

Abstract
Increased activity of transforming growth factor β (TGF-β), which binds to and stimulates cell surface receptors, contributes to cancer progression and fibrosis by driving epithelial cells toward a migratory mesenchymal phenotype and increasing the abundance of extracellular matrix proteins. The abundance of TGF-β receptors at the cell surface determines cellular responsiveness to TGF-β, which is often produced by the same cells that have the receptors, and thus serves as an autocrine signal. We found that Akt-mediated phosphorylation of AS160, a RabGAP [guanosine triphosphatase (GTPase)-activating protein] promoted the translocation of TGF-β receptors from intracellular stores to the plasma membrane of mouse embryonic fibroblasts (MEFs) and NMuMG epithelial cells. Consequently, insulin, which is commonly used to treat hyperglycemia and activates Akt signaling, increased the amount of TGF-β receptors at the cell surface, thereby enhancing TGF-β responsiveness. This insulin-induced increase in autocrine TGF-β signaling contributed to insulin-induced gene expression responses, attenuated the epithelial phenotype, and promoted the migration of NMuMG cells. Furthermore, the enhanced delivery of TGF-β receptors at the cell surface enabled insulin to increase TGF-β-induced gene responses. The enhancement of TGF-β responsiveness in response to Akt activation may help to explain the biological effects of insulin, the progression of cancers in which Akt is activated, and the increased incidence of fibroses in diabetes.
INTRODUCTION
Among the extracellular factors that control signaling pathways and cell behavior, transforming growth factor-β (TGF-β) is a potent regulator of cell proliferation and differentiation of many cell types by directing the expression of hundreds of target genes. As the prototype of a family of TGF-β-related proteins, the control of cell physiology by TGF-β signaling provides the basis to understand the roles of TGF-β family proteins in tissue differentiation and homeostasis. Pathologically, increased TGF-β signaling drives aspects of fibrosis and carcinoma progression (1–3). In both contexts, increased TGF-β signaling promotes epithelial cells to acquire a more migratory, mesenchymal phenotype. This process, known as epithelial-mesenchymal transdifferentiation (EMT), contributes to fibrosis, promotes cancer cell invasion and dissemination, and enhances the generation of cancer stem cells with tumor reseeding capacity (4–6). Increased TGF-β signaling also increases the abundance of extracellular matrix proteins, which contributes to cancer stroma formation (7) and fibrosis, such as in diabetic nephropathy (8, 9).
TGF-β signaling is initiated at the cell membrane through cell surface complexes of two pairs of transmembrane receptors with dual specificity kinase specificity: the type I and type II TGF-β receptors, commonly named TβRI and TβRII. Upon ligand binding, the TβRII receptors phosphorylate and thus activate the TβRI receptors that then phosphorylate the C-terminus of Smad2 and Smad3, thereby activating these effectors and enabling them to form trimeric complexes with Smad4. Following translocation into the nucleus, the Smad complexes cooperate with DNA-binding transcription factors, such as AP-1 complexes and ETS proteins, and with coregulators to activate or repress transcription of TGF-β target genes (10–12). TGF-β receptors also activate non-Smad signaling pathways, such as MAPK pathways and PI3K-Akt signaling (13, 14).
TGF-β signaling and, in particular the Smad pathway, are extensively regulated by kinases and signaling pathways that help define the cellular TGF-β response. In addition to signaling crosstalk, cells have developed strategies to regulate the availability of TGF-β receptors at the cell surface, and control in this way the sensitivity to TGF-β and TGF-β responsiveness. Ectodomain shedding by the transmembrane metalloprotease TACE, which is activated by the Erk or p38 MAPK pathways, decreases the amount of TβRI receptors at the cell surface, and thus decreases the cell’s TGF-β responsiveness (15). Additionally, association of the decoy receptor BAMBI with TGF-β family receptors inhibits type I receptor activation in response to TGF-β family proteins (16, 17). Furthermore, high glucose at 25 mM induces a rapid increase of TβRI and TβRII at the cell surface without changing their total expression, and thus confers increased TGF-β responsiveness (18). The rapid mobilization of TGF-β receptors in response to high glucose implies the existence of an intracellular pool of TGF-β receptors, which raises the question of which signaling pathways control the TGF-β receptor availability at the cell surface and enhance TGF-β responsiveness.
Because high glucose induces Akt activation (19), we asked whether insulin, which also activates the PI3K-Akt pathway and controls glucose homeostasis (20, 21), regulates the cell surface abundance of TGF-β receptors. Insulin is a small polypeptide hormone that is released by pancreatic beta cells and induces glucose uptake by mobilizing glucose transporters from intracellular stores to the cell membrane, thus lowering the glucose levels in blood (22, 23). Because of its central role in glucose homeostasis, insulin administration is the standard therapy for hyperglycemia in diabetic patients. Following activation of the insulin receptor (InsR), a receptor tyrosine kinase (RTK), insulin induces PI3K-Akt and Erk MAPK pathway signaling (21), with Akt activation driving the translocation of the glucose transporter Glut4 from cytoplasmic vesicles to the cell surface to facilitate glucose import (24, 25). Activation of the PI3K-Akt pathway also promotes the translocation of other transmembrane proteins to the cell surface (26, 27), Insulin signaling also controls cell survival, proliferation, and protein synthesis, and leads to activation or repression of a variety of genes (28, 29).
We now show that insulin induces a rapid increase of the TGF-β receptors, TβRI and TβRII, at the cell surface, without an immediate effect on their overall abundance. This increase was driven by Akt activation and controlled by AS160, an endosomal membrane-associated Rab GTPase activating protein that is directly phosphorylated by Akt (30, 31). The insulin-induced increase in TGF-β receptor cell surface abundance enhanced the cell responsiveness to autocrine TGF-β, with increased TGF-β/Smad signaling consequently participating in the cellular response to insulin. Thus, increased TGF-β responsiveness and signaling is integral to the insulin-responsive gene expression response, insulin-induced attenuation of epithelial integrity and insulin-induced cell migration, and allows for cooperation between TGF-β and insulin signaling. These findings highlight a role of Akt activation in controlling the TGF-β receptor availability and TGF-β responsiveness, and may be relevant for the interpretation of the effects of insulin in cultured cells and in vivo.
RESULTS
Insulin induces an increase in TGF-β receptors at the cell surface
We reported that 25 mM glucose, as seen in hyperglycemia, induces a rapid increase in cell surface levels of both the TβRI and TβRII receptors (18). Thus, replacing 4 mM glucose with 25 mM glucose in cell culture medium resulted in increased cell surface TβRI and TβRII abundance by 15 min after treatment, as detected by biotinylation of cell surface proteins, in NMuMG epithelial cells and mouse embryonic fibroblasts (MEFs) (Fig. 1A). To study the underlying signaling pathway(s) that mediate(s) this increase, we tested the effect of insulin, which activates PI3K-Akt signaling, similarly to high glucose (19, 32). Treatment of NMuMG cells or MEFs with insulin induced rapid increases in the TβRI and TβRII abundance at the plasma membrane, without changes in their total abundance (Fig. 1B). These increases in the cell surface abundance of TβRI and TβRII were not significantly enhanced by the addition of 25 mM glucose (Suppl. Fig. S1).
Fig. 1. Insulin enhances TGF-β receptor abundance at the plasma membrane and induces Smad2/3 activation through autocrine TGF-β signaling.
(A, B) Immunoblot analyses of the TβRI and TβRII receptors in NMuMG epithelial cells and mouse embryonic fibroblasts (MEFs), treated with 25 mM glucose (A) or insulin (B) for 15 or 30 min. The top two panels show the cell surface receptors isolated through Neutravidin adsorption after cell surface protein biotinylation. The lower panels show TβRI and TβRII in total cell lysates, and β-actin or GAPDH as loading control. Immunoblots in (A) and (B) are representative of three independent experiments. Blots were analyzed by densitometry and the ratios of cell surface TβRI to total lysate TβRI, or cell surface TβRII to total lysate TβRII, were compared at different time points. Pairwise comparison of relative abundance across time was performed using Wilcoxon rank-sum test; *p < 0.05. (C) MEFs were treated with 100 nM insulin with or without SB431542 for the indicated times, and Smad2 and Smad3 activation were assessed by immunoblotting for phosphorylated Smad2 (pSmad2) or phosphorylated Smad3 (pSmad3) immunoblotting. Blots were analyzed by densitometry and the ratios of phosphoSmad2 to total lysate Smad2, or phosphoSmad3 to total Smad3, at different time points are shown by scatter plot. (D) Immunoblots of nuclear and cytoplasmic extracts from insulin-treated NMuMG cells show phosphoSmad2 and phosphoSmad3 accumulation in the nucleus. Histone H3 and glyceralde-3-phosphate dehydrogenase (GAPDH) mark the nuclear and cytoplasmic compartments. Immunoblots in (C) and (D) are representative of two independent experiments. (E) Smad3-mediated luciferase expression in NMuMG cells transfected with the 4xSBE (left panel) or 3TP (right panel) luciferase reporters and treated with insulin for 24 h show insulin-induced luciferase expression that was inhibited in the presence of SB431542. Luciferase activities were normalized against the co-transfected Renilla-Lux reporter. Error bars indicate standard errors, based on three independent experiments. Statistical analysis was performed using Wilcoxon rank-sum test; *p < 0.05**, p<0.0083***, p<0.001. AP, affinity precipitation; IB, immunoblot; Ctrl, control; SB, SB431542.
Insulin enhances autocrine TGF-β/Smad activation
The amount of functional TGF-β receptors at the cell surface is a key determinant of cellular sensitivity to TGF-β and Smad-activated TGF-β signaling. Inhibition of ectodomain shedding of TβRI results in increased TβRI cell surface amounts and increased TGF-β-induced Smad signaling (15). Additionally, the increased TβRII and TβRI abundance at the plasma membrane in response to high glucose enhance TGF-β-induced Smad activation and autocrine TGF-β responsiveness (18). We therefore evaluated whether insulin enhanced autocrine TGF-β signaling.
To study the effect of insulin on TGF-β-induced Smad activation, cells were treated with insulin for different times. Insulin induced Smad2 and Smad3 activation, as assessed by immunoblotting for C-terminally phosphorylated Smad2 or Smad3, a response that was blocked by the TβRI kinase inhibitor SB431542 (SB) (Fig. 1C). Because Smad activation results in nuclear translocation (10, 33), we evaluated whether insulin induced the nuclear translocation of activated Smad2 and Smad3 in cells. Insulin induced a gradual increase of C-terminally phosphorylated Smad2 and Smad3 in nuclear fractions (Fig. 1D). These results indicate that insulin activates autocrine Smad2/3 activation through the TGF-β receptor TβRI.
Smad3 activation in response to TGF-β results in transcriptional regulation at Smad binding DNA sequences (10, 11). We therefore evaluated the effect of insulin on luciferase expression from tandem Smad3 binding sites in the 4xSBE transcription reporter (34) in NMuMG cells and MEFs. The basal expression of luciferase from this reporter, which resulted from autocrine TGF-β signaling, was enhanced by insulin, and blocked by SB431542 (Fig. 1E, left). Similarly, the 3TP-luciferase reporter, which combines Smad3 binding with AP-1-responsive DNA sequences (35), showed enhanced transcriptional activity in response to insulin, which was also blocked by SB431542 (Fig. 1E, right). Together, these data illustrate that insulin activates autocrine TGF-β signaling that requires the kinase activity of TβRI and results in Smad2/3 activation and Smad-dependent transcription. Because insulin treatment did not induce an increase in the expression of mRNAs encoding TGF-β or activation of TGF-β during the time windows of the experiments (Suppl. Fig. S2), the increased autocrine TGF-β signaling resulted from increased sensitivity of the cells to autocrine TGF-β signaling through enhanced cell surface abundance of TGF-β receptors.
