

Abstract
A computational docking study of a series of de novo structural analogs of the highly potent, non-nitrogen containing, acetylcholinesterase inhibitor (+)-arisugacin A is presented. In direct comparison to the recently reported X-ray single-crystal structure of (+)-territrem B bound hAChE, the modeling suggests that there is a unique conformational preference for the E-ring that is responsible for the superior inhibitory activity of (+)-arisugacin A against hAChE relative to (+)-territrem B, and that substitutions on the E-ring also play an important role in the protein-ligand interaction.
Keywords: (+)-Arisugacin A, E-ring conformation, E-ring substitutions, Territrems, Acetylcholinesterase, Alzheimer's disease, Computational modeling
Graphical abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
We recently disclosed a computational study of the highly potent, non-nitrogen atom containing, acetylcholinesterase [AChE] inhibitor (+)-arisugacin A,1 which was isolated from Penicillium Sp. Fo-425 with an impressive IC50 of 1.0 nM against AChE [Figure 1].2,3 Our modeling results obtained using AutoDock Vina4 reveal that (+)-arisugacin A is a potential dual binding site and possibly a covalent inhibitor of AChE, thereby representing a novel mode of action. As highlighted in Figure 1, a set of predicted non-covalent interactions (a)–(e) between (+)-arisugacin A and AChE [Torpedo Californica AChE or tcAChE numbering] are quite consistent with the only available SAR data reported by Peng5a using (+)-territrem B,5b a close structural relative of (+)-arisugacin A [vide infra].5b-1 More significantly, we identified two additional key and likely critical features that render (+)-arisugacin A unique in its mode of binding.
Figure 1.
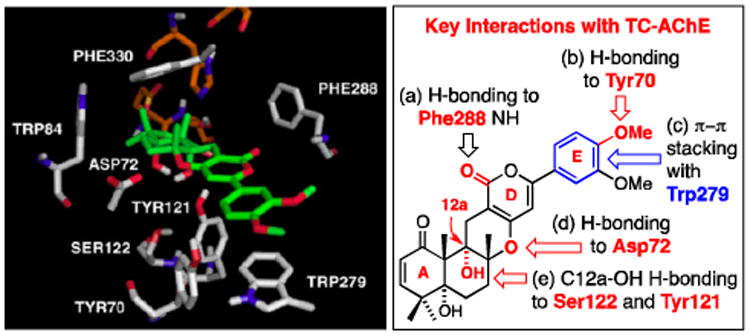
Predicted Signature Interactions in the Binding of (+)-Arisugacin A to Torpedo Californica AChE (tcAChE) Using AutoDock Vina.
Firstly, a close view of the catalytic site near the classical serine protease triad, His440, Ser200, and Glu327, evokes the potential for the engagement of Ser200 with the A-ring enone through either 1,2-addition of Ser200-OH to the C1-carbonyl or 1,4-addition to C3, thereby suggesting a possible reversible covalent inhibition as the mode of action.1,6-8 Although this possibility may be dependent on the ability of (+)-arisugacin A to move within the binding pocket leading to C1 or C3 proximity to Ser200, it is consistent with Peng's observation5a that the α,β-unsaturated ketone motif in the A-ring is significant. Secondly, we demonstrated that (+)-arisugacin A mimics donepezil [Aricept™],9 which is a dual binding site inhibitor of AChE. These two ligands share a H-bonding interaction between the Phe288-NH and the respective ligand carbonyls, a π-stacking interaction between Trp279 and aromaticity in the respective ligands, and H-bonding between Tyr70 and the respective methoxy-aryl groups [Figure 2B].
Figure 2.
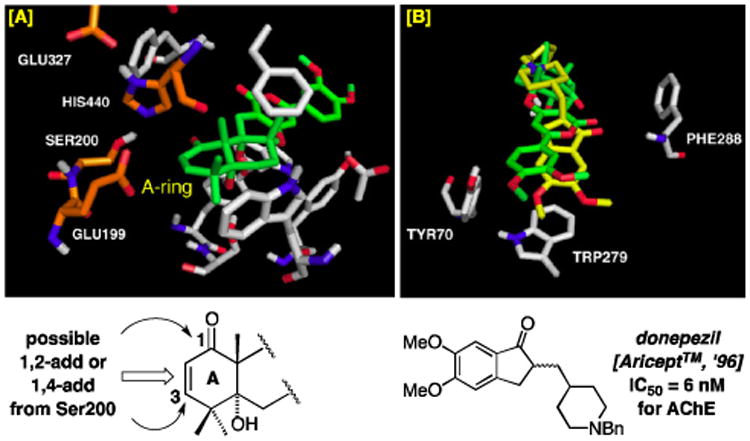
[A] Possible Covalent Interactions Between tcAChE and the A-Ring of (+)-Arisugacin A. [B] Overlay of Binding of (+)-Arisugacin A (in Green) to tcAChE with Donepezil (in Yellow).
