adhesion, and migration [3–6]. ABL1 was first discovered as the oncogene in the Abelson murine leukemia virus (v-Abl) and subsequently identified as an oncogene associated with chromosome translocations in BCR-ABL1-positive human leukemias. The Gag-Abl and BCR-ABL1 fusion proteins are constitutively active and drive cellular transformation. In contrast, the kinase activities of ABL1 and ABL2 are tightly regulated by intra- and inter-molecular interactions as well as phosphorylation [3, 7].
ABL1 and ABL2 share amino (N)-terminal regulatory and catalytic domains that are over 90% identical and include the Src homology 3 (SH3), SH2 and SH1 (tyrosine kinase) domains (Figure 1). The carboxy (C)-terminus of both ABL kinases contains a conserved filamentous (F) actin-binding domain. ABL1 contains a G-actin (globular actin)-binding domain upstream of the F-actin-binding domain, whereas ABL2 has a second internal F-actin-binding domain and a microtubule-binding domain, which are not found in ABL1 (Figure 1). The ABL kinases share conserved PXXP motifs that mediate binding to SH3 domain-containing proteins. ABL1 has three nuclear localization signal (NLS) motifs and one nuclear export signal (NES) in its C-terminus, which mediates its nuclear-cytoplasmic shuttling. In contrast, ABL2, which lacks the NLS motifs, localizes primarily to the cytoplasm and preferentially accumulates at F-actin-rich sites in the cell periphery, focal adhesions, adherens junctions, invadopodia and phagocytic cups [6]. Alternative splicing of the first exons produces various ABL1 and ABL2 isoforms with distinct N-terminal sequences (Figure 1). The 1b isoforms of both ABL kinases contain an N-terminal glycine that is myristoylated, while the 1a variants lack this site and the corresponding modification.
Figure 1. Mechanisms for activation of ABL family kinases in Leukemia and Solid Tumors.
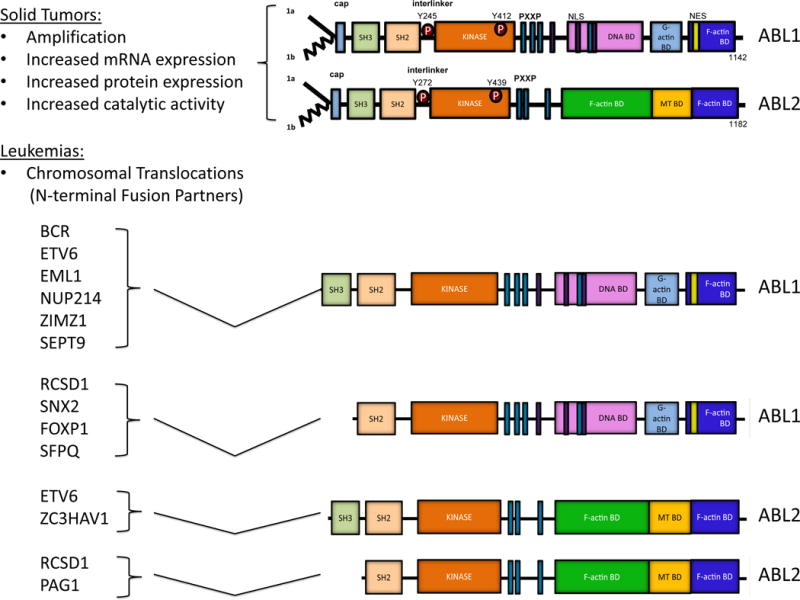
Schematic representation of ABL1, ABL2, and the various ABL1 and ABL2 fusion proteins that arise as a consequence of chromosome translocations in leukemias. In solid tumors, ABL kinases are up-regulated through various mechanisms including amplification, increased mRNA expression, enhanced protein expression, and/or hyper-activation of catalytic activity. In leukemias, Abl kinases are activated mainly through chromosomal translocation events. Various N-terminal fusion partners generate chimeric proteins that retain both the SH3 and SH2 domains, or just the SH2 domain of ABL1 and ABL2 as indicated. The distinct partner sequences fused to the N terminus of the ABL kinases promote enhanced kinase and transforming activities by disrupting inhibitory intramolecular interactions, providing sequences that facilitate oligomerization, enhancing tyrosine phosphorylation and or by recruiting the chimeric kinases to distinct subcellular sites and protein complexes.
Multiple intra-molecular interactions mediate ABL autoinhibition and include the binding of the SH3 domain to the polyproline-containing linker sequence connecting the SH2 and kinase domains, as well as interactions of the SH2 domain with the C-terminal lobe of the kinase domain (SH1), leading to the formation of a SH3-SH2-SH1 clamp structure [8]. The autoinhibited conformation of ABL kinases is stabilized by the binding of the myristoylated residue in the ABL N-terminus to a hydrophobic pocket within the C-lobe of the kinase domain in the myristoylated 1b isoform of the ABL kinases (Figure 1). Additionally, inter-molecular interactions with distinct binding partners can negatively or positively regulate ABL kinase activity [3]. Inter-molecular interactions that disrupt autoinhibitory interactions result in stabilization of the active conformation of the ABL kinases and increased enzymatic activity. In contrast, inter-molecular interactions that stabilize the inactive conformation of the ABL kinases inhibit enzymatic activity and downstream signaling. The activity of the ABL kinases can also be modulated by interactions with lipids such as phosphatidylinositol 4,5-bisphosphate (PIP2), which inhibits the ABL kinases in vitro and in cells; experimentally decreasing cellular PIP2 levels stimulates ABL kinase activity [9].
The enzymatic activity of the ABL kinases can also be regulated by tyrosine phosphorylation [3]. This modification occurs in trans for both ABL1 and ABL2, and it is referred to as “autophosphorylation”. ABL family kinases are also phosphorylated by SRC family kinases and receptor tyrosine kinases such as PDGFR. Phosphorylation of critical residues in the activation loop located at the interface between the small and large lobes of the catalytic domain of protein tyrosine kinases is required to achieve high catalytic activity. Among the tyrosine residues phosphorylated on ABL1 are Y412 in the activation loop (corresponds to ABL2 Y439) and Y245 in the SH2-kinase domain linker (corresponds to ABL2 Y272) (Figure 1). Phosphorylation of these sites stabilizes the active ABL conformation leading to enhanced signaling.
The presence of common and unique domains in ABL1 and ABL2 suggests that these kinases may exhibit overlapping as well as unique functions. Consistent with redundant roles for the murine Abl kinases, mice with global inactivation of both Abl1 and Abl2 die before embryonic day 11 [10], while the Abl1 single knockout mice are viable or exhibit perinatal lethality depending on the strain and display distinct phenotypes from those presented by the viable Abl2 global knockout mice (Box 1) [11–17]. Analysis of mice with tissue-specific deletion of Abl1 and/or Abl2 revealed they have unique and overlapping roles, depending on the cell type (Box 1) [18–21]. The unique domains present in Abl1 and Abl2 control their differential subcellular localization and/or association with distinct protein complexes leading to diverse functional roles for these kinases in various cell types.
Box 1. Physiological roles of murine Abl kinases.