Insulin-induced Akt signaling drives TGF-β receptor translocation to the cell surface
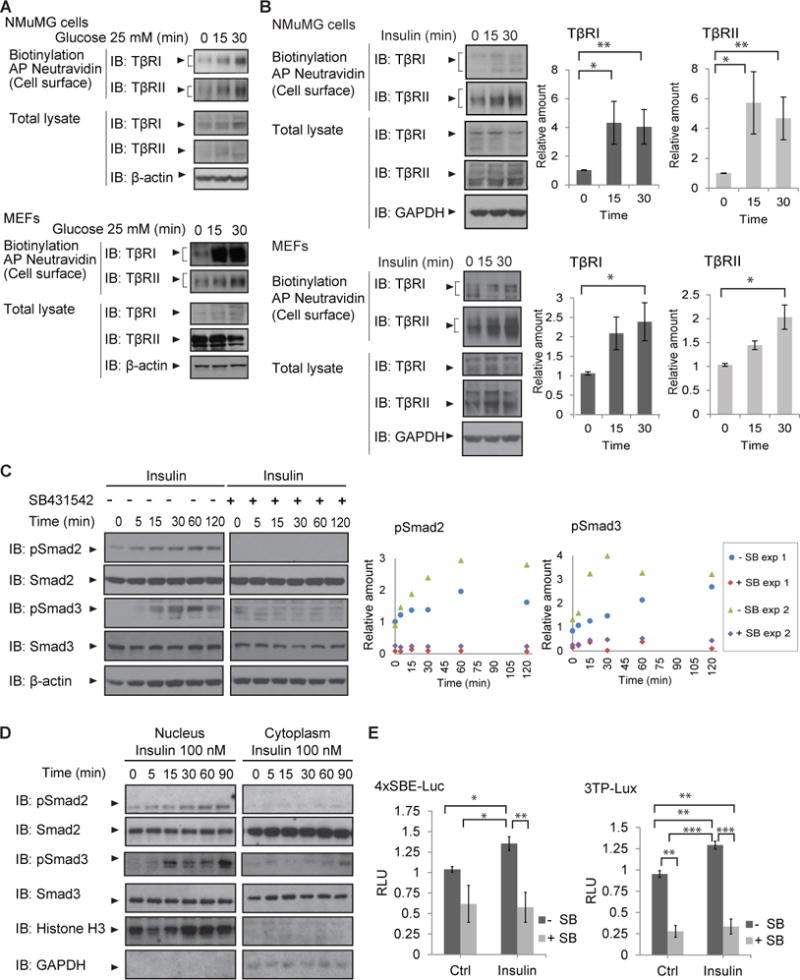
Insulin can activate both the PI3K-Akt and the MEK1/2-Erk MAPK pathways (21). To identify the signaling pathway that contributes to or drives the insulin-induced increase in cell surface TβRII and TβRI receptors, we tested the effects of pharmacological inhibition of either pathway. Treatment with GDC0941, a PI3K inhibitor that preferentially inhibits the α and δ p110 isoforms of PI3K (36, 37), enhanced the cell surface abundance of TβRI and TβRII in the absence of insulin, and prevented the insulin-induced increase in their cell surface abundance, as determined by cell surface protein biotinylation (Fig. 2A, B). GDC0941 also prevented Akt activation, as shown by immunoblotting for Akt phosphorylation on Ser473 (Fig. 2A,B), consistent with the dependence of insulin-induced activation of Akt on PI3K activation (38). AktVIII, a selective inhibitor of Akt1 and Akt2 activation (39–41), also prevented the insulin-induced increase in the abundance of TβRI and TβRII at the cell surface (Fig. 2A, B). In contrast, UO126, which inhibits activation of MEK1/2 and thus, of the MAPK Erk, had little or no effect on the insulin-induced increase of TGF-β receptor abundance at the cell surface (Fig. 2C, D). These results suggest that Akt activation is required for the increase of TβRI and TβRII at the plasma membrane in response to insulin.
Fig. 2. Role of PI3K-Akt signaling in insulin-induced cell surface presentation of TGF-β receptors.
(A, B) Inhibition of PI3K or Akt activation using GDC0941 or AktVIII prevents the insulin-induced increase in TβRI or TβRII abundance at the cell surface, as assessed by affinity purification of cell surface proteins using Neutravidin, and immunoblotting. Total lysates were immunoblotted to detect phospho Akt-Ser473, Akt, TβRI, TβRII, and β-actin as loading control. (C, D) Inhibition of MEK1/2 activation using U0126 does not prevent the insulin-induced increase in TβRI or TβRII abundance at the cell surface, as assessed using cell surface protein biotinylation as in A and B. Total protein lysates were immunoblotted to detect TβRI, TβRII, or phosphoErk1/2 or Erk1/2 (panel D), and β-actin as loading control. (A–D) Fold change of cell surface protein abundance is represented in bar graphs with error bars. (E) Expression of Myr-Akt2 increases TβRI and TβRII abundance at the cell surface, and this increase is not further enhanced by insulin. Cell surface TGF-β receptors were biotinylated, affinity purified, and immunoblotted with antibodies to detect TβRI or TβRII. Total lysates were immunoblotted for TβRI, TβRII, phospho Akt-Ser473, Akt2, and β-actin as loading control. Error bars in (A) to (E) indicate standard errors, based on three independent experiments. Comparisons were performed using Wilcoxon rank-sum test; *p < 0.05. AP, affinity precipitation; IB, immunoblot; Ctrl, control.
Localization to the plasma membrane plays an important role in Akt activation (42, 43). To validate a role for Akt activation in the cell surface presentation of TβRI and TβRII, we expressed a form of Akt2 with a myristoylation sequence fused to its NH2-terminus (Myr-Akt2) that confers constitutive activation (44). A role for Akt2 in insulin-stimulated glucose transport has been established (45, 46). Consistent with our results on the role of Akt in TGF-β receptor transport, expression of Myr-Akt2 resulted in an increase of TGF-β receptors at the plasma membrane that was comparable to the effect of insulin treatment (Fig 2E).
Growth factors that act through receptor tyrosine kinases also induce Akt activation, with the extent of activation depending on the ligand-receptor combination and cell context. We therefore evaluated the effect of insulin-like growth factor 1 (IGF-1). Treatment of NMuMG cells with IGF-1 resulted in Akt activation and enhanced the cell surface abundance of TβRI and TβRII (Suppl. Fig. S3). Furthermore, inhibition of Akt activation using AktVIII prevented the increase in cell surface abundance of TβRI and TβRII in response to 25 mM glucose (Suppl. Fig. S4), supporting a central role of Akt activation in controlling the amount of TGF-β receptor at the cell surface.
Akt1 and Akt2, which are encoded by distinct genes, are present in most cells. Insulin treatment of NMuMG cells or MEFs resulted in activation of both Akt1 and Akt2, assessed by phosphorylation at Ser473 and Thr408, with Akt1 activation seeming more prominent than Akt2 activation due to much higher total amounts of Akt1 (Suppl. Fig. S5A, B). Selective silencing using siRNAs suggested that both Akt1 and Akt2 contributed to the insulin-induced increase in cell surface abundance of both TβRI and TβRII (Suppl. Fig. S5C), although Akt2 appeared to have a more prominent role than Akt1 in attenuating the cell surface abundance of TGF-β receptors. Silencing of Akt2, but of not Akt1, enhanced TβRI and TβRII amounts at the cell surface in the absence of insulin, and prevented their increase in response to insulin (Suppl. Fig. S5C).
AS160 controls insulin-induced transport of TβRI and TβRII to the cell surface
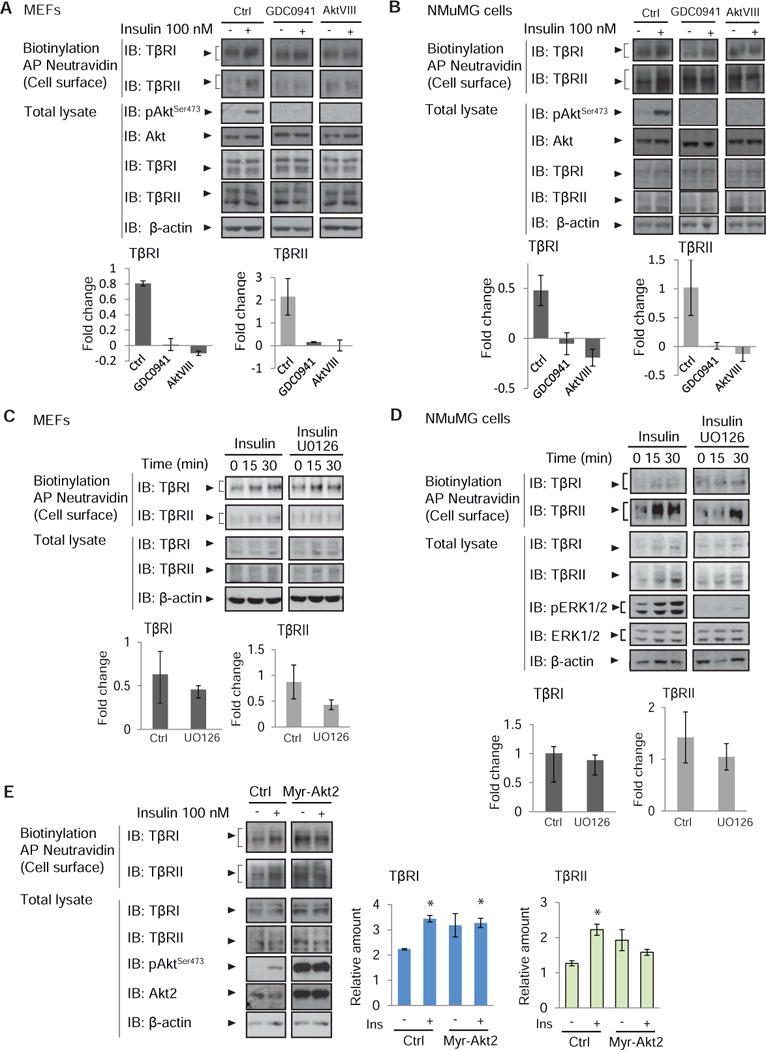
Because Akt activation was required for the insulin-induced cell surface presentation of TβRI and TβRII, we explored the role of the Rab-GTPase activating protein AS160, a kinase substrate of Akt that has been implicated in selective cell surface transport of Glut4 in muscle and fat cells (31, 47–50), the intracellular routing of the vasopressin-regulated water channel aquaporin-2, (51) and the aldosterone-regulated sodium channel ENaC (26) in an epithelial duct cell line, as well as the translocation of the fatty acid receptor CD36 to the cell surface of cardiomyocytes (52). Phosphorylation of AS160 by Akt overcomes the intracellular retention of Glut4-containing vesicles by AS160, allowing transport of Glut4 to the cell surface (31, 48, 53). As shown in adipose cells and myoblasts (31, 54, 55), insulin induced the phosphorylation of AS160 at Thr642 in NMuMG cells and MEFs (Fig 3A; Suppl. Fig. S6).
Fig. 3. AS160 controls the insulin-induced cell surface presentation of TβRI and TβRII.
(A) Increased phosphorylation of AS160 in response to insulin accompanies increased cell surface abundance of TβRI and TβRII. MEFs were treated with insulin and cell surface proteins were biotinylated. Neutravidin-affinity purified biotinylated proteins were immunoblotted for TβRI and TβRII. Phosphorylation of AS160 was assessed by immunoblotting for phosphoAS160-Thr642. Whole cell lysates were immunoblotted for AS160, TβRI, TβRII, and GAPDH as loading control. Error bars indicate standard errors, based on three independent experiments. (B) siRNA-mediated decrease of AS160 in NMuMG cells results in increased TβRI and TβRII abundance at the cell surface, assessed by immunoblotting of affinity-purified biotinylated cell surface proteins. Whole cell lysates were immunoblotted for AS160, TβRI and TβRII, and GAPDH as loading control. Error bars indicate standard errors, based on three independent experiments. Pairwise comparison of relative abundance was performed using Wilcoxon rank-sum test. * p<0.05 (C) NMuMG cell lysates were subjected to sequential centrifugations to generate the LDM fraction of cell membranes, which was then fractionated by centrifugation in a 10–36% sucrose gradient. Twelve equal fractions spanning the gradient were analyzed by immunoblotting for AS160, TβRI, TβRII, EEA1, Rab4 and Rab11. Immunoblots are representative of three independent experiments. (D) Immunofluorescence of NMuMG cells expressing Flag-tagged AS160 and TβRI-EGFP, double stained with anti-Flag and anti-GFP. Scale bar, 10 µm. Images are representative of two independent experiments. (E) 293T cells expressing Flag-tagged AS160 and/or Myc-tagged TβRI were subjected to chemical crosslinking using DSP, and analyzed by immunoprecipitation and immunoblotting using the antibodies shown. Immunoprecipitation of TβRI resulted in co-immunprecipitation of AS160. Immunoblots are representative of two independent experiments. (F, G) AS160 silencing enhances autocrine TGF-β/Smad-induced transcription in NMuMG cells. In (F), cells were transfected with the Smad reporter plasmids 4xSBE or 3TP-Lux, and luciferase expression of cells, treated or not with SB431542, was normalized against the co-transfected Renilla-Lux reporter. In (G) Serpine1 mRNA (which encodes PAI-1) and Smad7 mRNA, expressed by cells treated or not with SB431542, were quantified by qRT-PCR and normalized against Rpl19 mRNA. Error bars indicate standard errors, based on three independent experiments. Statistical analysis was performed using Wilcoxon rank-sum test (* p<0.05, ** p<0.0083). In panels A, B, C and E: AP, affinity purification, IB, immunoblot, IP, immunoprecipitation. Ctrl, control.