These findings can be significant in the context of developing disease-modifying therapeutics targeting symptomatic aspects of dementia diseases with a combined long-term efficacy in neuroprotection. Alzheimer's disease [AD]10 affects well over 24 million people worldwide11,12 mostly in the geriatric population. With the exact pathogenic mechanism of this degenerative and devastating syndrome remaining unknown, the current leading theory in disease-modifying therapeutic approaches is the amyloid cascade hypothesis. It focuses on β-amyloid peptides [Aβ], which is a primary component of the neurotoxic senile plaques, with specific intents of (a) inhibiting its production,13 (b) preventing its aggregation, and (c) altering its metabolism and clearance.14 On the other hand, the long standing cholinergic deficiency hypothesis12,15 represents almost the only rational design that has led to FDA approved drugs such as tacrine, rivastigmine, galanthamine, and huperzine A [Figure 3] as well as donepezil [see Figure 2B].16-18
Figure 3.
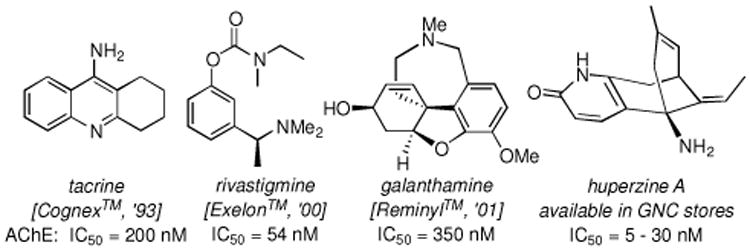
AChE Inhibitors Approved as Therapeutic drugs for Combating AD. Respective Trade Names and the Year of Release are in Brackets.
While the two hypotheses appear to be parallel, recent findings believe that they are in fact not mutually exclusive but strongly interconnected. Firstly, acetylcholine [ACh] activation of the muscarinic receptors leads indirectly to increases in the non-amyloidogenic α-secretase processing of Aβ precursor protein (APP),19 thereby linking simple ACh-hydrolysis inhibition to the diversion of APP away from Aβ production. Secondly, pro-aggregating ability of AChE on Aβ formation has been recognized20 to involve the enzyme's peripheral anionic site (PAS) located at the entrance of the active site gorge. Thus, AChE inhibitors able to bind to PAS have been found to inhibit this secondary enzyme activity.20a Consequently, there has been a re-emerging interest in AChE inhibitor-based drug development, leading to a new paradigm of developing disease-modifying drugs that target multiple pathways associated with AChE with modulations of both the cholinergic and amyloid targets through dual binding site inhibition. The goal is not only impeding ACh-hydrolysis, but also (a) aiding or improving PAS binding, leading to enhanced inhibition of Aβ pro-aggregating activity of AChE,21,22 and/or (b) concomitantly inhibiting β-secretase (BACE-1) activity, the enzyme involved in the rate-limiting step of APP processing into Aβ.23
In light of these findings, our earlier modeling suggests that designing dual binding site ligands based on (+)-arisugacin A can be significant in identifying novel AD therapeutics capable of targeting early stages of the neurotoxic cascade while mitigating the cholinergic deficiency. Recently, our computational evidence of the mode of binding of (+)-arisugacin A in tcAChE was confirmed by Cheung et al.'s seminal X-ray crystal structure of human AChE [hAChE] bound with (+)-territrem-B [TB].24 Their reported structure is almost in exclusive agreement with our model [vide infra].1 Encouraged by this success, we expanded our modeling studies to the arisugacin and territrem families of natural products as well as a series of de novo analogs focusing on the E-ring. We wish to report here computational evidence in which the E-ring plays a significant role in the binding of (+)-arisugacin A to hAChE.
Cheung's published X-ray crystal structure of (+)-territrem B-bound-hAChE [TB–hAChE] provided the foundation for the study of the binding affinities of the arisugacins and territrems. Specifically, the reported TB–hAChE24 bound structure appears to better accommodate arisugacin-like structures with a large degree of reorganization around the catalytic active site and protein backbone in the peripheral site that led to an altered gorge shape.24 Our modeling study employed AutoDock v4.2 and the more recent version in AutoDock Vina,4 made available by The Scripps Research Institute under a General Public License. While these models do allow for flexibility in selected protein residues, these degrees of freedom do not account for the conformation changes seen between the donepizil-tcAChE (protein structure source in our 2011 study1) and TB–hAChE24 active site gorge conformations. As shown in Figure 4, our docking of TB in hAChE matched perfectly the reported structure.
Figure 4.
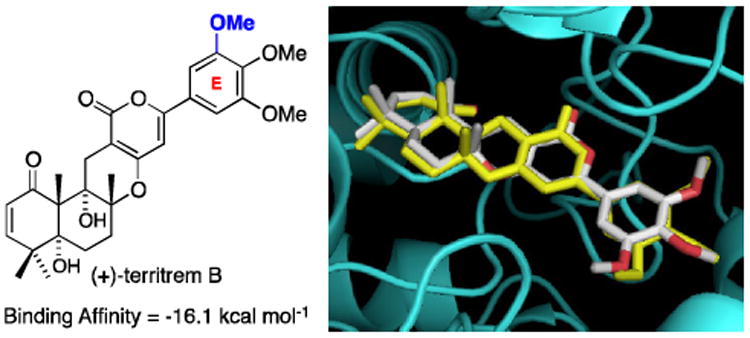
An Overlay of the Predicted Binding of TB to hAChE using AutoDock (in White) and the Observed Binding of TB to hAChE in the Actual X-Ray Structure obtained by Cheung et al. (in Yellow).