Abl1 and Abl2 function redundantly in some cellular contexts, but also have unique roles during mouse development and physiology in the adult. Disruption of murine Abl1 on a mixed (129/SvEv and C57BL/6J) genetic background resulted in neonatal lethality in about 50% of the mice, a phenotype that was more severe on the C57BL/6J genetic background [11, 12]. The Abl1 knockout mice exhibit splenic and thymic atrophy, reduced numbers of B and T cells, cardiac abnormalities, and osteoporosis linked to defective osteoblast proliferation and premature senescence[11–13, 15, 72]. In contrast, Abl2 (Arg) knockout mice are viable and exhibit neuronal defects that include age-related dendrite destabilization and regression [10, 14, 17, 73]. Functional overlap by Abl1 and Abl2 kinases is supported by the embryonic lethality (by embryonic day 11) of Abl1−/− Abl2−/− mice [10]. Conditional knockout mice with tissue-specific deletion of the Abl kinases revealed unique and overlapping roles for these kinases in neuronal cells, immune cells (T cells and myeloid cells), smooth muscle cells, and cardiovascular development and function [10, 18, 20, 21, 74, 75]. For example, Abl kinases have redundant roles in mature T cells as deletion of both Abl1 and Abl2 was required to inhibit TCR-induced proliferation and cytokine production, as well as chemokine-induced T cell migration [19, 74]. In contrast, Abl1 alone is required for thymocyte differentiation and chemokine-induced migration [76]. Abl1 has a unique role in airway smooth muscle as disruption of Abl1 in these cells attenuated airway hyper-responsiveness and remodeling in a mouse model of allergen-induced asthma [75]. Distinct cellular functions of Abl1 and Abl2 might be mediated by their unique domains, differential subcellular localization and/or association with distinct protein complexes.
ABL activation in leukemias and development of targeted therapies
Chromosomal translocations are the hallmark of oncogenic activation of the ABL kinases in human leukemias [22]. Disruption of inhibitory ABL1 intra-molecular interactions in Philadelphia-positive (Ph+) human leukemias occurs as a consequence of the t(9;22)(q34;q11) chromosome translocation that generates BCR-ABL1 fusion proteins with constitutive tyrosine kinase activity (Figure 1). CML begins with a chronic phase (CP-CML) that is characterized by expansion of the myeloid lineage and retention of hematopoietic differentiation [1]. This early phase can progress to a blastic phase (BP-CML) characterized by reduced cellular differentiation and displacement of mature cells with immature blasts. The majority of BP-CML patients harbor several genetic alterations in addition to BCR-ABL1. Three different BCR-ABL1 proteins have been identified that differ in the amount of BCR sequences retained in the fusion protein leading to distinct types of leukemia: P210 BCR-ABL1 is causal in chronic myelogenous leukemia (CML); P185 BCR-ABL1 is found in 20–30% of adult and 3–5% of childhood B-cell acute lymphocytic leukemia (B-ALL); and P230 BCR-ABL1 is associated with neutrophilic-CML and rare cases of CML [23]. Oncogenic activation of ABL1 in the BCR-ABL1 fusion protein is dependent on the presence of the BCR N-terminal coiled-coil (CC) oligomerization domain. Multiple signaling pathways have been identified that function to mediate the oncogenic activity of BCR-ABL1, and include the RAS, NF- κB, PI3K-AKT, JUN, β-catenin, and STAT signaling pathways [22].
The most successful example of molecular targeted therapy to date has been the development of tyrosine kinase inhibitors (TKIs) against BCR-ABL1 for the treatment of CML in chronic phase (Table 1). The majority of CP-CML patients treated with the BCR-ABL1 inhibitor imatinib (Gleevec; STI571) as first-line therapy have durable remissions with five-year overall and progression free-survival rates approaching 90% [24]. However, imatinib is less effective for the treatment of blast crisis CML and Ph+ B cell-ALL patients. Several second- and third-generation TKIs targeting BCR-ABL1 have been approved or are under development for CML patients who are resistant or intolerant to imatinib (Table 1). Among these are dasatinib and nilotinib, which have been FDA-and European Medicines Agency (EMA)-approved as both frontline and second-line therapies, and bosutinib and ponatinib which have been FDA- and EMA-approved for second-line therapy to treat Ph+ leukemia patients with BCR-ABL1 kinase domain mutations [2]. Recently, axitinib, a vascular endothelial growth factor receptor (VEGFR) kinase inhibitor approved for second-line therapy of refractory renal cell carcinoma, was reported to potently inhibit the BCR-ABL1 (T315I) gate-keeper mutation, which confers resistance to imatinib, dasatinib and nilotinib [25]. Threonine (T) 315 is known as the gatekeeper residue, because it is found at the periphery of the nucleotide-binding site of the ABL1 kinase within the hinge region of the enzymatic cleft [26]. T315 stabilizes the binding of imatinib, dasatinib and nilotinib through a hydrophobic pocket in the active site, and thus the T315I mutation elicits complete insensitivity to these ATP-competitive inhibitors. Interestingly, axitinib preferentially inhibits the BCR-ABL1 (T315I) mutant over wild-type BCR-ABL1 [25]. Thus, axitinib might be useful for the treatment of BCR-ABL1 (T315I)-driven CML and Ph+ B-cell ALL. Ponatinib also inhibits the BCR-ABL1 (T315I) mutant. Notably, the ATP-competitive kinase inhibitors imatinib, dasatinib, nilotinib, bosutinib and ponatinib have broad target specificity and inhibit multiple tyrosine kinases in addition to ABL kinases (Table 1). Axitinib has a more restricted target specificity compared to other FDA-approved ATP-competitive inhibitors, as it only targets KIT, PDGFRα, and VEGFRs in addition to the BCR-ABL1 (T315I) mutant kinase.
Table 1.