To evaluate whether AS160 controls Akt-mediated cell surface transport of TGF-β receptors, we decreased the AS160 abundance using siRNA (Fig. 3B). This decrease resulted in increased TβRI and TβRII abundance at the cell surface in the absence of insulin that were comparable to those in insulin-stimulated cells transfected with control siRNA (Fig. 3B). Additionally, insulin treatment of AS160 siRNA-transfected cells conferred no further increase in TGF-β receptor abundance at the plasma membrane (Fig. 3B). Similar results were obtained with cells that were lentivirally infected to express shRNA to decrease the AS160 abundance (Suppl. Fig. S7). These data suggest that, in the absence of insulin, AS160 retains the TGF-β receptors intracellularly and prevents them from moving to the plasma membrane.
The role of AS160 in insulin-induced TGF-β receptor presentation at the cell surface suggests that the TGF-β receptors may localize in the same compartment as AS160. Cell fractionation studies have shown that the bulk of AS160 that is not associated with the plasma membrane resides in a low-density microsomal (LDM) fraction (56). We therefore isolated the LDM fraction and separated it from the high-density microsomal (HDM) fraction, the plasma membrane and the cytosol. The LDM fraction of NMuMG cells was then fractionated over a 10–36% sucrose gradient. Immunoblotting of gradient fractions revealed that TβRI and TβRII were present in the LDM fraction, and were found in gradient fractions 4–12, with peaks in fractions 10–12, coinciding with the presence of AS160 in fractions 5–12 (Fig. 3C). These data indicate that TGF-β receptors and AS160 co-exist in an LDM subpopulation. EEA1, an early endosomal and LDM marker (57), was present in most fractions (Fig. 3C). The co-fractionation of EEA1 with the TGF-β receptors is consistent with the recycling of TGF-β receptors to the plasma membrane (58–60). These fractions also contained Rab4 and Rab11 (Fig. 3C), two Rab GTPases that have been implicated in endosomal recycling (61) and that colocalize in endosomes (62).
The colocalization of TGF-β receptors with AS160 was also supported by immunofluorescence and coimmunoprecipitation. For immunofluorescence, NMuMG cells were transfected to express Flag-tagged AS160 and HA-tagged TβRI. As has been previously reported (63), AS160 staining was diffuse throughout the cytoplasm with more concentrated, punctate staining adjacent to nucleus, partially overlapping with the punctate TβRI immunostaining in the cytoplasm and adjacent to the nucleus (Fig. 3D). The limitations of available antibodies did not allow verification of the colocalization of endogenous AS160 with TβRI or TβRII. In coimmunoprecipitation analyses using transfected 293T cells, we found TβRI in association with AS160 (Fig. 3E). This low-level association was apparent only after chemical stabilization of pre-existing complexes using dithiobis succunimidylpropionate (DSP), which covalently links adjacent Lys residues. However, we failed to detect the interaction of AS160 with TβRII using available antibodies. Collectively, these data show that AS160 and the TGF-β receptors colocalize in the same vesicles, and raise the possibility of their association in protein complexes destined to be routed to the plasma membrane.
AS160 controls autocrine TGF-β signaling
Because AS160 silencing resulted in increased TGF-β receptor abundance at the cell surface, we addressed whether AS160 controls autocrine TGF-β responsiveness. We therefore evaluated the effect of AS160 siRNA on Smad-dependent gene expression in the absence or presence of SB431542, which blocks TGF-β-dependent Smad activation. In Smad-dependent transcription assays using the 4xSBE (34) and 3TP luciferase reporters (35), decreasing the AS160 abundance resulted in increased transcription that was largely inhibited by SB431542 (Fig. 3F). Consistent with the increased Smad activation, decreasing AS160 resulted in enhanced expression of TGF-β/Smad target genes, such as Smad7 and Serpine1, which encodes plasminogen activator inhibitor type I (PAI-1), although these increases were not statistically significant (Fig. 3G). These results show that decreasing AS160 increases TGF-β responsiveness. We conclude that AS160 limits TGF-β responsiveness by preventing cell surface presentation of the TGF-β receptors, similarly to the effect of insulin.
Insulin-induced TGF-β receptor delivery reveals mutual dependence of TβRI and TβRII
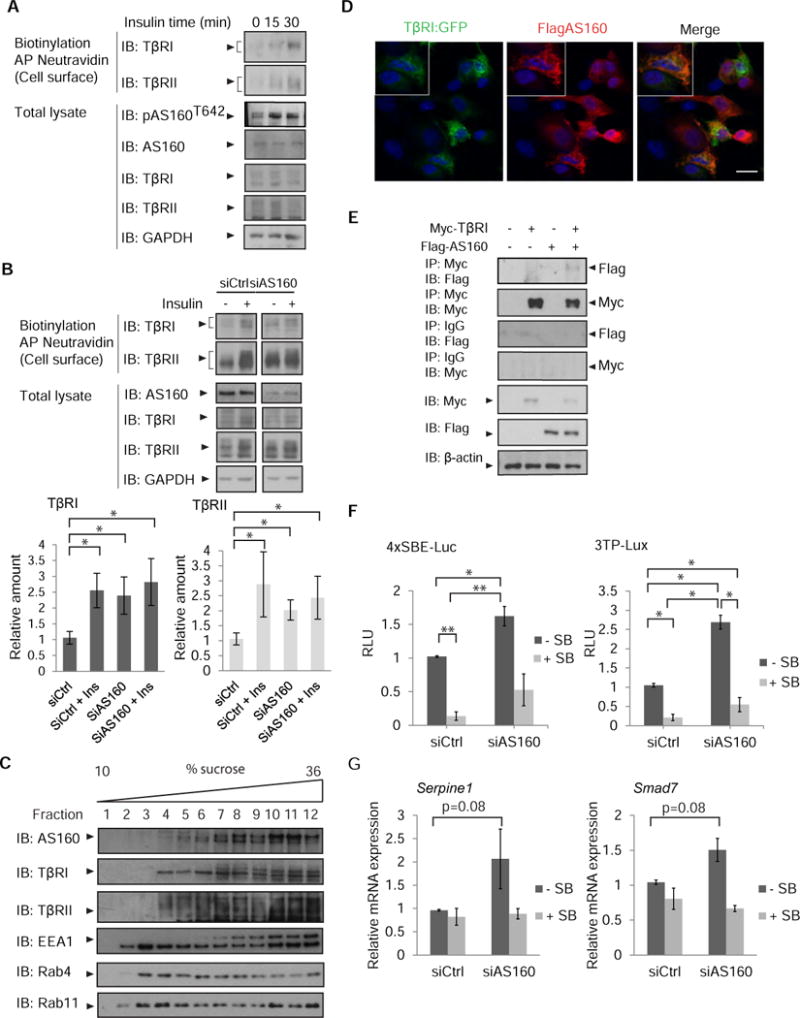
Because insulin induced an increase in cell surface abundance of both TβRI and TβRII, and the TGF-β receptors dynamically interact with each other in the absence of ligand (64, 65), we examined whether the TβRI and TβRII receptors depended on each other in the insulin-induced increase of cell surface TGF-β receptors. Silencing TβRI in NMuMG cells enhanced the cell surface abundance of TβRII but attenuated the insulin-induced increase in cell surface TβRII (Fig. 4A). Additionally, insulin did not induce an increase in cell surface abundance of TβRII in Tgfbr1−/− MEFs (Fig. 4B). Reconstituting TβRI in these cells restored the insulin-induced increase in TβRI and TβRII amounts at the cell surface, although not always in a statistically significant manner, similarly to what was seen in wild-type MEFs (Fig. 4B).
Fig. 4. Requirement of TβRI and TβRII in insulin-stimulated presentation of the TGF-β cell surface receptors.
(A) siRNA-mediated silencing of TβRI in NMuMG cells enhances TβRII abundance and attenuates the insulin-induced increase in surface TβRII. Cells were treated with insulin, and cell surface proteins were biotinylated. Neutravidin-affinity purified biotinylated proteins were immunoblotted for TβRI and TβRII. Whole cell lysates were immunoblotted for TβRI and TβRII, and β-actin as loading control. Immunoblots in (A) are representative of three independent experiments. Fold change of cell surface protein abundance is presented in bar graphs with error bars. (B) Effects of insulin on the cell surface abundance of TβRI and TβRII in matched wild-type MEFs, Tgfbr1−/− MEFs and MEFs with TβRII silencing. Tgfbr1−/− MEFs were reconstituted with HA-tagged TβRI (third column), and MEFs with TβRII silencing were reconstituted with siRNA-resistant Flag-tagged TβRII (fifth column). Cell surface TβRI and TβRII were analyzed as in (A). Error bars indicate standard errors, based on three independent experiments. (C) siRNA-mediated silencing of TβRII in NMuMG cells impairs the increase in surface TβRI in response to insulin. Cells were treated with insulin, and cell surface TβRI and TβRII were analyzed as in (A). Error bars indicate standard errors, based on three independent experiments. Pairwise comparisons were performed using Wilcoxon rank-sum test *p < 0.05. AP, affinity precipitation; IB, immunoblot; Ctrl, control.
To evaluate whether insulin-stimulated transport of TβRI conversely depends on TβRII, we silenced the expression of TβRII in NMuMG cells and MEFs using an siRNA that targets the 3′UTR of its mRNA. In MEFs, silencing TβRII enhanced the cell surface amount of TβRI, yet still allowed for insulin to induce enhanced cell surface TβRI abundance (Fig. 4B), whereas in NMuMG cells, the decrease in TβRII impaired the increase of cell surface TβRI in response to insulin (Fig. 4C). Restoration of TβRII rescued the insulin-induced increased cell surface abundance of TβRI and TβRII (Fig. 4C). These analyses suggest a mutual dependence of TβRI and TβRII on each other in insulin-induced presentation of the TGF-β receptors at the plasma membrane, with some difference between NMuMG cells and MEFs, and perhaps a stronger dependence for the cell surface presentation of TβRII on TβRI. These results are consistent with the dynamic interactions of TβRI and TβRII in the absence of ligand resulting in heteromeric association of a fraction of TβRI and TβRII at any given time (64).
Enhanced autocrine TGF-β signaling participates in the insulin gene expression response
The increased TGF-β-dependent, autocrine Smad activation in response to insulin (Fig. 1C) suggested that insulin might mildly induce TGF-β target gene expression. Indeed, insulin induced the expression of TGF-β target genes, including those encoding Smad7 and PAI-1 (Fig. 5A). As demonstrated by Smad3 reporter assays (Fig. 1E), the basal expression of these genes was enhanced by insulin, an increase that was blocked by SB431542 (Fig. 5A). Because IGF-1, similarly to insulin, induced Akt activation and enhanced TβRI and TβRII abundance at the cell surface (Suppl. Fig. S3), we also evaluated the effect of IGF-1 on TGF-β target gene expression in NMuMG cells and MEFs. Also IGF-1 induced the expression of mRNAs encoding Smad7 and PAI-1, and this induction was blocked by SB431542 (Suppl. Fig. S8).
Fig. 5. Contribution of TGF-β signaling to insulin-induced gene expression.