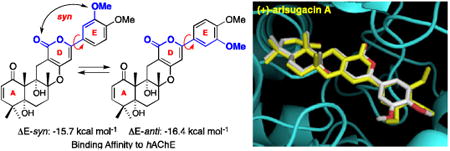
When examining the predicted binding of (+)-arisugacin A with hAChE and comparing these docking events with the X-ray structure of TB–hAChE, we uncovered an intriguing as well as significant observation. As shown in Figure 5, while the X-ray structure of TB-hAChE nails the predicted binding of (+)-arisugacin A to hAChE, there is a minor structural difference between (+)-arisugacin A and TB: (+)-Arisugacin A has one less m-OMe group on the E-ring. This minor difference disrupts the E-ring symmetry seen in TB, thus rendering two possible E-ring conformations for (+)-arisugacin A. The D and E-rings of (+)-arisugacin A could be regarded as being syn when the m-OMe group on the E-ring is syn to the α-pyrone-carbonyl of D-ring, while being anti when the m-OMe group is anti to the α-pyrone-carbonyl group. Intriguingly, these two conformations pose a real difference in their respective binding energies. Specifically, we found that the conformer (+)-arisugacin A-DE-syn has a binding affinity of -15.7 kcal mol-1, which is inferior to that of TB [-16.1 kcal mol-1]; while (+)-arisugacin A-DE-anti has a superior binding affinity at -16.4 kcal mol-1
Figure 5.
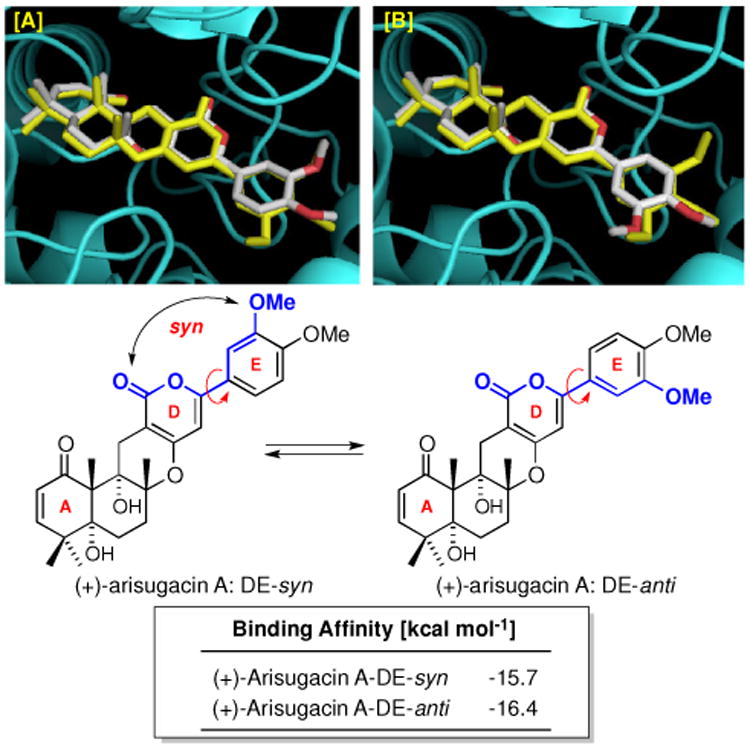
[A] An Overlay of the Predicted Binding of (+)-Arisugacin A-DE-syn to hAChE using AutoDock and the Observed Binding of TB–hAChE in the X-Ray Structure. [B] An Overlay of the Predicted Binding of (+)-Arisugacin A-DE-anti with the Observed Binding of TB.
This difference in binding affinities aroused our interest because TB is reported by Õmura,2a,2d to possess an inferior inhibitory power against AChE versus that of (+)-arisugacin A. We recognized that our observation appears to be more than just a coincidence, and that binding of (+)-arisugacin A to hAChE may very well favor the anti-conformation with respect to the DE-ring. Consequently, we proceeded to obtain binding affinities of all known members from both arisugacin and territrem families. As shown in Table 1, we compare the predicted binding affinities with their respective IC50 values reported by Õmura,2a,2d and tabulated Δ affinities against the analog we named as (+)-arisugacin-Ph in which the E-ring contains no OMe substitutions [entry 1].
Table 1.