Selective and Non-Selective ABL Kinase Inhibitors
| Name | Alternative name | Targets | Inhibitor type | Regulatory status | Year of approval | Company |
|---|---|---|---|---|---|---|
| Imatinib | Gleevec/STI571 | ABL1, ABL2, BCR-ABL1, CSF1R, DDR1, DDR2, KIT, NQO2, PDGFR1 | ATP-site, Type II | FDA approved for CML, Ph+ ALL, MDS/MPD, ASM, HES/CEL, DFSP, GIST | 2001 | Novartis |
| Dasatinib | Sprycel/BMS-354825 | ABL1, ABL2, BCR-ABL1, BLK, BTK, CSK, CSR1R, DDR1, DDR2, EGFR, ERBB2, FGR, FRK, FYN, GAK, GCK, HCK, ILK, KIT, LCK, LIMK1, LIMK2, LYN, MAP2K, MAP3K, MAP4K, PDGFR, RIPK2, SLK, SRC, SYK, TEC, TYK2, YES1, | ATP-competitive, Type I | FDA approved for CML, Ph+ ALL | 2006 | Bristol-Myers Squibb Company |
| Nilotinib | Tasigna/AMN107 | ABL1, ABL2, BCR-ABL1, CSF1R, DDR1, DDR2, KIT, NQO2, PDGFR | ATP-site, Type II | FDA approved for CML | 2007 | Novartis |
| Bosutinib | Bosulif/SKI-606 | ABL1, ABL2, BCR-ABL1, CAMK2G, CDK2, HCK, LYN, MAPKK1, MAPKK2, MAPKKK2, SRC | ATP-competitive, Type I | FDA approved for CML | 2012 | Pfizer Inc. |
| Ponatinib | Iclusing/AP24534 | ABL1, ABL2, BCR-ABL1, BLK, CSFR1, DDR1, DDR2, EPHRs, FGFR1, FGFR2, FGR, FLT3, FRK, FYN, HCK, LCK, LYN, RET, SRC, TEK, TIE2, TRKA, TRKB, TRKC, PDGFR, VEGFR1, VEGFR2, VEGFR3, YES1 | ATP-site, Type II | FDA approved for CML, Ph+ ALL | 2012 | Ariad Pharmaceuticals Inc. |
| Axitinib | Inlyta/AG013736 | BCR-ABL1 (T315I), KIT, PDGFR, VEGFR1, VEGFR2, VEGFR3 | ATP-competitive, Type I | FDA approved for Renal Cell Carcinoma | 2012 | Pfizer Inc. |
| Vandetanib | Caprelsa/ZD-6474 | ABL1, EGFR, RET, VEGFR | ATP-site, Type II | Thyroid Cancer | 2011 | AstraZeneca |
| GNF2, GNF5 | ABL1, ABL2, BCR-ABL1 | Allosteric | Not FDA approved | Novartis | ||
| ABL001 | ABL1, ABL2, BCR-ABL1 | Allosteric | Phase I Trial for CML and Ph+ ALL | Novartis |
ABL TKIs can be classified into three main classes based on their mechanism of action. The ATP-competitive inhibitors can be sub-classified into type 1 inhibitors targeting the active conformation of the kinase domain (dasatinib, bosutinib), and type 2 inhibitors targeting the inactive conformation of the kinase domain (imatinib, nilotinib, ponatinib). The third class includes the allosteric inhibitors which do not target the ATP-binding pocket, but instead bind to regulatory domains to inhibit kinase activity. Among allosteric TKIs targeting ABL are GNF2 and GNF5, which bind to the myristoyl-binding pocket in the C-lobe of the ABL kinase domain (Table 1) [27]. In contrast to ATP-competitive inhibitors that target multiple kinases, the allosteric inhibitors are highly selective for the ABL kinases. These allosteric inhibitors were shown to inhibit BCR-ABL1-driven leukemogenesis in mice and sensitize mutant BCR-ABL1 to inhibition by ATP-competitive TKIs [27]. A phase I, multi-center clinical trial with a novel allosteric inhibitor of BCR-ABL1 (ABL001) that targets the myristoyl-binding pocket is currently ongoing for patients with refractory CML or Ph+ B cell ALL (http://clinicaltrials.gov/show/NCT02081378) (Table 1).
Oncogenic activation of the ABL kinases via chromosomal translocations has also been shown to occur in Ph-negative human leukemias [28–30]. ABL1 has been identified as a fusion partner with a number of genes in T cell acute lymphoblastic leukemia (T-ALL), B-ALL, AML and other leukemias (Figure 1). The ABL kinase fusions identified in a precursor B-ALL subtype lacking the BCR-ABL1 fusion (designated Ph-like ALL) are associated with poor outcome among children and adolescents [28]. Similar to BCR-ABL1, several translocations retain the ABL1 SH3 and SH2 domains. Among these are the N-terminal fusion partners: ETV6 (TEL), EML1, NUP214, ZMIZ1, and SEPT9 [28–30]. Other translocations fuse N-terminal sequences present in RCSD1, SFPQ, FOXP1 and SNX2 to the ABL1 SH2 domain and lack the SH3 domain (Figure 1). Chimeric fusions involving the ABL2 gene have also been identified in rare leukemias. ETV6 and ZC3HAV1 are fused to ABL2 sequences upstream of the SH3 and SH2 domains, while RCSD1 and PAG1 are fused to the ABL2 SH2 domain (Figure 1). Some fusion partners encode proteins that contain coiled-coil or helix-loop-helix motifs that promote oligomerization of the resulting chimeric proteins leading to enhanced ABL kinase activity. However, the NUP-ABL1 fusion requires localization to the nuclear pore complex rather than oligomerization for enhanced transforming activity [31]. The effectiveness of the ABL TKIs for the treatment of Ph-negative leukemias associated with multiple ABL fusion partners remains to be established.
ABL kinases in solid tumors
Activation of ABL kinases in solid tumors is not linked to chromosome translocation events as found in human leukemias, but rather is driven by enhanced ABL1 or ABL2 expression and/or activation due to amplification, increased gene expression, enhanced protein expression, and/or increased enzymatic activity in response to stimulation by oncogenic tyrosine kinases, chemokine receptors, oxidative stress, metabolic stress, and/or inactivation of negative regulatory proteins [32–38].
The Cancer Genome Atlas (TCGA) and other large-scale sequencing projects report ABL amplification, somatic mutations and/or increased mRNA expression in multiple solid tumors (www.cbioportal.org). These genomic alterations are more common in ABL2 than ABL1, with ABL2 alterations observed in 24% of liver hepatocellular carcinomas, and to a lesser extent in uterine endometrioid carcinoma (20%), breast invasive carcinoma (19%), lung adenocarcinoma (15%), lung squamous cell carcinoma (12%), and kidney renal clear cell carcinoma (6%) (www.cbioportal.org). These findings are consistent with reports of elevated ABL2 expression in advanced high-grade breast, colorectal, pancreatic, renal and gastric tumors [34, 37–40]. While ABL2 amplification and increased mRNA levels are genomic alterations found in a subset of human cancers, somatic mutations of ABL1 and ABL2 in solid tumors are rare, but have been reported in lung cancer and uterine corpus endometrioid carcinoma among other cancers (www.cbioportal.org). The role of these mutations in regulating ABL oncogenic activity remains to be determined.
Oncogenic activation of ABL proteins in solid tumors is due to increased ABL tyrosine kinase activity. Enhanced activation of the ABL kinases downstream of multiple receptor tyrosine kinases (RTKs) including the platelet-derived growth factor receptor (PDGFR), the ERBB family member EGF receptor (EGFR), and the hepatocyte growth factor receptor (MET) has been reported by multiple groups [4, 41, 42]. Cancer cells expressing activated ERBB receptors exhibited rapid EGF-induced ABL kinase stimulation [43]. Subsequent studies demonstrated that ABL kinases are tyrosine phosphorylated and activated in breast, lung, colorectal, gastric, renal, and prostate cancer cells as well as in melanoma [4, 33]. The catalytic activity of the ABL kinases can be up-regulated by ligand-dependent and ligand-independent activation of RTKs in cancer cells. Activation of ABL kinases in breast cancer cells has been reported to occur downstream of the EGFR, Her2 (ERBB2), insulin-like growth factor receptor (IGFR), and the CXCR4 chemokine receptor [4, 33]. ABL1 activation downstream of ligand-activated MET was shown in gastric carcinoma and hepatocellular carcinoma cells [44], and ABL1 activation in human anaplastic thyroid carcinoma cells was induced by a constitutively active form of the receptor tyrosine kinase RET [45].