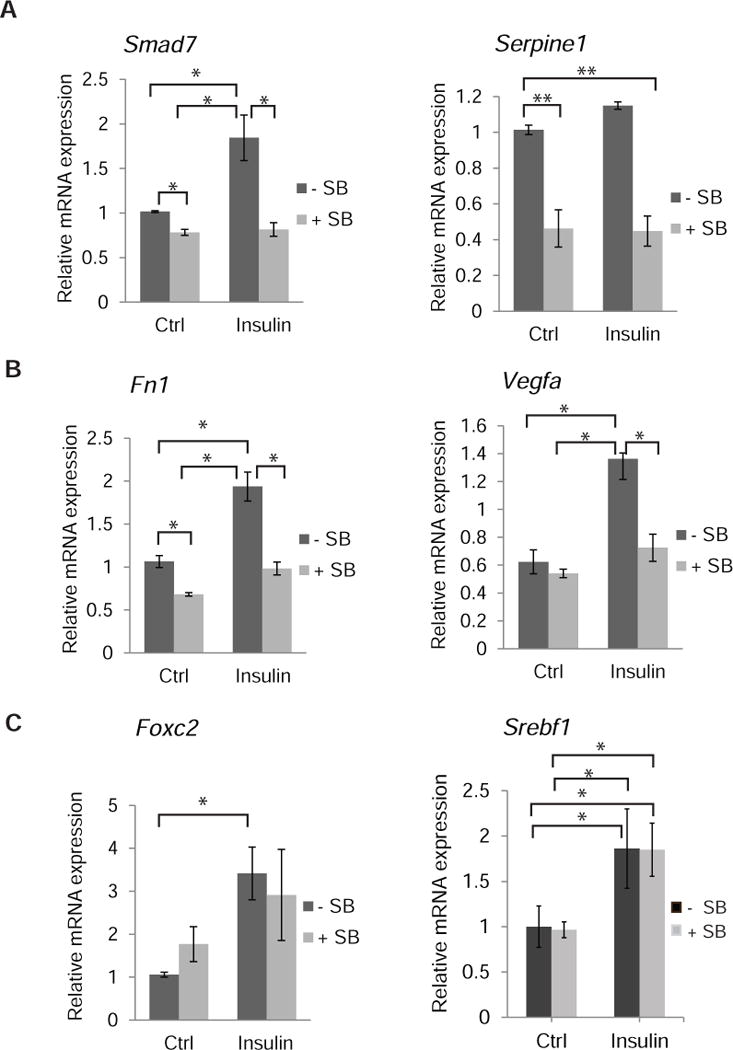
(A to C) NMuMG cells were stimulated for 1.5 (A, C) or 24 h (B) with insulin, in the presence or absence of SB431542, and mRNA for the indicated genes was quantified by qRT-PCR and normalized against RPL19 mRNA expression. (A) Induction of direct TGF-β/Smad3 target genes encoding Smad7 and plasminogen activator type 1 (Serpine1 mRNA) by insulin. (B–C) Induction of insulin-responsive genes by insulin. Induction of Vegfa mRNA and Fn1 mRNA (which encodes fibronectin) was repressed by SB431542 (B), and Foxc2 and Srebf1 mRNA induction was not affected by SB431542 (C). Error bars indicate standard errors, based on three independent experiments. Statistical analysis was performed using Wilcoxon rank-sum test; *p < 0.05; **p<0.0083.
In addition to insulin inducing TGF-β target genes, the increased autocrine TGF-β signaling in response to insulin may participate in the insulin-induced gene expression response. To determine whether autocrine TGF-β signaling contributes to insulin-induced gene expression changes, we treated NMuMG cells with or without insulin, in the presence or absence of the TGF-β signaling inhibitor SB431542. Quantification of insulin target genes with qRT-PCR revealed that autocrine TGF-β signaling participated in insulin-induced activation of some genes but not others. For example, insulin induces the expression of Vegfa and Fn1 mRNA (66–68), but this induction was prevented when TGF-β signaling was blocked using SB431542 (Fig. 5B). Additionally, siRNA-mediated knockdown of AS160, which results in enhanced TGF-β receptor abundance at the cell surface (Fig. 3B), also enhanced Vegfa and Fn1 mRNA expression (Suppl. Fig. 9), providing further support for a functional link between AS160 and increased autocrine TGF-β signaling in the insulin-induced gene response. In contrast to the induction of Vegfa and Fn1 mRNA expression, SB431542 did not affect the insulin-induced expression of Foxc2 and Srebf1 mRNAs. Both FOXc2 and SREBF1, the latter of which is also known as SREBP-1c, are transcription factors that control lipid homeostasis in response to insulin (69, 70) (Fig. 5C). We conclude that insulin induces a modest increase in the expression of TGF-β target genes by increasing TGF-β responsiveness, and that increased TGF-β signaling contributes to the expression of some insulin-responsive genes.
Insulin promotes an autocrine TGF-β-dependent epithelial plasticity response
The increased sensitivity of cells to autocrine TGF-β signaling and increased fibronectin abundance in response to insulin (Fig. 5B) suggested that insulin may impair epithelial integrity. TGF-β induces epithelial plasticity responses, normally in development and pathologically in carcinomas and fibrosis, that result in decreased epithelial and increased mesenchymal gene expression and properties, and can lead to EMT (1–3, 71). While increased fibronectin expression marks EMT, the epithelial plasticity response is driven by the EMT master transcription factors Snail1 or Snail2 (also known as Slug) (72, 73). In NMuMG cells, EMT is initiated by activation of Snai1 expression (74–76). Insulin induced the expression of Snai1 mRNA and protein, an induction that was blocked by SB431542 (Fig. 6A, B), without affecting Snai2 mRNA expression (Suppl. Fig. S10). Consistent with the role of AS160 in the insulin-induced increase in autocrine TGF-β responsiveness, decreasing the AS160 abundance also enhanced the expression of Snai1 mRNA in NMuMG cells (Suppl. Fig. S11). Besides increased fibronectin abundance at the mRNA and protein levels (Fig. 5B, 6B, C), which is a characteristic of EMT, insulin induced a modest increase in the mRNA and protein amounts of the mesenchymal marker N-cadherin (Fig. 6B, D). We did not see, after 24 h of insulin stimulation, decreased abundance of the epithelial junction protein E-cadherin or increased abundance of vimentin (Fig. 6B), as is often observed in EMT, suggesting that the epithelial plasticity response induced by short exposure to insulin is modest but depends on increased autocrine TGF-β signaling.
Fig. 6. Contribution of TGF-β signaling to insulin-induced epithelial plasticity and migration.
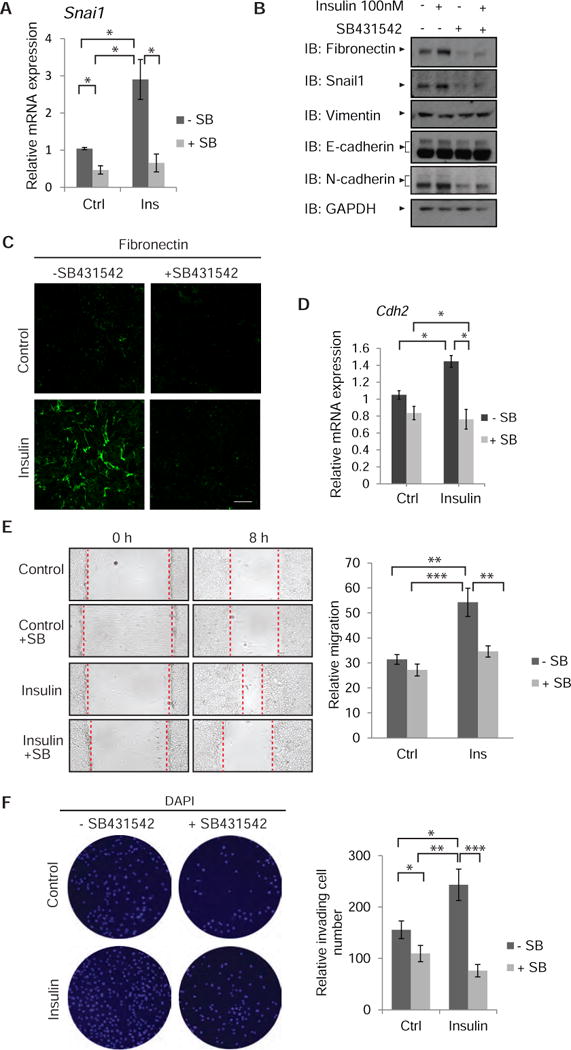
(A) Induction of Snai1 mRNA in NMuMG treated with insulin for 90 min, in the presence or absence of SB431542. mRNA was quantified by qRT-PCR and normalized against RPL19 mRNA expression. Error bars indicate standard errors, based on three independent experiments. (B) Immunoblot analysis of EMT-associated protein abundance in NMuMG cells, treated with or without insulin in the presence or absence of SB431542 for 24 h. The two N-cadherin bands are likely to correspond to the precursor (upper band) and mature form (lower band) of N-cadherin (105). Similarly, the upper E-cadherin band represents immature E-cadherin, and the more intense band represents its mature form (106, 107). GAPDH served as loading control. Immunoblots in (B) are representative of two independent experiments. (C) Immunofluorescence staining of fibronectin in NMuMG cells after insulin treatment for 24 h, in the absence or presence of SB431542. Images are representative of three independent experiments. Scale bar, 100 μm (D) Induction of Cdh2 mRNA (which encodes N-cadherin) in NMuMG treated with insulin for 40 h, in the presence or absence of SB431542. mRNA was quantified by qRT-PCR as in (A). (A, D) Error bars indicate standard errors, based on three independent experiments. The statistical significance was determined by Wilcoxon test: * p < 0.05. (E) Migration of NMuMG cells measured in a monolayer wounding assay. Confluent monolayers were scratched with a pipette tip at time 0 (left), and cells were allowed to migrate into the wounded area for 8 h (right). The migration distance is graphically presented. Error bars indicate standard error of the means of three experiments. * p < 0.05, **p< 0.0083, *** p < 0.0001. The statistical significance was determined by post hoc Tukey–Kramer HSD test. (F) Invasion of NMuMG cells in Transwell assays in the presence or absence of insulin, with or without SB431542. After 24 h, the invaded cells at the bottom filter surface were counted as shown in the graph. The results are averages of five randomly chosen microscopic fields from two separate experiments, each conducted in duplicate. Error bars indicate standard error of means. The statistical significance was determined by Wilcoxon method; * p < 0.05, **p< 0.0083, *** p < 0.0001.
Enhanced autocrine TGF-β signaling promotes insulin-induced cell migration
Epithelial plasticity and EMT result in increased cell motility (77). Because insulin induced a mild epithelial plasticity response, and promotes cell migration during wound healing (78, 79), we asked whether autocrine TGF-β signaling in response to insulin contributed to insulin-induced cell migration. We therefore performed monolayer wounding assays using confluent NMuMG cells, with the wounded monolayers exposed to insulin with or without SB431542. Insulin increased the migration of NMuMG cells to close the wound, when compared to control cells, an effect that was prevented by SB431542 (Fig. 6E). No differences in cell number were apparent after 8 h (Suppl. Fig. S12), the time interval used to score cell migration.
Cell invasion requires increased cell migration with increased extracellular matrix degradation and remodeling. Thus, we evaluated whether insulin induces NMuMG cell invasion. Transwell assays indicated that insulin increased cell invasion through extracellular matrix, an increase was prevented in the presence of SB431542 (Fig. 6F). Together, these data suggest that insulin-induced autocrine TGF-β signaling is required for insulin-stimulated cell migration and invasion.
Insulin enhances TGF-β-induced epithelial plasticity responses
The enhanced TGF-β responsiveness in response to insulin raises the possibility that insulin might potentiate TGF-β responses. To address this possibility, we evaluated TGF-β-induced EMT responses in NMuMG cells. Indeed, insulin enhanced TGF-β/Smad-induced gene expression responses at a low TGF-β concentration (0.2 ng/ml), as illustrated with the expression of Snai1, Serpine1 and Fn1 mRNA (Fig. 7A). The insulin-enhanced induction of these TGF-β-responsive genes was prevented by SB431542. Immunoblotting indicated that insulin also mildly enhanced the abundance of fibronectin, Snail1, and pro-N-cadherin (upper band), without affecting that of vimentin and E-cadherin (Fig. 7B). Consistent with the enhancement of TGF-β responsiveness by insulin, the elongated cell morphology induced by low concentrations of TGF-β was more pronounced in the presence of insulin, whereas cells treated with insulin alone (without adding TGF-β) retained a cuboidal epithelial phenotype (Fig. 7C). Decreasing AS160 abundance also promoted an elongated cell phenotype in the presence of a low concentration of TGF-β (Suppl. Fig. S13A), which was accompanied by increased fibronectin deposition (Suppl. Fig. S13B), supporting the notion that the insulin-Akt target AS160 controls the epithelial plasticity response to low concentrations of TGF-β.