A Comparison of Reported IC50 Values [All are taken from reports by Õmura: See References 2a and 2d] against AChE with Predicted Binding Affinity Values using AutoDock. Δ Affinity = Binding Affinity of the Inhibitor Subtracting Affinity of Arisugacin-Ph [entry 1].
| entry | AChE Inhibitors | IC50 [in μM] | binding affin it y [kcal mol-1] | Δ affinity |
|---|---|---|---|---|
| 1 | Arisugacin–Ph | -- | -15.4 | 0.0 |
| 2 | (+)-Arisugacin A-DE-syn | 0.001 | -15.7 | -0.3 |
| 3 | (+)-Arisugacin A-DE-anti | 0.001 | -16.4 | -1.0 |
| 4 | (+)-Arisugacin B | 0.0258 | -15.8 | -0.4 |
| 5 | (+)-Arisugacin C | 2.5 | -15.6 | -0.2 |
| 6 | (+)-Arisugacin D | 3.5 | -12.8 | +2.6 |
| 7 | (+)-Arisugacin E | >100.0 | -13.9 | +1.5 |
| 8 | (+)-Arisugacin F | >100.0 | -15.0 | +0.4 |
| 9 | (+)-Arisugacin G | >100.0 | -15.6 | -0.2 |
| 10 | (+)-Arisugacin H | >100.0 | -12.4 | +3.0 |
| 11 | (+)-Territrem A-DE-syn | -- | -16.4 | -1.0 |
| 12 | (+)-Territrem A-DE-anti | -- | -16.3 | -0.9 |
| 13 | (+)-Territrem B | 0.0076 | -16.1 | -0.7 |
| 14 | (+)-Territrem C | 0.0068 | -16.1 | -0.7 |
Our tabulated analysis distinctly reveals potential significance of not only the meta- but also the para-OMe group on the E-ring of arisugacins and territrems. Lack of an OMe substitution at the meta position, as shown with (+)-arisugacin B, appears to be associated with inferior binding affinity as well as IC50 value [entry 4 versus 2 or 3]. On the other hand, although the experimental IC50 value of (+)-arisugacin-Ph is not known, it appears that the binding affinity suffers when devoid of the p-OMe group [entry 1 versus 4]. Most notably, as we had eluded to, TB has an IC50 value of 7.6 nM2a,2d [entry 13] versus that of 1 nM for (+)-arisugacin A [entry 2 and 3].2a However, only when the DE-ring is in the anti conformation is the predicted binding affinity consistent with this lower IC50 value [entry 3].
In addition, intriguingly, (+)-territrem A [TA] also exhibits a binding affinity difference between DE-syn and anti [entries 11 and 12], although to the best of our knowledge, the IC50 value of TA against AChE has not been documented. Lastly, our predicted binding affinities should correlate to much lower IC50 values for (+)-arisugacins C-H [entries 5-10 – all in blue]. These analogs involve structural alterations on the A-ring5 and/or deletions of the angular OH groups. Discrepancies in IC50 values and predicted binding affinities may be due to physical properties differences (ex. solubility, compare Arisugacin B to G with one less degree of unsaturation and two less hydroxyl groups), although we are not certain at this point as to the real rationale behind these differences.
Intrigued by these observations, we attempted through modeling to identify possible interactions that would be consistent with the DE-ring conformational preference as well as benefit of the p-OMe group. As shown in Figure 7, we found three key interactions that could rationalize our finding. Firstly, the presence of an m-OMe group at the syn position has an energetic cost of 0.1 to 0.3 kcal mol-1. This cost is likely due to possible steric interactions between the syn m-OMe group with the TYR341 and SER293 carbonyl groups. Secondly, the presence of m-OMe at the anti position actually provides an energetic benefit of 0.5 kcal mol-1. Possible H-bonding between the anti m-OMe group and the TYR72-OH group could lead to such benefit. Thirdly, the presence of p-OMe appears to have an energetic benefit of 0.4 kcal mol-1. This could be a result of possible interaction between the p-OMe group and the TRP286 methylene unit through van der Waal forces.
Figure 7.
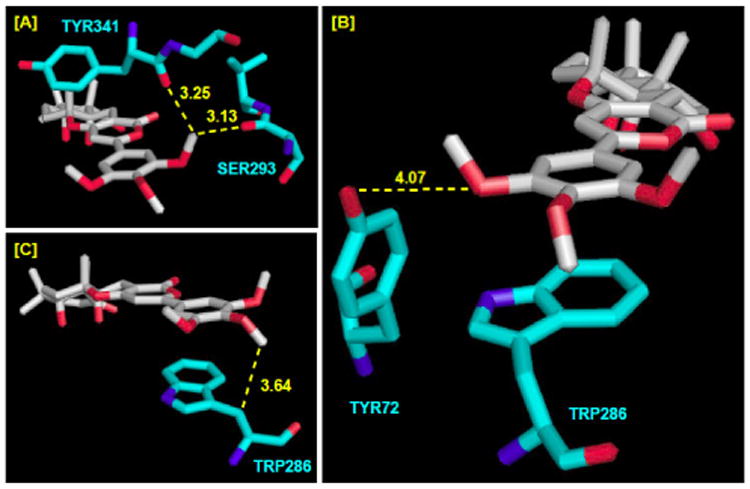
Close Views of [A] TB's m-OMe-syn. [B] TB's m-OMe-anti. [C] TB's p-OMe. Interatomic distances (Å) in yellow dotted lines. All are hAChE numberings.
Recognizing the potential significance of our finding, we proceeded to examine a series of de novo arisugacin structural analogs with a focus on the E-ring substitution patterns through modeling and found striking evidence that further accentuates the impact of the E-ring on binding.25 Firstly, we observed with the arisugacin analog (+)-arisugacin-m-OMe with the E-ring containing just one OMe group at the meta position, the anti conformer again possesses superior binding affinity than its respective syn conformer [Figure 8]. On the other hand, the analog with bis-m-OMe groups provides no further improvement on binding affinity than (+)-arisugacin-m-OMe-DE-anti – until an OMe group is re-enlisted at the para position, which is of course TB itself! This study is in agreement with the three key interactions described above.