The cellular consequences of ABL family kinase activation and signaling are diverse and include changes in cell proliferation, survival, migration, and invasion. Below we provide examples of ABL-dependent regulation of these processes in solid tumors, focusing primarily on studies that use knockout/knockdown strategies or specific allosteric inhibitors targeting the ABL kinases, rather than relying on the use of ATP-competitive TKIs (imatinib, dasatinib, nilotinib, bosutinib, and ponatinib) which inhibit multiple tyrosine kinases (Table 1) [4]. Several studies have reported inhibitory and, in some cases, stimulatory effects of imatinib, nilotinib, dasatinib and other TKIs, on cancer cell proliferation, survival and motility [33, 46, 47]. However, the cellular responses to these compounds cannot be solely attributed to inhibition of the ABL kinases as these compounds target numerous kinases and some non-kinase enzymes. Furthermore, TKIs such as nilotinib, imatinib and dasatinib were shown to have off-target effects leading to the formation of BRAF/RAF1 dimers and ERK activation in several cancer cell types [48]. In contrast, paradoxical activation of RAF-ERK signaling was not induced by treatment of these cancer cells with allosteric inhibitors targeting the unique myristate-binding site in the ABL kinase domain.
ABL-dependent regulation of cancer cell proliferation
The EphB2 receptor tyrosine kinase can function as oncogene during adenoma development and as tumor suppressor in the progression of invasive colorectal cancer [49, 50]. Genetic studies with Abl1-null mice showed that Abl1 is required for EphB2-mediated proliferation in the small intestine and epithelium, as deletion of Abl1 reduced the number of proliferating cells in these tissues [49]. Inactivation of Abl1 in the Apcmin/+ mouse model of intestinal adenoma impaired EphB2-mediated tumor promotion without affecting its tumor suppressor function [49, 51]. Further, Abl1 inactivation inhibited tumor initiation by intestinal stem cells, decreased tumor load, and extended the life span of Apcmin/+ mice [51]. Interestingly, Abl1 knockdown or pharmacological inhibition in some human colon carcinoma cell lines expressing low levels of EphB2 resulted in decreased levels of cyclin D1 and impaired cell proliferation (Figure 2). Thus, Abl activity and function may become dissociated from EphB2 signaling at later stages of adenocarcinoma progression.
Figure 2. Kinase activation and signaling of ABL kinases in solid tumors.
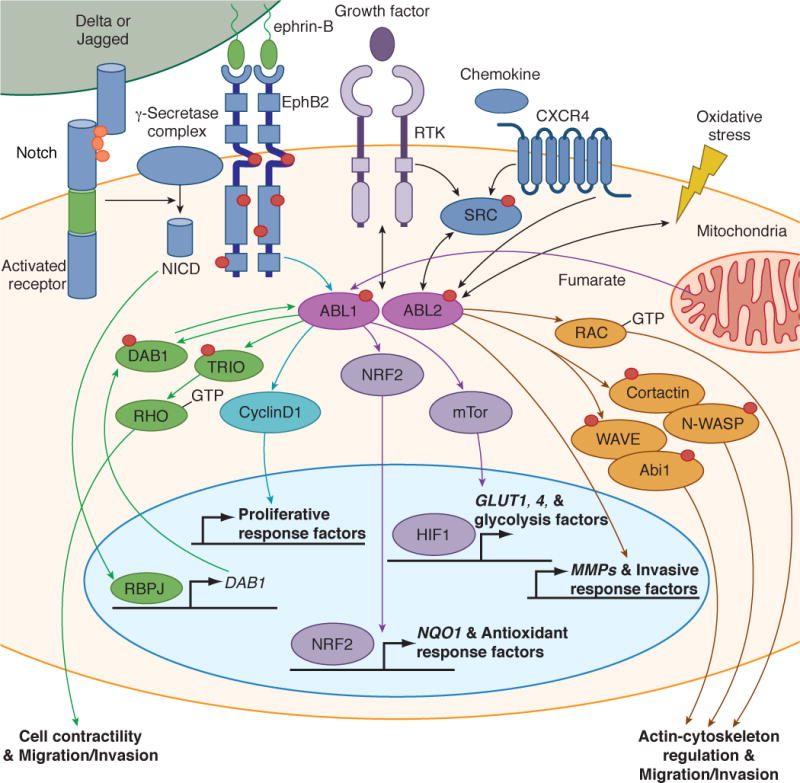
ABL kinases are activated downstream of hyperactive receptor tyrosine kinases (RTKs), chemokine receptors and SRC family kinases, or in response to oxidative and metabolic stress pathways. The activated ABL kinases promote cancer cell migration and invasion by activating multiple MMPs and actin-regulatory proteins such as Rac, cortactin, N-WASP, Abl interactor 1 (ABI1) and WAVE [59, 74, 83, 84] (orange pathways). ABL1 functions downstream of the EphB2 receptor to regulate CyclinD1 signaling to promote activation of proliferative response factors in intestinal epithelium and adenomas [49, 51] (blue pathway). The ABL1 kinase is hyperactive in FH-deficient renal cancer cells (HLRCC) in response to high fumarate levels; the activated ABL1 promotes aerobic glycolysis through activation of the mTOR- HIF1α pathway and also induces nuclear localization of the transcription factor NRF2 to induce expression of NQO1 and other antioxidant response factors in HLRCC [55] (purple pathway). Activation of NOTCH in the intestinal epithelium of Apc+/Δ716 polyposis mice promoted RBPJ-mediated transcription leading to increased levels of DAB1, a substrate and activator of the ABL kinases; the activated ABL in colorectal cancer cells induced tyrosine phosphorylation of TRIO on Y2681, leading to enhanced TRIO Rho-GEF activity [61] (green pathway).
ABL1 and ABL2 may have distinct roles in the regulation of breast cancer cell proliferation. Pharmacological inhibition or knockdown of ABL1 alone in MDA-MB-231 breast cancer cells and human mammary epithelial cells overexpressing nuclear geminin, a protein implicated in the regulation of chromosomal integrity, markedly decreased the growth of orthotopic mammary tumors [52]. In contrast, knockdown of ABL2 alone in MDA-MB-231 breast cancer cells increased primary tumor size due to enhanced cell proliferation [53]. These results suggest that ABL1 and ABL2 may have opposing effects in the regulation of cell proliferation in some breast tumor types.