Fig. 7. Cooperation of insulin and TGF-β signaling in epithelial plasticity and migration.
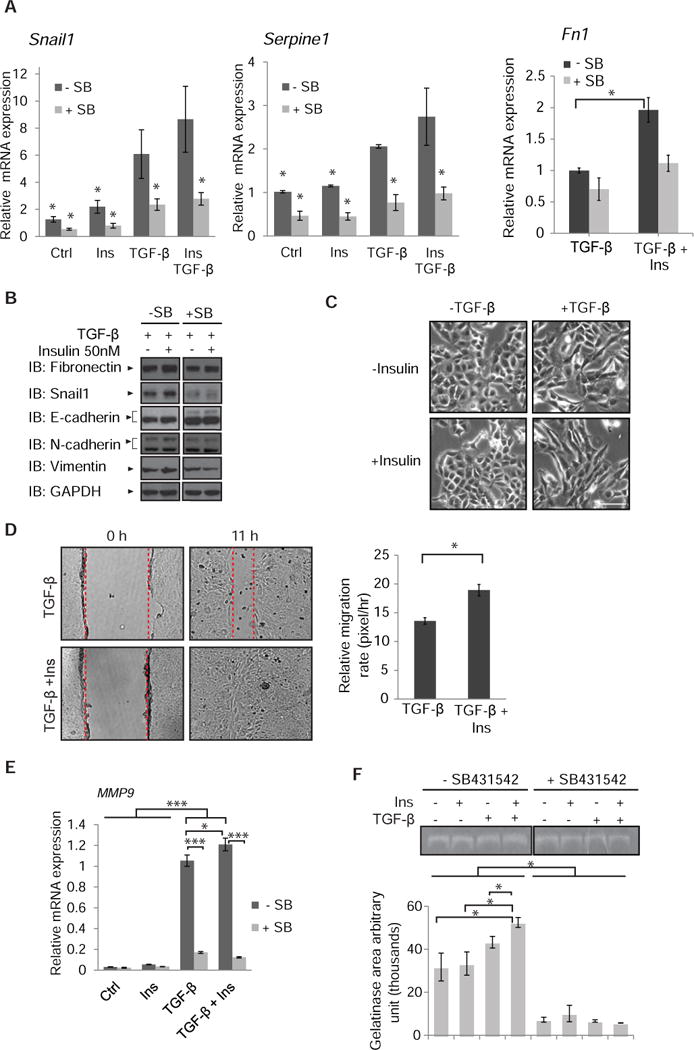
(A) TGF-β-induced Snai1, Serpine1 and Fn1 mRNA expression in NMuMG cells was enhanced after 90 min of insulin exposure. mRNA was quantified by qRT-PCR and normalized against RPL19 mRNA. Error bars indicate standard errors, based on three independent experiments with duplicates. * p < 0.05, compared with the TGF-β group by Wilcoxon test. (B) Immunoblot analysis of EMT-associated protein abundance of NMuMG cells treated with TGF-β or TGF-β and insulin for 24 h in the presence or absence of SB431542. GAPDH served as loading control. Immunoblots are representative of two independent experiments. (C) Phase contrast images of NMuMG cells treated with TGF-β, insulin, or both for 24 h. Images are representative of three independent experiments. Scale bar, 100 µm. (D) Migration of NMuMG cells, in the presence or absence of TGF-β with or without insulin, measured in a monolayer wounding assay. Confluent monolayers were scratched with a pipette tip at time 0 (left), and cells were allowed to migrate into the wounded area for 11 h (right). The migration distance was measured and is graphically presented. Error bars indicate standard error of the means of three experiments. Statistical significance was determined by Wilcoxon test; *p<0.05. (E) TGF-β-induced Mmp9 mRNA expression in NMuMG cells is enhanced in the presence of insulin. NMuMG cells were treated for 24 h with or without insulin in the presence or absence of TGF- β. Error bars indicate standard errors, based on three independent experiments. (F) Gelatinase zymogram of equal amount of protein extracted from the media of cells treated with insulin, TGF-β, or insulin and TGF-β, in the presence or absence of SB431542. Densitometric analyses represent MMP9 activities. Error bars indicate standard errors, based on three independent experiments. Statistical analysis was carried out using non parametric Wilcoxon test; *p<0.05, ***p<0.001.
The ability of insulin to enhance TGF-β responses was also apparent in the increased cell migration of NMuMG cells, when compared with TGF-β treatment alone. Insulin enhanced TGF-β-induced cell migration, resulting in more rapid monolayer wound closure (Fig. 7D). Consistent with the role of AS160 in this aspect of the insulin response, siRNA-mediated knockdown of AS160 also conferred a modest increase in migratory behavior in response to low levels of TGF-β (Suppl. Fig. S13C). Finally, insulin also enhanced the expression and gelatinolytic activity of MMP-9, which contributes to extracellular matrix degradation (80, 81) (Fig 7E, F). Together, these results illustrate that insulin enhances TGF-β responses, which based on our results above, results from increased amounts of TGF-β receptors at the plasma membrane.
DISCUSSION
Our findings reveal a strategy that cells employ to control TGF-β responsiveness through increased TGF-β receptor presentation at the cell surface, and provides insight into the nature of the cell response to insulin. Specifically, we have shown that (i) insulin induces a rapid increase in the cell surface abundance of TGF-β receptors, thus enhancing autocrine TGF-β responsiveness, (ii) Akt activation leads to increased cell surface abundance of TGF-β receptors through its control of the Rab GTPase activating protein AS160, (iii) insulin-induced autocrine TGF-β responsiveness contributes to and is integral to the cellular response to insulin, (iv) autocrine TGF-β signaling is an essential component of insulin-induced cell migration, (v) the increased TGF-β responsiveness in response to insulin allows for cooperation between TGF-β and insulin signaling.
Insulin enhances the abundance of TGF-β receptors at the cell surface through Akt activation
We have come to appreciate that cells regulate their TGF-β responsiveness by controlling the functional receptor abundance at the cell surface. Activation of the Erk or p38 MAPK pathways by growth factors or inflammatory cytokines results in ectodomain shedding, thus decreasing the functional TβRI abundance at the cell surface and TGF-β responsiveness (15). High glucose, in contrast, rapidly increases the TGF-β receptor abundance at the cell surface end enhances TGF-β responsiveness (18). We now present evidence that insulin increases TGF-β responsiveness by mobilizing intracellular TGF-β receptors and enhancing their cell surface abundance. Thus, the cells enhance their basal autocrine TGF-β signaling, which is often considered as negligible.
With the bulk of TGF-β receptors residing intracellularly (18), we sought to identify a signaling mechanism that mediates the rapid increase of TGF-β receptors at the cell surface. We found that Akt activation was required in this response to insulin, and that its constitutive activation was sufficient to induce increased TGF-β receptor presentation. Therefore, the PI3K-Akt pathway plays a central role in mobilizing the TGF-β receptors to the cell surface and in increasing the cell responsiveness to TGF-β. We surmise that other growth factors and stimuli that activate Akt signaling similarly induce TGF-β hyper-responsiveness through Akt. Indeed, the growth factor IGF-1, which also confers Akt activation, induces increased autocrine TGF-β responsiveness, and inhibition of Akt activation prevents the increase in cell surface TGF-β receptor abundance in response to IGF-1 or high glucose, as we observed with insulin.
The widely distributed Rab GTPase activating protein AS160 is an Akt kinase substrate, and Akt-mediated phosphorylation inactivates its ability to retain intracellularly a subpopulation of endosomes that harbor the transmembrane Glut4 glucose transporters. Insulin-induced Akt activation results in AS160 phosphorylation and a rapid increase of Glut4 at the cell surface, thus conferring increased glucose uptake (31, 82–84). The activity of AS160 is, however, not restricted to these transporters, because it also controls endosomal delivery of several other proteins, such as the epithelial sodium channel ENaC, a Na+/K+-ATPase, the water channel aquaporin-2 and the glycoprotein receptor CD36, to the plasma membrane (26, 51, 52, 63). As seen with Glut4, silencing AS160 resulted in increased TGF-β receptor abundance at the cell surface, consistent with attenuated intracellular retention (48). A role of AS160 in the regulated transport of TGF-β receptors to the cell surface is also supported by their subcellular colocalization and the presence of the TβRI receptor in AS160 protein complexes. Moreover, the mutual dependence between TβRI and TβRII in the translocation to the cell surface is consistent with the dynamic interaction of TβRI and TβRII in the absence of ligand (61). Hence, on the basis of our data, we propose a model for the insulin-induced increase of TGF-β receptors at the cell surface (Fig. 8). Activation of Akt in response to insulin allows intracellular vesicles with TGF-β receptors to translocate to the plasma membrane, thus increasing the TGF-β receptor abundance at the cell surface, which in turn permits increased TGF-β stimulation and enhanced autocrine TGF-β/Smad signaling. Although fibroblasts and epithelial cells differentially activate mTOR complexes through Akt in response to TGF- β (85), our analyses of NMuMG epithelial cells and MEFs suggest an overall common pathway in both cell types.
Fig 8. Model for regulation of the cell surface presentation of TGF-β receptors by Akt.
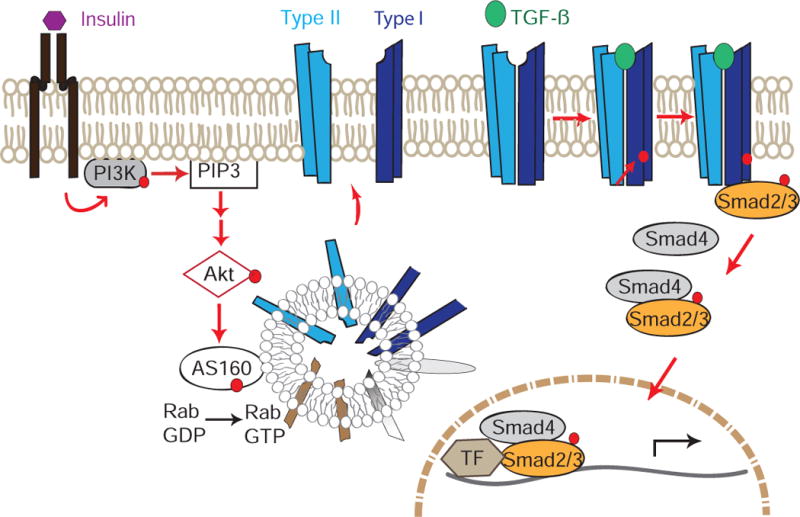
Insulin-induced PI3K activation increases PI(3,4,5)P3 concentrations, which leads to Akt activation. Activated Akt phosphorylates AS160, thus promoting GTP-loading of Rab proteins and increasing the translocation of intracellular vesicles containing TβRII and TβRI to the plasma membrane. The increased abundance of TGF-β receptors at the cell surface confer increased TGF-β responsiveness. Increased TGF-β ligand binding to TβRII/TβRI complexes results in enhanced activation of Smad2 and Smad3 through phosphorylation by the activated TβRI receptors. Activated Smad2 and Smad3 form complexes with Smad4 that then translocate into the nucleus and cooperate with other transcription factors to regulate TGF-β/Smad target gene expression.
Increased TGF-β responsiveness is integral to the cell response to insulin
The increased TGF-β responsiveness in response to insulin may have substantial physiological relevance, especially because most if not all cells respond to insulin, and insulin is therapeutically used to repress hyperglycemia in diabetic patients. The increased autocrine TGF-β signaling in response to insulin results in a low, basal activation of TGF-β target genes, such as those encoding Smad7 and PAI-1. This is particularly relevant because many TGF-β target genes, such as those encoding fibronectin, collagens, integrins, metalloproteases or protease inhibitors, contribute to increased extracellular matrix deposition and turnover, as seen in fibrosis. Considering the key role of TGF-β signaling in activating fibroblasts and driving fibrosis (3, 11), continued exposure of cells to insulin may confer a TGF-β-mediated profibrotic tissue response that perpetuates the response to high glucose. Indeed, both insulin and high glucose induce Akt activation, and increased cell surface TGF-β receptor abundance and autocrine TGF-β signaling. It should be noted that fibrosis with extensive extracellular matrix deposition commonly associates with increased TGF-β/Smad activity in rodent models and diabetic patients, including those with prolonged insulin treatment (8, 86). Whether insulin administration promotes or enhances fibrosis development in mouse models remains to be studied.