Figure 8.
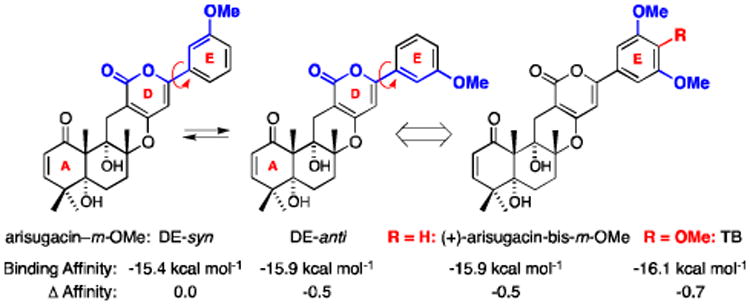
(+)-Arisugacin Analogs Containing Either Mono m-OMe Group or Bis-m-OMe Groups on the E-Ring. Δ Affinity = Binding Affinity of the Analog Subtracting Affinity of Arisugacin-Ph [-15.4 kcal mol-1].
The second study stems from the observation that (+)-territrem B and (+)-territrem C [TC] differ only by the group at the para position on the E-ring: OMe in B versus OH in C. Their binding to hAChE and IC50 values appear to be very comparable with TC being slightly lower. This prompted us to systematically examine if changes from OMe to OH would improve or diminish binding. As shown in Table 2, a general trend with some additive value can be summarized as following: (a) When the syn m-OMe group is replaced by an OH group, an enhancement in binding was found [entry 4 vs. 3; 12 vs. 11; and 15 vs. 13]. This enhancement is likely very much related to diminished steric interaction between the syn m-OMe group and the TYR341 and SER293 carbonyl groups. (b) When an m-OMe group is replaced with OH group at the anti position, diminish binding is observed [entry 8 vs. 7 and 16 vs. 13]. One exception is shown in entry 14 in comparison to TB in entry 11. (c) A p-OH group also leads to diminish binding affinities [entry 2 vs. 1; 5 vs. 3; and 9 vs. 7].
Table 2.
Impact of E-Ring Substitutions on Predicted Binding Affinities. Δ Affinity = Binding Affinity of the Analog Subtracting Affinity of Arisugacin-Ph [-15.4 kcal mol-1].
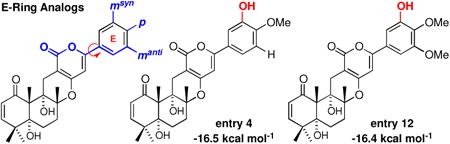
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | NPs and Analogs | msyn | p | manti | binding affinity [kcal mol-1] | Δ affinity |
| 1 | (+)-Arisugacin B | H | OMe | H | -15.8 | -0.4 |
| 2 | (+)-Arisugacin B-OH | H | OH | H | -15.5 | -0.1 |
| 3 | (+)-Arisugacin A-DE-syn | OMe | OMe | H | -15.7 | -0.3 |
| 4 | OH | OMe | H | -16.5 | -1.1 | |
| 5 | OMe | OH | H | -15.6 | -0.2 | |
| 6 | OH | OH | H | -16.1 | -0.7 | |
| 7 | (+)-Arisugacin A-DE-anti | H | OMe | OMe | -16.4 | -1.0 |
| 8 | H | OMe | OH | -16.3 | -0.9 | |
| 9 | H | OH | OMe | -16.1 | -0.7 | |
| 10 | H | OH | OH | -15.9 | -0.5 | |
| 11 | (+)-Territrem B | OMe | OMe | OMe | -16.1 | -0.7 |
| 12 | OH | OMe | OMe | -16.4 | -1.0 | |
| 13 | (+)-Territrem C | OMe | OH | OMe | -16.1 | -0.7 |
| 14 | OMe | OMe | OH | -16.3 | -0.9 | |
| 15 | OH | OH | OMe | -16.3 | -0.9 | |
| 16 | OMe | OH | OH | -15.9 | -0.5 | |
| 17 | OH | OMe | OH | -16.3 | -0.9 | |
| 18 | OH | OH | OH | -15.9 | -0.5 | |
Overall, we seem to have hit a plateau at -16.5 kcal mol-1 for binding affinities through simple changes of OMe groups on the E-ring. This however unveils a structure that represents the best arisugacin analog [entry 4]. Another plateau is at -16.4 kcal mol-1 for the best TB structural analog [entry 12]. It is noteworthy that with a switch of the m-OMe group to OH, the respective arisugacin analogs tend to have a higher binding affinity in the syn DE-ring conformation based on our definition rather than the anti conformer, a reversal from that observed with (+)-arisugacin A [compare entries 3-7 with 4-8, 12 to 14, and 15 to 16]. This improvement is likely associated with both relief of steric interactions between the OMe group and the TYR341 and SER293 carbonyl groups, and introduction of at least a new H-bonding interaction between the m-OH and the SER293 carbonyl with maintained proximity to the TYR341 carbonyl [Figure 9].