ABL-mediated metabolism and oxidative stress in cancer
A recent breakthrough study revealed a critical role for ABL1 in an aggressive form of hereditary kidney cancer [54]. Patients with a germline mutation in the enzyme fumarate hydratase (FH) are susceptible to the development of hereditary leimyomatosis and renal cell carcinoma (HLRCC). FH-deficient renal tumors are highly glycolytic, accumulate high levels of fumarate, lactate, and hypoxia-stimulated transcription factor (HIF1α), and have decreased activity of AMP-activated kinase (AMPK) [55]. The ABL1 kinase was found to be hyperactive in FH-deficient renal cancer cells in response to high fumarate levels (Figure 2). Mechanistically, activation of ABL1 in HLRCC functions to promote aerobic glycolysis through activation of the mTOR-HIF1α pathway and also induces nuclear localization of the antioxidant response transcription factor NRF2 (Figure 2). Thus, high ABL1 activity enables these tumors to simultaneously meet their high energetic needs and to neutralize the elevated levels of oxidative stress generated by excess fumarate accumulation in HLRCC. Importantly, ABL1 knockdown or inhibition with either imatinib or vandetanib (an inhibitor that also targets EGFR, RET and VEGFR; Table 1), was cytotoxic to FH-deficient HLRCC [54]. The anti-tumor activity of vandetanib in these cells was shown to be ABL1-dependent. Moreover, vandetanib was shown to potently inhibit the ABL1 kinase (IC50= 15 nM) in vitro and in cells. Vandetanib alone markedly inhibited the growth of HLRCC xenografts, and combination of low-dose vandetanib with the AMPK activator metformin induced complete regression of the HLRCC tumors in 100% of the treated mice [54]. ABL kinases have been shown to be activated in response to oxidative stress and reactive oxygen species (ROS) [56]. Elevated levels of ROS are a feature characteristic of many solid tumors and are also an inevitable by-product of cellular metabolism. Thus, the data on the role for ABL1 in HLRCC suggest that ABL1 kinase inhibitors could be developed for the treatment of FH-deficient tumors and other cancers with high levels of oxidative and metabolic stress.
Role of ABL kinases in cancer cell invasion and metastasis
The progression of solid tumors require invasion of primary tumor cells into the surrounding tissue, followed by intravasation, migration, extravasation, and formation of metastases at distant sites [57]. The various steps in the metastatic cascade require dynamic remodeling of the actin cytoskeleton. ABL kinases have been shown to engage the actin polymerization machinery to promote formation of membrane protrusions, morphological changes, altered cell adhesion, migration and invasion of diverse cell types [6]. Among the various functions of the ABL kinases, regulation of cell motility has been shown to be a predominant and evolutionarily conserved role for these kinases. A requirement for ABL kinases in cancer cell motility and invasion was shown downstream of IGF-1, EGF, serum and chemokines [4]. This requirement is consistent with the localization of ABL2 to invadopodia, which are actin-rich, protrusive membrane structures that promote remodeling of the extracellular matrix during tumor invasion [58, 59]. ABL kinases promote maturation of invadopodia and are required for matrix degradation and invasion in some but not all breast cancer types [58–60]. Among the actin cytoskeleton-regulatory proteins targeted by ABL kinases at invadopodia are cortactin, N-WASP, WAVE, and the Abl interactor 1 (Abi1) adaptor protein (Figure 2). Importantly, ABL kinases regulate the expression, localization and activity of matrix metalloproteinase (MMP) during invadopodia maturation. Active ABL2 interacts with and promotes phosphorylation of the membrane type 1-matrix metalloproteinase (MT1-MMP, MMP14) and is required for its localization and function at invadopodia [58]. Both ABL1 and ABL2 kinases were shown to regulate MMPs expression through STAT3-dependent and independent pathways in melanoma cells [33]. Knockdown of ABL2 alone decreased cancer cell invasion and intravasation following implantation of MDA-MB-231 cells in the mammary fat pad [53]. A requirement for ABL kinases for invasion and metastasis of melanoma cells was also shown, which may be mediated in part by the NM23-H1 metastasis suppressor [42]. Active ABL kinases induced cathepsin-dependent lysosomal degradation of NM23-H1 in melanoma and breast cancer cells.
A recent report demonstrated a novel role for ABL kinases in promoting colorectal cancer cell invasion and metastasis by linking the activation of the NOTCH to the phosphorylation of TRIO (pY2681) leading to enhanced TRIO Rho-GEF activity and corresponding increase of Rho-GTP levels [61]. Activation of NOTCH by homozygous deletion of Aes (Amino-terminal enhancer of split) in the intestinal epithelium of Apc+/Δ716 polyposis mice resulted in enhanced RBPJ-mediated transcription leading to increased levels of DAB1, a substrate and activator of the ABL kinases. Activated ABL in colorectal cancer cells induced tyrosine phosphorylation of TRIO on Y2681, leading to enhanced TRIO Rho-GEF activity (Figure 2). Rho activation in colorectal cancer cells promoted invasion, extravasation and metastasis. Importantly, inhibition of ABL kinases in Apc/Aes compound knockout mice dramatically suppressed both invasion and intravasation incidence without affecting tumor size. These findings suggest that ABL kinases may function to link activation of other cell surface receptors to Rho signaling in different tumors. In this regard, it has also recently been shown that ABL kinases link the ligand-activated MET receptor tyrosine kinase to Rho activation required for cell scattering, tubulogenesis, migration and invasion [41].
Role for ABL kinases in cancer drug resistance
Enhanced activation of the ABL kinases has been reported in some cancers that have intrinsic or acquired resistance to chemotherapy. Hyper-activation of both ABL1 and the PDGFR was detected in aromatase inhibitor (AI)-resistant breast cancer patient specimens [62]. ABL1 expression increased at the point of relapse in AI treated patients and correlated with increased expression of the Ki67 proliferation marker. In vitro studies showed that estrogen deprivation of MCF7 breast cancer cells, which became AI resistant, was accompanied by up-regulation of PDGFR and ABL1 signaling [62]. Treatment of these cells with nilotinib, a PDGFR and ABL inhibitor, suppressed proliferation and estrogen receptor (ER)-mediated transcription, in part by destabilizing the ER protein. Down-regulation of ABL1 in some human breast cancer cell lines by RNA interference or imatinib treatment was reported to overcome resistance to fulvestrant, a compound that down-regulates ERα levels and activity [63]. Furthermore, in vitro studies using breast cancer cells resistant to lapatinib, an EGFR and ErbB2 inhibitor, showed that imatinib treatment or ABL1 depletion restored lapatinib sensitivity to these breast cancer cells [64]. While the mechanisms by which decreased ABL1 signaling sensitizes breast cancer cells to various compounds was not reported, these studies suggest that inhibition of the ABL kinases may be effective for overcoming cancer cell resistance to diverse therapeutic agents.
A role for ABL kinase inhibitors in reversing resistance to doxorubicin in breast cancer (BT-549 and MDA-MB-468) and melanoma (WM3248) cell lines has been linked to at least two pathways [65]. Imatinib blocked intrinsic resistance to doxorubicin by inhibiting STAT3-mediated cell survival and repressing NF-kB target gene expression. Additionally, imatinib prevented acquired resistance by inhibiting the increased expression of the ABCB1 drug transporter, which mediates efflux of chemotherapeutic compounds such as doxorubicin. Similar to imatinib, other ATP-competitive inhibitors (nilotinib and dasatinib) have been reported to sensitize cancer cells to cytotoxic chemotherapies and targeted TKI therapies. However, the majority of these studies were carried out with ABL TKIs and did not evaluate whether these effects were mediated specifically by inactivation of the ABL1 and/or ABL2 kinases in the cancer cells or associated cells in the tumor microenvironment (see Box 2).