Because TGF-β-activated Smads cooperate with various transcription factors in regulating gene responses (10, 11), we also evaluated whether increased autocrine TGF-β signaling contributes to the insulin-induced gene expression program. This was indeed the case. Insulin activates various genes, including some that are targeted by Smads in response to TGF-β (87, 88), like those encoding fibronectin and VEGF-A (66–68). The insulin-induced expression of these genes was attenuated if not blocked when TGF-β signaling was prevented. The striking contribution of autocrine TGF-β signaling to insulin-induced increases in VEGF-A production may be important considering the key role of VEGF-A in angiogenesis, including in cancer progression (89). Other insulin-responsive genes, however, namely those encoding FoxC2 (69) and SREBP1F (90), were not affected by blocking autocrine TGF-β signaling. Together, increased autocrine TGF-β signaling appears to be integral to insulin-induced changes in gene expression. A comprehensive transcriptome analysis will better define the participation of autocrine TGF-β signaling in the response to insulin.
Because TGF-β signaling represses the epithelial phenotype and can induce a partial or complete EMT, we also assessed whether the increased autocrine TGF-β signaling in response to insulin results in an epithelial plasticity response. Whereas insulin did not repress the epithelial phenotype, some epithelial plasticity responses that are involved in EMT were noted, which depended on autocrine TGF-β signaling. Thus, insulin slightly increased the abundance of the EMT transcription factor Snail1 and mesenchymal fibronectin and N-cadherin, and blocking TGF-β signaling prevented these increases. Furthermore, the enhanced TGF-β responsiveness in response to insulin allowed for cooperation of TGF-β and insulin signaling in inducing EMT, as apparent by the expression of mesenchymal genes and a conversion from epithelial into more elongated cell morphology.
Insulin promotes cell motility in vitro (78, 91). However, attenuation of the epithelial phenotype also enables increased cell motility, which can lead to invasion, and these responses are apparent in TGF-β-induced EMT (87, 92). We now show that insulin-induced motility depends on autocrine TGF-β signaling. Furthermore, as in gene expression responses, the increased TGF-β responsiveness allowed for cooperation between TGF-β and insulin signaling in the induction of cell migration. Similarly to the cell motility response, insulin promoted cell invasion in vitro, which was blocked in the absence of TGF-β signaling. These observations further support the participation of autocrine TGF-β signaling in the cellular response to insulin, and raise the possibility that continued exposure to insulin in vivo may affect epithelial and/or endothelial integrity through increased TGF-β signaling, which then may contribute to the progression of fibrosis.
Linking insulin signaling and Akt activation with TGF-β signaling in cancer progression
The linkage of Akt activation with TGF-β responsiveness may be relevant in cancer progression. Glioblastomas and carcinomas commonly show increased Akt activation (93, 94), and increased TGF-β signaling leads to epithelial plasticity or EMT, thus promoting cancer invasion and dissemination, and possibly cancer stem cell generation (1, 2, 6). Based on our results, we now propose that increased Akt signaling, resulting in AS160 phosphorylation, may enhance the TGF-β responsiveness of cancer cells, thus promoting invasion and dissemination. In support of this notion, Akt activation and AS160 phosphorylation in carcinomas correlate with cancer progression (95, 96). Although Akt activation promotes cell proliferation, glucose uptake and protein synthesis, its role in increasing TGF-β signaling, thus promoting cell invasion and dissemination, is also worth considering.
The increased TGF-β responsiveness in response to insulin or IGF-1, and cooperation between insulin and TGF-β signaling may be of further relevance, because carcinomas commonly show increased abundance of the IGF-1 receptor which can be activated by either IGF-1 or insulin (97). Thus, continued exposure of cancer cells to insulin may promote epithelial plasticity, cell invasion and migration, and cancer progression and dissemination, through cooperation of insulin with enhanced TGF-β signaling. Although insulin has multiple effects on tumor cells, the proposed mechanism of signaling cooperation may help understand the clinical and epidemiological association of increased cancer risk and cancer progression with insulin therapy in diabetic patients (98–101).
MATERIALS AND METHODS
Cell culture and transfections
NMuMG epithelial cells and wild-type and Tgfbr1−/− (102) mouse embryonic fibroblasts (MEFs) were cultured in DMEM with 4 mM glucose and 10% fetal bovine serum (FBS) at 37°C and 5% CO2 in a humidified incubator. Cells were transfected using Lipofectamine 2000 (Invitrogen) or RNAiMAX (Invitrogen) for double-stranded siRNA, according to the manufacturer’s protocol.
Expression plasmids encoding Flag- or Myc-tagged human TβRI or TβRII have been described (103). An expression plasmid encoding Flag-tagged AS160 was from Gustav E. Lienhard (Dartmouth Medical School, USA) (30), and a plasmid encoding NH2-terminally myristolated Akt2 was from Kevan Shokat (University California San Francisco, USA).
Insulin from bovine pancreas (Sigma) was used at 100 nM, human IGF-1(Sigma) was used at 50 ng/ml, and TGF-β1 from HumanZyme was used at 0.2 ng/ml. The TβRI kinase inhibitor SB4315242 (Sigma) was used at 5 μM and added 30 min before treatment. GDC0941 (Selleck Chemicals), Akt VIII (EMD Milipore), and U0126 (Sigma) were used at 10 μM, 5 μM, or 10 μM, respectively.
Determination of protein concentrations
Protein concentrations were quantified using Bio-Rad Bradford Assay (Biorad) with bovine serum albumin as standard, and a SpectraMax M5 microplate reader.
Immunoprecipitations and immunoblotting
293T cells were transfected, and lysed after 36 h in 150 mM NaCl, 25 mM Tris-Cl pH 8, 1 mM EGTA, 1% NP-40, 50 mM NaF, 5 mM glycerol phosphate, 5 mM Na4O7P2, 5 mM Na3VO4, and complete protease inhibitor cocktail (Roche). Protein associations were chemically stabilized by crosslinking using dithiobis succinimidyl propionate (DSP) (ThermoScientific), the lysates were precleared and subjected to immunoprecipitations with anti-Myc (9E10) antibody and Protein G Sepharose (GE Healthcare) for 4 h at 4°C. Beads were washed three times with 200 mM NaCl, 25 mM Tris-Cl pH 8, 1 mM EGTA, 50 mM NaF, 5 mM glycerol phosphate, 5 mM Na4O7P2, 5 mM Na3VO4, complete protease inhibitor cocktail (Roche), 1% glycerol. Specifically adsorbed proteins were eluted from the beads in LDS sample buffer (Invitrogen), analyzed by SDS-PAGE and immunoblotting.
For immunoblotting, we used the following antibodies: rabbit anti-human fibronectin, mouse anti-GAPDH, anti-Flag (M2) and anti-β-actin from Sigma; rabbit monoclonal antibodies to Smad2, phospho-Smad2 (Ser465/467), Smad3, Akt, Akt1, Akt2 phospho-Akt (Ser473), phospho-Akt (Thr308), Erk1/2, phospho-Erk1/2 (Thr202/Tyr204), AS160, phopho-AS160 (Thr642), Rab4, Rab11 (XP) vimentin, E-cadherin, N-cadherin, and Snail1 from Cell Signaling, rabbit polyclonal anti-AS160 from Milipore, rabbit polyclonal anti-GFP from Invitrogen, rabbit polyclonal anti-TGF-β receptor I and anti-phosphoSmad3 (Ser423/425) from Abcam, and rabbit polyclonal anti-TGF-β receptor II from Santa Cruz Biotechnology.
Cell surface protein biotinylation
Cell surface proteins were isolated as described (18). Cells were grown to 80–90% confluence before treatment, washed with ice-cold PBS, and incubated for 30 min with EZ-Link-Sulfo-NHS-LC-Biotin (Thermo Scientific). After blocking the non-reacted biotin with 0.1 mM glycine for 20 min, cells were lysed in MLB buffer (20 mM Tris-Cl, 200 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1% NP-40, complete protease inhibitor (Roche)), and the lysate was removed from the plate by scraping, then incubated on ice for 10 min, and centrifuged for 15 min (13,000 rcf, 4 °C) to remove the pelleted insoluble material. Supernatant proteins were incubated for 4 h to overnight with Neutravidin beads (ThermoScientific). Beads were washed three times with MLB with 1% glycerol, eluted with LDS sample buffer (Invitrogen) and subjected to SDS PAGE and followed by immunoblotting.
TGF-β Bioassay
Active TGF-β was measured using TMLC reporter cells (104). MEFs or NMuMG cells were starved with serum-free medium, stimulated with insulin for 30 min or 60 min, or left untreated. Conditioned media samples were then heated at 80°C for 10 min or kept at 4°C until assayed. TMLC cells were incubated with conditioned media or serial dilutions of a TGF-β standard for 12–24 hr. Unconditioned media without serum was used as a control (0 min). To increase the concentration of TGF-β in the media, conditioned media from MEFs or NMuMG cells were concentrated using Pierce Concentrator 7K (Pierce).
Immunofluorescence and cell imaging
Cells were fixed in 3.7% paraformaldehyde for 5 min, permeabilized in PBS with 0.2% Triton-X100 (PBS-T) for 5 min, and blocked with 5% BSA in PBS for 2 h at room temperature. Cells were incubated with rabbit-anti GFP (1:100, Invitrogen) and mouse anti-Flag (M2) at 1:100 diluted in 5% BSA/PBS overnight at 4°C, washed with PBS at room temperature, and then incubated with secondary Alexa Fluor-conjugated secondary (Invitrogen) in 5% BSA/PBS for 2 h at room temperature in the dark. Slides were mounted with Vectorshield and stained with DAPI. Immunofluorescence images were obtained using a Leica SP5 AOBS Upright 2 laser scanning confocal microscope. Changes in cell morphology were observed by phase contrast microscopy using a Leica microscope DMI5000, and analyzed using Photoshop CS5.
Luciferase reporter assays
Cells were transfected with the luciferase reporter plasmid 4xSBE-Luc (34) and the Renilla luciferase vector pRL-TK (Promega). Luciferase activities were quantified using a SpectraMax M5 microplate reader and the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s protocol. Data were normalized to Renilla luciferase and expressed as relative luminescence units (RLU).
Isolation of nuclear and cytoplasmic extracts
Nuclear and cytoplasmic extracts were isolated according to the nuclear extraction protocol from Life Technologies. Hypotonic buffer (20 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, Roche protease inhibitor Mini Complete) was used to lyse the cells, while keeping the nuclei intact, and thus extract the cytoplasmic proteins. RIPA extraction buffer was used to resuspend the nuclei and extract nuclear proteins. Protein concentrations in the nuclear and cytoplasmic extracts were determined using the Bradford assay.
Low density microsome subcellular fractionation
NMuMG cells, grown to 80–90% confluence in two 150 mm plates, were washed with ice-cold PBS, and harvested by scraping in 20 mM HEPES pH 7.4, 1 mM EDTA, 250 mM sucrose, 10 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and complete protease inhibitor cocktail (Roche). The separation of the high density microsomal (HDM) and low density microsomal (LDM) fractions was adapted from published work (84) with some changes. The cells were homogenized with 25 to 30 strokes using a tight-fitting Dounce homogenizer, and the homogenates were centrifuged at 500 rcf for 10 min at 4°C to remove unbroken cells. Equal supernatant protein amounts were centrifuged at 10,080 rcf for 20 min at 4°C. The cytosol (supernatant) fraction was then centrifuged using Sorvall RC6T at 15,750 rcf and 4°C for 20 min, and the resultant supernatant was re-centrifuged in 70Ti rotor (Beckman) at 175,000 rcf for 120 min at 4°C to separate the cytosol and pelleted LDM fractions. The resulting pellets (LDM) were resuspended in extraction buffer, loaded on a layered 10 – 36 % (wt/wt) discontinuous sucrose gradient cushion, and centrifuged using SW38 rotor (Beckman) for 12 h at 4°C at 284000 rpm. Twelve fractions were collected, starting from the top of the gradient, and equal volumes of each fraction were analyzed by SDS-PAGE followed by immunoblotting.