Figure 9.
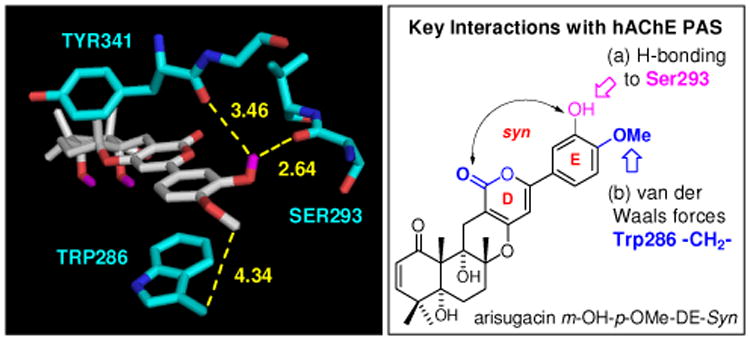
Predicted Interactions of Arisugacin m-OH-p-OMe-DE Syn with the hAChE PAS. Interatomic distances (Å) in yellow dotted lines. All are hAChE numberings.
We note here that we are aware of the fact that binding affinity differences may not arise solely from positioning of E-ring substituents, as any substitution change could impact the position of the entire scaffold.24 In addition, as much as one would expect to gain in binding affinity by improving or building in new non-covalent interactions, there is an empirical basis to expect non-additive and/or overall diminishing returns. This relates to the empirical limits of achievable binding affinities of small molecule ligands.26,27 However, while Table 2 may be presenting a simplistic view, its general trend, as summarized, can serve as a useful aid for designing potent inhibitors of AChE based on how presence or lack of a substituent could impact binding. Our current study demonstrates that it is possible to identify more powerful inhibitors than (+)-arisugacin A.
In summary, we have examined computational dockings of a series of de novo structural analogs of the highly potent, non-nitrogen containing, acetylcholinesterase inhibitor (+)-arisugacin A. Aided by the X-ray crystal structure of (+)-territrem B bound hAChE, our modeling suggests that there is a unique conformational preference for the E-ring that is responsible for the superior affinity of (+)-arisugacin A to hAChE in comparison to (+)-territrem B. In addition, substitutions on the E-ring appear to play a critical role in binding to hAChE. These computational assessments present useful parameters for future designs of potential disease-modifying therapeutics based on (+)-arisugacin A. Synthetic efforts toward some of these computationally designed structural analogs are underway.
Supplementary Material
Figure 6.
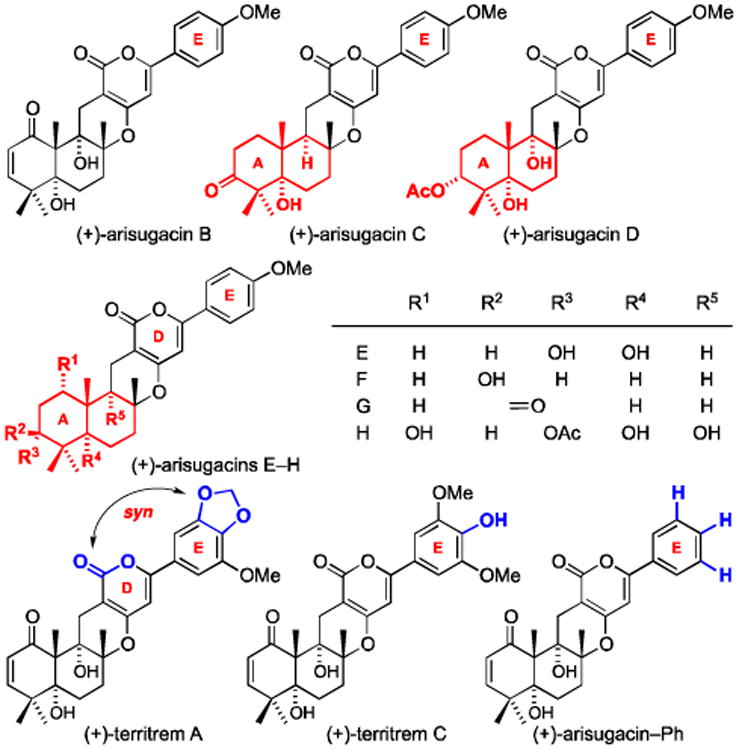
Arisugacins A-H and Territrems A-C.
Acknowledgments
We thank the NIH [NS38049] for financial support, and The Scripps Research Institute for making available the AutoDock docking program and supporting literature.
Footnotes
Supplementary Material: Supplementary material presenting AChE docking model development and validation has been provided.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Al-Rashid ZF, Hsung RP. Bioorg Med Chem Lett. 2011;21:2687. doi: 10.1016/j.bmcl.2010.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For leading references on arisugacins, see: Otoguro K, Shiomi K, Yamaguchi Y, Arai N, Sunazuka T, Masuma R, Iwai Y, Õmura S. J Antibiot. 2000;53:50. doi: 10.7164/antibiotics.53.50.Otoguro K, Kuno F, Õmura S. Pharmacol Ther. 1997;76:45. doi: 10.1016/s0163-7258(97)00093-4. and leading references cited within. For biological activities of arisugacin, see: Kuno F, Otoguro K, Shiomi K, Iwai Y, Õmura S. J Antibiot. 1996;49:742. doi: 10.7164/antibiotics.49.742.Õmura S, Kuno F, Otoguro K, Sunazuka T, Shiomi K, Masuma R, Iwai Y. J Antibiot. 1995;48:745. doi: 10.7164/antibiotics.48.745..