Box 2. Targeting Abl kinases in endothelial cells and fibroblasts.
Endothelial cells (ECs) and cancer-associated fibroblasts contribute to tumor progression and metastasis. TKIs such as imatinib have anti-angiogenic activity. For example, imatinib treatment of a mouse model of cervical cancer impaired angiogenesis in part by blocking the function of cancer-associated fibroblasts [77]. The anti-angiogenic effects of imatinib have been largely attributed to inhibition of the PDGFR. However, Abl kinases, which are also targeted by imatinib, regulate diverse cellular processes in both ECs and fibroblasts. Conditional deletion of Abl1 in ECs in Abl2-null mice resulted in late-stage embryonic and perinatal lethality [20]. Loss of Abl kinases led to increased endothelial cell apoptosis. Abl kinases play a dual role in angiopoietin (Angpt)/Tie2 signaling by regulating both Tie2 expression and activation of Tie2-mediated pathways required for cell survival. Abl kinases are also required for induction of endothelial permeability by VEGF and other factors [78]. Inactivation of the Abl kinases with pharmacological inhibitors or genetic inactivation in mice impaired VEGF-induced vascular permeability. Recently, ABL1 was shown to interact with Neuropilin (NRP1) in human dermal microvascular ECs and link fibronectin-dependent activation of NRP1 to paxillin phosphorylation, actin remodeling and EC motility [79]. Moreover, Abl kinases regulate signaling downstream of multiple cell-surface receptors in fibroblasts. Abl kinases are activated by ligand-activated PDGF receptor leading to fibroblast proliferation and motility [80]. Abl1 can also be activated downstream of the lipid second messenger sphingosine 1 phosphate (S1P) and its receptor leading to Rac activation and cytoskeletal remodeling required for fibroblast migration and invasion. Abl1 promotes S1P-dependent reciprocal signaling between stellate cells and pancreatic cancer cells required for NF-kB activation and MMP9 production [81]. Abl1 also functions downstream of NRP1 in stromal myofibroblasts to induce integrin activation and fibronectin fibril assembly in the tumor microenvironment [82]. Thus, pharmacological inhibitors target Abl signaling not only the in tumor cells but also in the various cell types populating the tumor stroma, including ECs and fibroblasts, and may function to blunt angiogenesis through multiple pathways.
While imatinib sensitizes some breast cancer cells to apoptosis by treatment with cisplatin and other chemotherapeutic agents [66], imatinib or GNF2 treatment was reported to protect mouse oocytes to cisplatin-induced cell death [67]. The disparate responses by germ cells versus cancer cells to DNA damaging agents in the presence of ABL kinase inhibitors may be due to differential roles for ABL1 in the regulation of double-strand breaks and DNA damage signaling [68]. Further, different cellular responses may be elicited depending on the status of TP53 or its homolog TAp63, ABL1 enzymatic activity levels, ABL1 nuclear versus cytoplasmic localization, and the extent of DNA damage.
In contrast to the success of ATP-competitive inhibitors imatinib, nilotinib and dasatinib in treating BCR-ABL1-induced leukemias, treatment of diverse solid tumors with these compounds has not achieved similar success [5, 69]. The variable clinical responses to these TKIs may be due to the lack of the relevant oncogenic target, the presence of additional mutations driving the tumor, tumor heterogeneity, and/or dynamic reprograming of signaling networks in response to TKI treatment [70, 71]. An alternative mechanism that underlies the poor response to TKI therapy is the paradoxical activation of proliferative pathways as a result of unintended targeting of other kinases. This was demonstrated by the activation of BRAF/ RAF1 complexes leading to enhanced activation of the MEK-ERK pathway by nilotinib, imatinib and dasatinib in melanoma, lung, colorectal, pancreatic carcinoma cells and BCR-ABL1 TKI-resistant leukemic cells expressing activated RAS [48]. In contrast to the ABL-targeted ATP-competitive TKIs, the ABL allosteric inhibitors do not induce formation of BRAF/RAF1 dimers, and fail to elicit paradoxical activation of the MEK-ERK pathway. Thus, it is critical to identify those tumors that may benefit from therapies with selective ABL TKIs in combinations to prevent the emergence of therapy resistance.
Concluding remarks and future perspectives
The development of TKIs to treat patients with BCR-ABL1-positive leukemias is the best example of the successful application of targeted therapy. However, it is clear that the use of these drugs is inadequate for the treatment of solid tumors as monotherapies due to the complexity of mutations even in early-stage tumors, and the potential for inappropriate activation (rather than inhibition) of proliferative pathways by some TKIs with multiple protein targets. Biomarkers are needed to select those tumors dependent on ABL kinases for survival, proliferation and/or invasion (Outstanding Questions Box). Cancer cell types with hyperactivation of the ABL kinases as a consequence of amplification, enhanced expression and/or elevated kinase activity would be more likely to rely on ABL signaling for tumor progression and metastasis. Thus, these cancer subtypes might benefit from treatment with ABL-selective TKIs such as the new allosteric inhibitors, resulting in the inhibition of ABL-dependent pathways in the tumor and associated stromal cells including endothelial cells, fibroblasts and infiltrating myeloid cells (Box 2 and Outstanding Questions Box). The use of specific ABL-dependent signatures (genomic, transcriptional, or phospho-proteomic) in various tumors and associated stroma might be useful for the identification of those solid tumor types that might benefit from the use of ABL TKIs in combination with other agents to impair metastatic progression and block the development of chemo-resistance.
Outstanding questions box.
-
-
Do the ABL1 and ABL2 kinases have distinct functional roles in tumor progression and metastasis? If so, are these roles different in distinct tumor types?
-
-
What are the biomarkers that predict ABL-dependent cancer types that could benefit from the use of specific ABL kinase inhibitors?
-
-
Do ABL kinase inhibitors interfere with angiogenesis and tumor-promoting effects of cells (myeloid, fibroblasts) in the tumor microenvironment?
-
-
Can inhibition of the ABL kinases sensitize resistant solid tumors to chemotherapeutic drugs and targeted therapies in some cancer patients? If so, which pathways mediate these effects?
TRENDS BOX.
The ABL family of tyrosine kinases links diverse stimuli to signaling pathways controlling cell growth, survival, invasion, adhesion, and migration.
While the ABL kinases were originally identified as drivers of leukemia, emerging data suggest a role for these kinases in the progression of several solid tumors.
Large-scale sequencing projects report ABL amplification, somatic mutations and/or increased mRNA expression in multiple solid tumors.
Enhanced ABL activity is required for some tumors to meet their high energetic needs and to neutralize the elevated levels of oxidative stress characteristic of these tumors.
The use of ABL TKIs in combination with other agents represents a promising strategy to impair metastatic progression and prevent the development of chemoresistance in selected solid tumors.