RNA interference
Double-stranded RNAi to silence mouse AS160 was from QIAGEN and targeted the coding sequence -5′-CCGGACCCUGGACAAGAUAA-3′. The RNAi to silence mouse TβRII was from Thermo Scientific and targeted the 3′UTR sequence - 5′- GGATAGCGTTAGCACTTGACA-3′. RNAi to silence mouse Akt1 (SI04715235) or Akt2 (SI00895930) was from QIAGEN. Cells were transfected using Lipofectamine RNAiMAX (Invitrogen) for 24 h, media were then replaced, and the cells were further incubated for 12–24 h before analyses. To stably silence TβRI in NMuMG cells, cells were infected with lentivirus expressing shRNA targeting the 3′UTR (Sigma) -5′-ATATCTGCTCCTGGGTGTTTA- or the lentiviral empty vector pLKO.1. The cells were selected with puromycin for 1 week before use.
Real-time RT-PCR
Total RNA was prepared using RNeasy Mini Kit (QIAGEN), according to the manufacturer’s protocol, and quantified by UV absorption using NaNo Drop at 260nm. One μg of total RNA was reverse transcribed using an iScript cDNA synthesis Kit (Bio-Rad), according to the manufacturer’s protocol. cDNA samples were subjected to real-time PCR using Bio-Rad PCR and the double strand-specific dye SYBR Green (Bio-Rad) using the following mouse primer sequences (forward, reverse): Snai1, (5′- AAGATGCACATCCGAAGC-3′ and 5′- ATCTCTTCACATCCGAGTGG -3′); Smad7 (5′-TCGGACAGCTCAATTCGGAC-3′, 5′- GGTAACTGCTGCGGTTGTAA-3′); Serpine1, (5′-TGCATCGCCTGCCATTG-3′; 5′-GGACCTTGAGATAGGACAGTGCTT-3′); Tbc1d4 (encoding AS160), (5′-TATTACCCAGGTTTTTGAGATGG-3′, 5′-GGTCCATGTTCTGTCTTTTCAAT-3′); FOXc2 (5′- GCAACCCAACAGCAAACTTTC-3′, 5′- GACGGCGTAGCTCGATAGG-3′); Cdh2, (5′- AGC GCA GTCTTACCGAAGG-3′, 5′ -TCGCTGCTTTCATACTGAACTTT-3′); Cdh1, (5′- CAGGTCTCCTCATGGCTTTGC -3′, 5′- CTTCCGAAAAGAAGGCTGTCC -3′); Fn1, (5′-GCAGTGACCACCATTCCTG-3′, 5′- GGTAGCCAGTGAGCTGAACAC-3′); Srebf1, (5′-AAGCCATCGACTACATCCG-3′, 5′-TCCATAGACACATCTGTGCCTC-3′); Vegfa, (5′-AATGCTTTCTCCGCTCTGAA-3′, 5′-CTCACCAAAGCCAGCACATA-3′); Tgf-β1, (5′-GCAACAATTCCTGGCGTTACC-3′, 5′-CCCTGTATTCCGTCTCCTTGGT-3′); Tgf-β2, (5′-TCGACATGGATCAGTTTATGCG-3′, 5′-CCCTGGTACTGTTGTAGATGGA-3′); Tgf-β3, (5′-CCTGGCCCTGCTGAACTTG-3′, 5′-TTGATGTGGCCCGAAGTCCAAC-3′); Gapdh, (5′-GCACAGTCAAGGCCGAGAAT-3′, 5′-GCCTTCTCCATGGTGGTGAA-3′). The mRNA expression values were normalized to the expression of Rpl19 mRNA (5′ ATGAGTATGCTCAGGCTACAGA-3′ and 5′-GCATTGGCGATTTCATTGGTC-3′).
In vitro wound healing assay
In vitro wound-healing assays were performed as described (87). NMuMG cells were seeded on cell culture dishes and treated for 24–36 h. Confluent cells were then scratched with a pipette tip across the entire diameter of the dish. Cells were rinsed twice using PBS to remove cell debris, and new culture medium was added. The denuded surface was photographed immediately after wounding and the migration into the denuded surface at later times was documented using Leica DMI5000 camera, followed by analyses using Photoshop CS5. The migration rate was quantified by calculating the distance before and after the scratch divided over the duration of the experiment.
Invasion assays
For invasion assays, performed as previously described (87), 50,000 cells were added to rehydrated Matrigel-coated inserts (BioCoat Matrigel Invasion Chamber; Becton Dickinson) in DMEM supplemented with 1% FBS. These inserts were then placed in companion plates with DMEM supplemented with 10% FBS for 24 h. Upper chambers were removed and cleaned using cotton tips. The filters were fixed in methanol for 5 min at −20°C, and mounted using Prolong Gold antifade reagent with DAPI (Invitrogen). Experiments were performed in duplicates and quantified by averaging the number of DAPI nuclei of view counting 5 fields per chamber. Images were analyzed using CellProfiler 2.1.1 (Broad Institute, USA).
Gelatin zymography
Cells were cultured in serum-free media for 12 h before treatment, and media samples were collected and centrifuged for 5 min at 2000 rcf to remove cell debris. The cleared media were transferred to a 9K MWCO concentrator (Thermo Scientific), and centrifuged at 2100 rcf for 25 min. Equal volumes of each fraction were run in SDS-PAGE gel containing 0.1% gelatin (Invitrogen), washed and incubated overnight at 37°C in gelatinase developing buffer (50 mM Tris base, 40 mM HCl, 200 mM NaCl, 5 mM CaCl2, 0.02% (w/v) Brij 35) before staining with Bio-Safe Coomassie Stain (Bio-Rad).
Statistical analyses
All statistical analyses were done using JMP 10; the methods used are mentioned in the figure legends.
Supplementary Material
Fig S1. Effects of high glucose on the insulin-induced increases in cell surface abundance of TβRI and TβRII.
Fig S2. Short term effects of insulin on the mRNA amounts and activation of TGF-β ligands.
Fig S3. Effects of IGF-1 on TGF-β receptor abundance at the surface.
Fig S4. Role of Akt activation in the high glucose-induced increase of cell surface TβRI and TβRII.
Fig S5. Contributions of Akt1 and Akt2 to the insulin response.
Fig S6. Insulin-induced phosphorylation of AS160 at Thr642.
Fig S7. Effect of decreasing AS160 abundance on the cell surface abundance of TGF-β receptors in NMuMG cells.
Fig S8. IGF-1 induces the expression of TGF-β target genes.
Fig S9. AS160 controls the expression of the TGF-β target genes encoding fibronectin or VEGF-A.
Fig S10. Relative expression of Snai2 mRNA after insulin treatment.
Fig S11. AS160 controls Snai1 mRNA expression.
Fig S12. Insulin and SB431542 do not affect cell numbers.
Fig S13. AS160 controls the epithelial plasticity response in the presence of low concentrations of TGF- β.
Acknowledgments
We thank Dr. Liyu Wu for early experiments that led to this research, Dr. Mark Anderson and Dr. Daniel Hart for critical reading, Dr. Katja Brueckner and the Derynck lab members for valuable suggestions, and Dr. Iryna Lobach for help and advice with the statistical analyses.
Funding: This research was sponsored by NIH grant CA136690 to R.D, a Juvenile Diabetes Research Fund postdoctoral fellowship # 3-2012-214, UCSF PBBR, and NIH T32 training grant support to E.H.B.
Reagents: We thank Dr. Gustav E. Lienhard (Dartmouth Medical School, USA) for the expression plasmid encoding Flag-tagged AS160 and Dr. Kevan Shokat ((University California San Francisco, USA) for the NH2-terminally myristolated Akt2.
Footnotes
Author contributions: E.H.B and R.D. conceived, designed and wrote the manuscript. E.H.B. performed the experiments, with B. P.M contributing to the sucrose gradient experiments.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Massagué J. TGFβ in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ikushima H, Miyazono K. TGFβ signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 3.Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011;73:413–435. doi: 10.1146/annurev-physiol-012110-142225. [DOI] [PubMed] [Google Scholar]
- 4.Moustakas A, Heldin P. TGFβ and matrix-regulated epithelial to mesenchymal transition. Biochim Biophys Acta. 2014;1840:2621–2634. doi: 10.1016/j.bbagen.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 6.Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 7.Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hills CE, Squires PE. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011;22:131–139. doi: 10.1016/j.cytogfr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Garud MS, Kulkarni YA. Hyperglycemia to nephropathy via transforming growth factor β. Current Diabetes Reviews. 2014;10:182–189. doi: 10.2174/1573399810666140606103645. [DOI] [PubMed] [Google Scholar]
- 10.Feng XH, Derynck R. Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 11.Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, Imamura T, Miyazono K, Aburatani H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor β signaling. Mol Cell Biol. 2009;29:172–186. doi: 10.1128/MCB.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moustakas A, Heldin CH. Non-Smad TGF-β signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 14.Zhang YE. Non-Smad pathways in TGF-β signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu C, Xu P, Lamouille S, Xu J, Derynck R. TACE-mediated ectodomain shedding of the type I TGF-β receptor downregulates TGF-β signaling. Mol Cell. 2009;35:26–36. doi: 10.1016/j.molcel.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massagué J, Niehrs C. Silencing of TGF-β signalling by the pseudoreceptor BAMBI. Nature. 1999;401:480–485. doi: 10.1038/46794. [DOI] [PubMed] [Google Scholar]
- 17.Yan X, Lin Z, Chen F, Zhao X, Chen H, Ning Y, Chen YG. Human BAMBI cooperates with Smad7 to inhibit transforming growth factor-β signaling. J Biol Chem. 2009;284:30097–30104. doi: 10.1074/jbc.M109.049304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu L, Derynck R. Essential role of TGF-β signaling in glucose-induced cell hypertrophy. Dev Cell. 2009;17:35–48. doi: 10.1016/j.devcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. In: Oxidative Stress in Cancer Biology and Therapy. Spitz DR, Dornfeld KJ, Krishnan K, Gius D, editors. Humana Press; New York: 2012. pp. 21–46. chap. 2. [Google Scholar]
- 20.Nathan DM. Clinical practice. Initial management of glycemia in type 2 diabetes mellitus. The New England journal of medicine. 2002;347:1342–1349. doi: 10.1056/NEJMcp021106. [DOI] [PubMed] [Google Scholar]
- 21.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 22.Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13:383–396. doi: 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- 23.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter Glut4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 24.Quon MJ, Chen H, Ing BL, Liu ML, Zarnowski MJ, Yonezawa K, Kasuga M, Cushman SW, Taylor SI. Roles of 1-phosphatidylinositol 3-kinase and Ras in regulating translocation of GLUT4 in transfected rat adipose cells. Mol Cell Biol. 1995;15:5403–5411. doi: 10.1128/mcb.15.10.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong LN, Chen H, Li Y, Zhou L, McGibbon MA, Taylor SI, Quon MJ. Physiological role of Akt in insulin-stimulated translocation of GLUT4 in transfected rat adipose cells. Mol Endocrinol. 1997;11:1881–1890. doi: 10.1210/mend.11.13.0027. [DOI] [PubMed] [Google Scholar]
- 26.Liang X, Butterworth MB, Peters KW, Frizzell RA. AS160 modulates aldosterone-stimulated epithelial sodium channel forward trafficking. Mol Biol Cell. 2010;21:2024–2033. doi: 10.1091/mbc.E10-01-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viard P, Butcher AJ, Halet G, Davies A, Nurnberg B, Heblich F, Dolphin AC. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nature Neurosci. 2004;7:939–946. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- 28.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nature Rev Drug Disc. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 30.Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 31.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 32.Sheu ML, Ho FM, Chao KF, Kuo ML, Liu SH. Activation of phosphoinositide 3-kinase in response to high glucose leads to regulation of reactive oxygen species-related nuclear factor-kappaB activation and cyclooxygenase-2 expression in mesangial cells. Molecular pharmacology. 2004;66:187–196. doi: 10.1124/mol.66.1.187. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell Membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 34.Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 35.Carcámo J, Zentella A, Massagué J. Disruption of transforming growth factor β signaling by a mutation that prevents transphosphorylation within the receptor complex. Mol Cell Biol. 1995;15:1573–1581. doi: 10.1128/mcb.15.3.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raynaud FI, Eccles SA, Patel S, Alix S, Box G, Chuckowree I, Folkes A, Gowan S, De Haven Brandon A, Di Stefano F, Hayes A, Henley AT, Lensun L, Pergl-Wilson G, Robson A, Saghir N, Zhyvoloup A, McDonald E, Sheldrake P, Shuttleworth S, Valenti M, Wan NC, Clarke PA, Workman P. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009;8:1725–1738. doi: 10.1158/1535-7163.MCT-08-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJ, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 38.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546:108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME, Lindsley CW. Discovery of 2,3,5-trisubstituted pyridine derivatives as potent Akt1 and Akt2 dual inhibitors. Bioorg Med Chem Lett. 2005;15:905–909. doi: 10.1016/j.bmcl.2004.12.062. [DOI] [PubMed] [Google Scholar]