- 3.For reviews on arisugacin A, see: Hsung RP, Cole KP. Strategies and Tactics in Organic Synthesis. Vol. 4. Harmata, M Ed., Elsevier Science, Pergamon Press; Oxford, UK: 2004. pp. 41–70.Sunazuka T, Õmura S. Chem Rev. 2005;105:4559. doi: 10.1021/cr040628i. For total synthesis of arisugacin A, see: Hsung RP, Cole KP, Zehnder LR, Wang J, Wei LL, Yang XF, Coverdale HA. Tetrahedron. 2003;59:311.Sunazuka T, Handa M, Nagai K, Shirahata T, Harigaya Y, Otoguro K, Kuwajima I, Õmura S. Tetrahedron. 2004;60:7845..
- 4.For references on AutoDock, see: Version 4.2 Morris G, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson AJ. J Comp Chem. 1998;19:1639. Vina Trott O, Olson AJ. J Comp Chem. 2010;31:455. doi: 10.1002/jcc.21334..
- 5.Peng FC. J Nat Prod. 1995;58:857. doi: 10.1021/np50120a006. [DOI] [PubMed] [Google Scholar]; For original isolations of territrems A-C, see: Ling KH, Yang CK, Peng FT. Appl Environ-Microbiol. 1979;37:355. doi: 10.1128/aem.37.3.355-357.1979.Ling KH, Liou HH, Yang CM, Peng FT. Appl Environ-Microbiol. 1984;47:98. doi: 10.1128/aem.47.1.98-100.1984.Aranov VL, Ling KH, Chen RC, Tsai MC. Neurosci Lett. 1993;152:69. doi: 10.1016/0304-3940(93)90485-4.Dowd PF, Peng FC, Chen JW, Ling KH. Entomol Exp Appl. 1992;65:57. Also, see: Nong XH, Wang YF, Zhang XY, Zhou MP, Xu XY, Qi SH. Mar Drugs. 2014;12:6113. doi: 10.3390/md12126113. For some synthetic approaches toward territrem analogs, see: Peng FC, Cheng JS. Mycopathologia. 1997;132:125. doi: 10.1023/A:1006845621623.Peng FC, Ho CP. Mycotoxin Res. 1995;11:99. doi: 10.1007/BF03192071.Zhao Y, Ku Y, Hao XJ, Lee SS. Tetrahedron. 2000;56:8901.Zhao J, Zhao F, Wang Y, Lia H, Zhang Q, Guénard D, Ge Q, Wei E, Jiang H, Wu Y, Wang L, Jiang H, Guéritte F, Wu X, Cheng CHK, Lee SS, Zhao Y. Helv Chim Acta. 2004;87:1832.Jiang X, Ao L, Zhou C, Yang L, Zhang Q, Li H, Sun L, Wu X, Bai H, Zhao Y. Chem Biodivers. 2005;2:557. doi: 10.1002/cbdv.200590036.Yang Y, Sun L, Dong S, Wang X, Yang L, Wu X, Bai H, Zhao Y. Mendeleev Commun. 2008;18:186..
- 6.Reports emerging from clinical trials demonstrate a significant difference between AChE inhibitors that are rapidly reversible versus slowly reversible. Rapidly reversible inhibitors such as donepezil, tacrine, and galanthamine induce marked elevation in AChE activity and protein levels over time, whereas the slowly reversible inhibitors such as rivastigmine significantly decreases activity with no up-regulation in protein levels.7,8 The clinical relevance of these findings is yet to be determined. However, an escalation of AChE levels and therefore activity arguably would lead to loss of therapeutic efficacy with regards to ameliorating the cholinergic deficit.
- 7.For a clinical trial report comparing cholinesterase inhibitory effects on cholinesterase activities and protein levels, see: Nordberg A, Darreh-Shori T, Peskind E, Soininen H, Mousavi M, Eagle G, Lane R. Curr Alz Res. 2009;6:4. doi: 10.2174/156720509787313961..
- 8.For reviews of clinical trials comparing AChE inhibitor effects on AChE activity levels, see: Darreh-Shori T, Soininen H. Curr Alz Res. 2010;7:67. doi: 10.2174/156720510790274455.Lane RM, Kivipelto M, Greig NH. Clin Neuropharmacol. 2004;27:141. doi: 10.1097/00002826-200405000-00011..
- 9.For X-ray crystal structure data on Donepezil, see: Kryger G, Silman I, Sussman JL. Structure Fold Des. 1999;7:297. doi: 10.1016/s0969-2126(99)80040-9..
- 10.Alzheimer A. Gesamte Psychiatrie. 1907;64:1264. [Google Scholar]
- 11.For a census study on the prevalence of dimentia, see: Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Lancet. 2005;366:2112. doi: 10.1016/S0140-6736(05)67889-0..