Acknowledgments
The authors regret that due to space limitations, they could not directly cite the work of many investigators. The Pendergast laboratory is supported by grants from the National Cancer Institute, including NIH grants R01CA155160 and R01AI056266 to A.M.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wong S, Witte ON. The BCR-ABL story: bench to bedside and back. Annual review of immunology. 2004;22:247–306. doi: 10.1146/annurev.immunol.22.012703.104753. [DOI] [PubMed] [Google Scholar]
- 2.Eide CA, O’Hare T. Chronic Myeloid Leukemia: Advances in Understanding Disease Biology and Mechanisms of Resistance to Tyrosine Kinase Inhibitors. Current hematologic malignancy reports. 2015 doi: 10.1007/s11899-015-0248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 2010;3:re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greuber EK, et al. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013;13:559–571. doi: 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ganguly SS, Plattner R. Activation of abl family kinases in solid tumors. Genes & cancer. 2012;3:414–425. doi: 10.1177/1947601912458586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradley WD, Koleske AJ. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J Cell Sci. 2009;122:3441–3454. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panjarian S, et al. Structure and dynamic regulation of Abl kinases. The Journal of biological chemistry. 2013;288:5443–5450. doi: 10.1074/jbc.R112.438382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nature reviews. Molecular cell biology. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 9.Plattner R, et al. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1. Nat Cell Biol. 2003;5:309–319. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- 10.Koleske AJ, et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- 11.Schwartzberg PL, et al. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–1175. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 12.Tybulewicz VL, et al. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 13.Qiu Z, et al. c-Abl tyrosine kinase regulates cardiac growth and development. Proc Natl Acad Sci U S A. 2010;107:1136–1141. doi: 10.1073/pnas.0913131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moresco EM, et al. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:6105–6118. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li B, et al. Mice deficient in Abl are osteoporotic and have defects in osteoblast maturation. Nature genetics. 2000;24:304–308. doi: 10.1038/73542. [DOI] [PubMed] [Google Scholar]
- 16.Kua HY, et al. c-Abl promotes osteoblast expansion by differentially regulating canonical and non-canonical BMP pathways and p16INK4a expression. Nat Cell Biol. 2012;14:727–737. doi: 10.1038/ncb2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gourley SL, et al. Loss of dendrite stabilization by the Abl-related gene (Arg) kinase regulates behavioral flexibility and sensitivity to cocaine. Proc Natl Acad Sci U S A. 2009;106:16859–16864. doi: 10.1073/pnas.0902286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greuber EK, Pendergast AM. Abl Family Kinases Regulate FcOE≥R-Mediated Phagocytosis in Murine Macrophages. The Journal of Immunology. 2012;189:5382–5392. doi: 10.4049/jimmunol.1200974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu JJ, et al. Defective T cell development and function in the absence of Abelson kinases. Journal of immunology. 2007;179:7334–7343. doi: 10.4049/jimmunol.179.11.7334. [DOI] [PubMed] [Google Scholar]
- 20.Chislock EM, et al. Abl kinases are required for vascular function, Tie2 expression, and angiopoietin-1‚Äìmediated survival. Proceedings of the National Academy of Sciences. 2013;110:12432–12437. doi: 10.1073/pnas.1304188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wetzel DM, et al. The Abl and Arg kinases mediate distinct modes of phagocytosis and are required for maximal Leishmania infection. Molecular and cellular biology. 2012;32:3176–3186. doi: 10.1128/MCB.00086-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 23.Advani AS, Pendergast AM. Bcr-Abl variants: biological and clinical aspects. Leukemia research. 2002;26:713–720. doi: 10.1016/s0145-2126(01)00197-7. [DOI] [PubMed] [Google Scholar]
- 24.O’Hare T, et al. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer. 2012;12:513–526. doi: 10.1038/nrc3317. [DOI] [PubMed] [Google Scholar]
- 25.Pemovska T, et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature. 2015;519:102–105. doi: 10.1038/nature14119. [DOI] [PubMed] [Google Scholar]
- 26.Nagar B, et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571) Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
- 27.Zhang J, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature. 2010;463:501–506. doi: 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roberts KG, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawai H, et al. Functional analysis of the SEPT9-ABL1 chimeric fusion gene derived from T-prolymphocytic leukemia. Leukemia research. 2014;38:1451–1459. doi: 10.1016/j.leukres.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 30.De Braekeleer E, et al. ABL1 fusion genes in hematological malignancies: a review. European journal of haematology. 2011;86:361–371. doi: 10.1111/j.1600-0609.2011.01586.x. [DOI] [PubMed] [Google Scholar]
- 31.De Keersmaecker K, et al. Kinase activation and transformation by NUP214-ABL1 is dependent on the context of the nuclear pore. Molecular cell. 2008;31:134–142. doi: 10.1016/j.molcel.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–4391. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- 33.Ganguly SS, et al. c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene. 2012;31:1804–1816. doi: 10.1038/onc.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sos ML, et al. Predicting drug susceptibility of non-small cell lung cancers based on genetic lesions. J Clin Invest. 2009;119:1727–1740. doi: 10.1172/JCI37127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson L, et al. Renal medullary carcinoma and ABL gene amplification. J Urol. 2005;173:1883–1888. doi: 10.1097/01.ju.0000158448.56888.09. [DOI] [PubMed] [Google Scholar]
- 38.Behbahani TE, et al. Tyrosine kinase expression profile in clear cell renal cell carcinoma. World journal of urology. 2012;30:559–565. doi: 10.1007/s00345-011-0767-z. [DOI] [PubMed] [Google Scholar]
- 39.Crnogorac-Jurcevic T, et al. Expression profiling of microdissected pancreatic adenocarcinomas. Oncogene. 2002;21:4587–4594. doi: 10.1038/sj.onc.1205570. [DOI] [PubMed] [Google Scholar]
- 40.Wu CW, et al. Arg tyrosine kinase expression in human gastric adenocarcinoma is associated with vessel invasion. Anticancer research. 2003;23:205–210. [PubMed] [Google Scholar]
- 41.Li R, et al. Abl Kinases Regulate HGF/Met Signaling Required for Epithelial Cell Scattering, Tubulogenesis and Motility. PLoS One. 2015;10:e0124960. doi: 10.1371/journal.pone.0124960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fiore LS, et al. c-Abl and Arg induce cathepsin-mediated lysosomal degradation of the NM23-H1 metastasis suppressor in invasive cancer. Oncogene. 2014;33:4508–4520. doi: 10.1038/onc.2013.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones RB, et al. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 44.Furlan A, et al. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell death and differentiation. 2011;18:1608–1616. doi: 10.1038/cdd.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iavarone C, et al. Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the RET proto-oncogene. The Journal of biological chemistry. 2006;281:10567–10576. doi: 10.1074/jbc.M513397200. [DOI] [PubMed] [Google Scholar]
- 46.Matei D, et al. Imatinib mesylate (Gleevec) inhibits ovarian cancer cell growth through a mechanism dependent on platelet-derived growth factor receptor alpha and Akt inactivation. Clin Cancer Res. 2004;10:681–690. doi: 10.1158/1078-0432.ccr-0754-03. [DOI] [PubMed] [Google Scholar]