- 40.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 41.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 43.Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- 44.Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci U S A. 2009;106:7004–7009. doi: 10.1073/pnas.0901933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng Y, Ramm G, Lopez JA, James DE. Rapid activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3-L1 adipocytes. Cell Metab. 2008;7:348–356. doi: 10.1016/j.cmet.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 47.Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391:87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Thong FSL, Bilan PJ, Klip A. The Rab GTPase-Activating Protein AS160 Integrates Akt, Protein Kinase C, and AMP-Activated Protein Kinase Signals Regulating GLUT4 Traffic. Diabetes. 2007;56:414–423. doi: 10.2337/db06-0900. [DOI] [PubMed] [Google Scholar]
- 50.Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 51.Jung HJ, Kwon TH. Membrane Trafficking of Collecting Duct Water Channel Protein AQP2 Regulated by Akt/AS160. Electrolyte Blood Press. 2010;8:59–65. doi: 10.5049/EBP.2010.8.2.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J Lipid Res. 2012;53:709–717. doi: 10.1194/jlr.M023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chavez JA, Roach WG, Mîinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J. 2007;403:353–358. doi: 10.1042/BJ20061798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- 55.Chen S, Wasserman DH, MacKintosh C, Sakamoto K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 2011;13:68–79. doi: 10.1016/j.cmet.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Watson RT, Pessin JE. Bridging the GAP between insulin signaling and GLUT4 translocation. Trends Biochem Sci. 2006;31:215–222. doi: 10.1016/j.tibs.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 57.Ren W, Cheema S, Du K. The association of ClipR-59 protein with AS160 modulates AS160 protein phosphorylation and adipocyte Glut4 protein membrane translocation. J Biol Chem. 2012;287:26890–26900. doi: 10.1074/jbc.M112.357699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-β receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 59.Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent transforming growth factor-β receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell. 2004;15:4166–4178. doi: 10.1091/mbc.E04-03-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayes S, Chawla A, Corvera S. TGF-β receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158:1239–1249. doi: 10.1083/jcb.200204088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sönnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct Membrane Domains on Endosomes in the Recycling Pathway Visualized by Multicolor Imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–913. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alves DS, Farr GA, Seo-Mayer P, Caplan MJ. AS160 associates with the Na+,K+-ATPase and mediates the adenosine monophosphate-stimulated protein kinase-dependent regulation of sodium pump surface expression. Mol Biol Cell. 2010;21:4400–4408. doi: 10.1091/mbc.E10-06-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ehrlich M, Gutman O, Knaus P, Henis YI. Oligomeric interactions of TGF-beta and BMP receptors. FEBS Lett. 2012;586:1885–1896. doi: 10.1016/j.febslet.2012.01.040. [DOI] [PubMed] [Google Scholar]
- 65.Chen RH, Moses HL, Maruoka EM, Derynck R, Kawabata M. Phosphorylation-dependent interaction of the cytoplasmic domains of the type I and type II transforming growth factor-beta receptors. J Biol Chem. 1995;270:12235–12241. doi: 10.1074/jbc.270.20.12235. [DOI] [PubMed] [Google Scholar]
- 66.Guller S, Allen DL, Corin RE, Lockwood CJ, Sonenberg M. Growth hormone and fibronectin expression in 3T3 preadipose cells. Endocrinology. 1992;130:2609–2616. doi: 10.1210/endo.130.5.1572284. [DOI] [PubMed] [Google Scholar]
- 67.Miele C, Rochford JJ, Filippa N, Giorgetti-Peraldi S, Van Obberghen E. Insulin and insulin-like growth factor-I induce vascular endothelial growth factor mRNA expression via different signaling pathways. J Biol Chem. 2000;275:21695–21702. doi: 10.1074/jbc.M000805200. [DOI] [PubMed] [Google Scholar]
- 68.Lu M, Amano S, Miyamoto K, Garland R, Keough Karen, Qin W, Adamis AP. Insulin-induced vascular endothelial growth factor expression in retina. Invest Ophthalmol Vis Sci. 1999;40:3281–3286. [PubMed] [Google Scholar]
- 69.Gronning LM, Cederberg A, Miura N, Enerback S, Tasken K. Insulin and TNF α induce expression of the forkhead transcription factor gene Foxc2 in 3T3-L1 adipocytes via PI3K and ERK 1/2-dependent pathways. Mol Endocrinol. 2002;16:873–883. doi: 10.1210/mend.16.4.0803. [DOI] [PubMed] [Google Scholar]
- 70.Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrin Metab. 2012;23:65–72. doi: 10.1016/j.tem.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Connor JW, Gomez EW. Biomechanics of TGF-β-induced epithelial-mesenchymal transition: implications for fibrosis and cancer. Clin Transl Med. 2014;3:23. doi: 10.1186/2001-1326-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 74.Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005;118:873–887. doi: 10.1242/jcs.01634. [DOI] [PubMed] [Google Scholar]
- 75.Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci. 2012;125:1259–1273. doi: 10.1242/jcs.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thuault S, Tan EJ, Peinado H, Cano A, Heldin CH, Moustakas A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem. 2008;283:33437–33446. doi: 10.1074/jbc.M802016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y, Petreaca M, Yao M, Martins-Green M. Cell and molecular mechanisms of keratinocyte function stimulated by insulin during wound healing. BMC Cell Biol. 2009;10:1. doi: 10.1186/1471-2121-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shanley LJ, McCaig CD, Forrester JV, Zhao M. Insulin, not leptin, promotes in vitro cell migration to heal monolayer wounds in human corneal epithelium. Invest Ophthalmol Vis Sci. 2004;45:1088–1094. doi: 10.1167/iovs.03-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fischoeder A, Meyborg H, Stibenz D, Fleck E, Graf K, Stawowy P. Insulin augments matrix metalloproteinase-9 expression in monocytes. Cardiovasc Res. 2007;73:841–848. doi: 10.1016/j.cardiores.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 81.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J Biol Chem. 1999;274:13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 82.Ramm G. A Role for 14-3-3 in Insulin-stimulated GLUT4 Translocation through Its Interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- 83.Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell. 2004;15:4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tan SX, Ng Y, Burchfield JG, Ramm G, Lambright DG, Stockli J, James DE. The Rab GTPase-activating protein TBC1D4/AS160 contains an atypical phosphotyrosine-binding domain that interacts with plasma membrane phospholipids to facilitate GLUT4 trafficking in adipocytes. Mol Cell Biol. 2012;32:4946–4959. doi: 10.1128/MCB.00761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rahimi RA, Andrianifahanana M, Wilkes MC, Edens M, Kottom TJ, Blenis J, Leof EB. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-β. Cancer Res. 2009;69:84–93. doi: 10.1158/0008-5472.CAN-08-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ziyadeh FN. Mediators of diabetic renal disease: the case for TGF-β as the major mediator. J Am Soc Nephrol. 2004;15(Suppl 1):S55–57. doi: 10.1097/01.asn.0000093460.24823.5b. [DOI] [PubMed] [Google Scholar]
- 87.Lamouille S, Derynck R. Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–451. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nakagawa T, Li J, Garcia G, Mu W, Piek E, Böttinger EP, Chen Y, Zhu H, Kang D, Schreiner G, Lan H, Johnson R. TGF-β induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 2004;66:605–613. doi: 10.1111/j.1523-1755.2004.00780.x. [DOI] [PubMed] [Google Scholar]
- 89.Goel HL, Mercurio AM. VEGF targets the tumour cell. Nature Rev Cancer. 2013;13:871–882. doi: 10.1038/nrc3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Liepvre X, Berthelier-Lubrano C, Spiegelman B, Kim JB, Ferre P, Foufelle F. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shigematsu S, Yamauchi K, Nakajima K, Iijima S, Aizawa T, Hashizume K. D-Glucose and insulin stimulate migration and tubular formation of human endothelial cells in vitro. Am J Physiol. 1999;277:E433–438. doi: 10.1152/ajpendo.1999.277.3.E433. [DOI] [PubMed] [Google Scholar]
- 92.Xu J, Lamouille S, Derynck R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Network TC. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- 95.Jiang XH, Sun JW, Xu M, Jiang XF, Liu CF, Lu Y. Frequent hyperphosphorylation of AS160 in breast cancer. Cancer Biol Ther. 2014;10:362–367. doi: 10.4161/cbt.10.4.12426. [DOI] [PubMed] [Google Scholar]
- 96.Cheng JC, McBrayer SK, Coarfa C, Dalva-Aydemir S, Gunaratne PH, Carpten JD, Keats JK, Rosen ST, Shanmugam M. Expression and phosphorylation of the AS160_v2 splice variant supports GLUT4 activation and the Warburg effect in multiple myeloma. Cancer Metab. 2013;1:14. doi: 10.1186/2049-3002-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A, Vigneri R. The role of insulin receptors and IGF-I receptors in cancer and other diseases. Arch Physiol Biochem. 2008;114:23–37. doi: 10.1080/13813450801969715. [DOI] [PubMed] [Google Scholar]
- 98.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. Diabetes Care. 2010;33:1674–1685. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnson JA, Gale EA. Diabetes, insulin use, and cancer risk: are observational studies part of the solution-or part of the problem? Diabetes. 2010;59:1129–1131. doi: 10.2337/db10-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barone BB, Yeh HC, Snyder CF, Peairs KS, Stein KB, Derr RL, Wolff AC, Brancati FL. Long-term all-cause mortality in cancer patients with preexisting diabetes mellitus: a systematic review and meta-analysis. JAMA. 2008;300:2754–2764. doi: 10.1001/jama.2008.824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peairs KS, Barone BB, Snyder CF, Yeh HC, Stein KB, Derr RL, Brancati FL, Wolff AC. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol. 2011;29:40–46. doi: 10.1200/JCO.2009.27.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Larsson J, Blank U, Helgadottir H, Bjornsson JM, Ehinger M, Goumans MJ, Fan X, Leveen P, Karlsson S. TGF-β signaling-deficient hematopoietic stem cells have normal self-renewal and regenerative ability in vivo despite increased proliferative capacity in vitro. Blood. 2003;102:3129–3135. doi: 10.1182/blood-2003-04-1300. [DOI] [PubMed] [Google Scholar]
- 103.Feng XH, Filvaroff EH, Derynck R. Transforming growth factor-β (TGF-β)-induced down-regulation of cyclin A expression requires a functional TGF-β receptor complex. Characterization of chimeric and truncated type I and type II receptors. J Biol Chem. 1995;270:24237–24245. doi: 10.1074/jbc.270.41.24237. [DOI] [PubMed] [Google Scholar]
- 104.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-β using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 105.Thoumine O, Lambert M, Mege RM, Choquet D. Regulation of N-cadherin dynamics at neuronal contacts by ligand binding and cytoskeletal coupling. Mol Biol Cell. 2006;17:862–875. doi: 10.1091/mbc.E05-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang JY, Zong CS, Xia W, Wei Y, Ali-Seyed M, Li Z, Broglio K, Berry DA, Hung MC. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Mol Cell Biol. 2006;26:7269–7282. doi: 10.1128/MCB.00172-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Effects of high glucose on the insulin-induced increases in cell surface abundance of TβRI and TβRII.
Fig S2. Short term effects of insulin on the mRNA amounts and activation of TGF-β ligands.
Fig S3. Effects of IGF-1 on TGF-β receptor abundance at the surface.
Fig S4. Role of Akt activation in the high glucose-induced increase of cell surface TβRI and TβRII.
Fig S5. Contributions of Akt1 and Akt2 to the insulin response.
Fig S6. Insulin-induced phosphorylation of AS160 at Thr642.
Fig S7. Effect of decreasing AS160 abundance on the cell surface abundance of TGF-β receptors in NMuMG cells.
Fig S8. IGF-1 induces the expression of TGF-β target genes.
Fig S9. AS160 controls the expression of the TGF-β target genes encoding fibronectin or VEGF-A.
Fig S10. Relative expression of Snai2 mRNA after insulin treatment.
Fig S11. AS160 controls Snai1 mRNA expression.
Fig S12. Insulin and SB431542 do not affect cell numbers.
Fig S13. AS160 controls the epithelial plasticity response in the presence of low concentrations of TGF- β.