- 47.Stahtea XN, et al. Imatinib inhibits colorectal cancer cell growth and suppresses stromal-induced growth stimulation, MT1-MMP expression and pro-MMP2 activation. International journal of cancer. Journal international du cancer. 2007;121:2808–2814. doi: 10.1002/ijc.23029. [DOI] [PubMed] [Google Scholar]
- 48.Packer LM, et al. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011;20:715–727. doi: 10.1016/j.ccr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genander M, et al. Dissociation of EphB2 signaling pathways mediating progenitor cell proliferation and tumor suppression. Cell. 2009;139:679–692. doi: 10.1016/j.cell.2009.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cortina C, et al. EphB-ephrin-B interactions suppress colorectal cancer progression by compartmentalizing tumor cells. Nature genetics. 2007;39:1376–1383. doi: 10.1038/ng.2007.11. [DOI] [PubMed] [Google Scholar]
- 51.Kundu P, et al. An EphB-Abl signaling pathway is associated with intestinal tumor initiation and growth. Science translational medicine. 2015;7:281ra244. doi: 10.1126/scitranslmed.3010567. [DOI] [PubMed] [Google Scholar]
- 52.Blanchard Z, et al. Geminin overexpression promotes imatinib sensitive breast cancer: a novel treatment approach for aggressive breast cancers, including a subset of triple negative. PLoS One. 2014;9:e95663. doi: 10.1371/journal.pone.0095663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gil-Henn H, et al. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene. 2012 doi: 10.1038/onc.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sourbier C, et al. Targeting ABL1-Mediated Oxidative Stress Adaptation in Fumarate Hydratase-Deficient Cancer. Cancer Cell. 2014;26:840–850. doi: 10.1016/j.ccell.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y, et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS One. 2013;8:e72179. doi: 10.1371/journal.pone.0072179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun X, et al. Activation of the cytoplasmic c-Abl tyrosine kinase by reactive oxygen species. The Journal of biological chemistry. 2000;275:17237–17240. doi: 10.1074/jbc.C000099200. [DOI] [PubMed] [Google Scholar]
- 57.Fidler IJ. The pathogenesis of cancer metastasis: the ’seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 58.Smith-Pearson PS, et al. Abl kinases are required for invadopodia formation and chemokine-induced invasion. Journal of Biological Chemistry. 2010;285:40201–40211. doi: 10.1074/jbc.M110.147330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mader CC, et al. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011;71:1730–1741. doi: 10.1158/0008-5472.CAN-10-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chevalier C, et al. ABL Tyrosine Kinase Inhibition Variable Effects on the Invasive Properties of Different Triple Negative Breast Cancer Cell Lines. PLoS One. 2015;10:e0118854. doi: 10.1371/journal.pone.0118854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sonoshita M, et al. Promotion of colorectal cancer invasion and metastasis through activation of NOTCH-DAB1-ABL-RHOGEF protein TRIO. Cancer Discov. 2015;5:198–211. doi: 10.1158/2159-8290.CD-14-0595. [DOI] [PubMed] [Google Scholar]
- 62.Weigel MT, et al. Preclinical and clinical studies of estrogen deprivation support the PDGF/Abl pathway as a novel therapeutic target for overcoming endocrine resistance in breast cancer. Breast Cancer Res. 2012;14:R78. doi: 10.1186/bcr3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao H, et al. Overcoming resistance to fulvestrant (ICI182, 780) by downregulating the c-ABL proto-oncogene in breast cancer. Molecular carcinogenesis. 2011;50:383–389. doi: 10.1002/mc.20721. [DOI] [PubMed] [Google Scholar]
- 64.Lo YH, et al. Inhibition of c-ABL sensitizes breast cancer cells to the dual ErbB receptor tyrosine kinase inhibitor lapatinib (GW572016) Anticancer research. 2011;31:789–795. [PubMed] [Google Scholar]
- 65.Sims JT, et al. Imatinib reverses doxorubicin resistance by affecting activation of STAT3-dependent NF-kappaB and HSP27/p38/AKT pathways and by inhibiting ABCB1. PLoS One. 2013;8:e55509. doi: 10.1371/journal.pone.0055509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sims JT, et al. STI571 sensitizes breast cancer cells to 5-fluorouracil, cisplatin and camptothecin in a cell type-specific manner. Biochemical pharmacology. 2009;78:249–260. doi: 10.1016/j.bcp.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonfloni S, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nature medicine. 2009;15:1179–1185. doi: 10.1038/nm.2033. [DOI] [PubMed] [Google Scholar]
- 68.Gonfloni S. DNA damage stress response in germ cells: role of c-Abl and clinical implications. Oncogene. 2010;29:6193–6202. doi: 10.1038/onc.2010.410. [DOI] [PubMed] [Google Scholar]
- 69.Puls LN, et al. Current status of SRC inhibitors in solid tumor malignancies. The oncologist. 2011;16:566–578. doi: 10.1634/theoncologist.2010-0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stuhlmiller TJ, et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell reports. 2015;11:390–404. doi: 10.1016/j.celrep.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duncan JS, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kua HY, et al. c-Abl promotes osteoblast expansion by differentially regulating canonical and non-canonical BMP pathways and p16INK4a expression. Nature Cell Biology. 2012 doi: 10.1038/ncb2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng H, et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26:358–373. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu JJ, et al. Abl family kinases modulate T cell-mediated inflammation and chemokine-induced migration through the adaptor HEF1 and the GTPase Rap1. Sci Signal. 2012;5:ra51. doi: 10.1126/scisignal.2002632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cleary RA, et al. Role of Abl in airway hyperresponsiveness and airway remodeling. Respiratory research. 2013;14:105. doi: 10.1186/1465-9921-14-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trampont PC, et al. ShcA Regulates Thymocyte Proliferation through Specific Transcription Factors and a c-Abl-Dependent Signaling Axis. Molecular and cellular biology. 2015;35:1462–1476. doi: 10.1128/MCB.01084-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pietras K, et al. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS medicine. 2008;5:e19. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chislock EM, Pendergast AM. Abl family kinases regulate endothelial barrier function in vitro and in mice. PLoS One. 2013;8:e85231. doi: 10.1371/journal.pone.0085231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raimondi C, et al. Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. The Journal of experimental medicine. 2014;211:1167–1183. doi: 10.1084/jem.20132330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sirvent A, et al. Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biology of the cell/under the auspices of the European Cell Biology Organization. 2008;100:617–631. doi: 10.1042/BC20080020. [DOI] [PubMed] [Google Scholar]
- 81.Bi Y, et al. Sphingosine-1-phosphate mediates a reciprocal signaling pathway between stellate cells and cancer cells that promotes pancreatic cancer growth. The American journal of pathology. 2014;184:2791–2802. doi: 10.1016/j.ajpath.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yaqoob U, et al. Neuropilin-1 stimulates tumor growth by increasing fibronectin fibril assembly in the tumor microenvironment. Cancer Res. 2012;72:4047–4059. doi: 10.1158/0008-5472.CAN-11-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun X, et al. Abl interactor 1 regulates Src-Id1-matrix metalloproteinase 9 axis and is required for invadopodia formation, extracellular matrix degradation and tumor growth of human breast cancer cells. Carcinogenesis. 2009;30:2109–2116. doi: 10.1093/carcin/bgp251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–5655. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]