- 12.For general reviews on therapeutic treatment of Alzheimer's disease, see: Giacobini E. In: Cognitive Enhancing Drugs. Buccafusco JJ, editor. Birkhauser; Basel–Boston–Berlin: 2004. pp. 11–36.Giacobini E, Spiegel R, Enz A, Veroff AE, Cutler RE. J Neural Transm. 2002;109:1053. doi: 10.1007/s007020200089.Giacobini E, Becker R, editors. Alzheimer's Disease: From Molecular Biology to Therapy, Birkhauser: Boston. 1997. John V, Lieberburg I, Thorsett ED. Ann Rep Med Chem. 1993;28:197..
- 13.For a review of the amyloid cascade hypothesis, see: Hardy J, Selkoe DJ. Science. 2002;297:353. doi: 10.1126/science.1072994..
- 14.For references on -amyloid targeting, see: Melnikova I. Nat Rev Drug Discov. 2007;6:341. doi: 10.1038/nrd2314.Skovronsky DM, Lee VMY, Trojanowski JQ. Annu Rev Pathol Mech Dis. 2006;1:151. doi: 10.1146/annurev.pathol.1.110304.100113.Saido TC, Iwata N. Neurosci Res. 2006;54:235. doi: 10.1016/j.neures.2005.12.015..
- 15.For leading references on the cholinergic deficiency hypothesis, see: Recanatini M, Cavalli A. Expert Opin Ther Pat. 2002;12:1853.Hogan DB, Patterson C. Can J Neurosci. 2002;29:306. doi: 10.1017/s031716710000216x.Farlow M. Int Psychogeriatr. 2002;14:93. doi: 10.1017/s1041610203008688.Emre M. Int J Clin Pract. 2002:64. For an earlier review, see: Perry EK. Br Med Bull. 1986;42:63. doi: 10.1093/oxfordjournals.bmb.a072100..
- 16.For recent reviews of clinical trials involving AChE inhibitors, see: Raina P, Santaguida P, Ismaila A, Patterson C, Cowan D, Levine M, Booker L, Oremus M. Ann Intern Med. 2008;148:379. doi: 10.7326/0003-4819-148-5-200803040-00009.Lanctôt KL, Herrmann N, Yau KK, Khan LR, Liu BA, LouLou MM, Einarson TR. CMAJ. 2003;169:557..
- 17.For a comparison of long-term effects of AChE inhibitors, see: Giacobini E. Neurochem Res. 2003;28:515. doi: 10.1023/a:1022869222652..
- 18.For tacrine, see: Summers WK, Majovski LV, Marsh GM, Tachiki K, Kling A. N Engl J Med. 1986;315:1241. doi: 10.1056/NEJM198611133152001. For donepezil, see: Inoue A, Kawai T, Wakita M, Iimura Y, Sugimoto H, Kawakami Y. J Med Chem. 1996;39:4460. doi: 10.1021/jm950596e. For huperzine A, see: Kozikowski AP, Campiani G, Nacci V, Sega A, Saxena A, Doctor BP. J Chem Soc, Perkin Trans I. 1996:1287.Bai DL, Tang XC, He XC. Curr Med Chem. 2000;7:355. doi: 10.2174/0929867003375281. For total syntheses of huperzine A, see: Xia Y, Kozikowski AP. J Am Chem Soc. 1989;111:4116.Qian L, Ji R. Tetrahedron Lett. 1989;30:2089. For rivastigimine, see: Ogura H, Kosasa T, Kuriya Y, Yamanishi Y. Meth Find Exp Clin Pharmacol. 2000;22:609. doi: 10.1358/mf.2000.22.8.701373. For galanthamine, see: Fulton B, Benfield P. Drugs Aging. 1996:60. doi: 10.2165/00002512-199609010-00006..
- 19.For a lead reference linking cholinergic activation with APP processing, see: Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Science. 1992;258:304. doi: 10.1126/science.1411529..
- 20.For lead references on AChE's ability to promote Aβ-aggregation, see: Inestrosa NC, Alvarez A, Pérez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Neuron. 1996;16:881. doi: 10.1016/s0896-6273(00)80108-7.Alvarez A, Opazo C, Alarcón R, Garrido J, Inestrosa NC. J Mol Biol. 1997;272:348. doi: 10.1006/jmbi.1997.1245..
- 21.For reviews on dual binding site AChE inhibitors as disease-modifying therapeutics, see: Muñoz-Torrero D. Curr Med Chem. 2008;15:2433. doi: 10.2174/092986708785909067.Castro A, Martinez A. Curr Pharm Des. 2006;12:4377. doi: 10.2174/138161206778792985.Recanatini M, Valenti P. Curr Pharm Des. 2004;10:3157. doi: 10.2174/1381612043383313..
- 22.For a report of a covalent dual binding site inhibitor of AChE, see: Belluti F, Rampa A, Piazzi L, Bisi A, Gobbi S, Bartolini M, Andrisano V, Cavalli A, Recanatini M, Valenti P. J Med Chem. 2005;48:4444. doi: 10.1021/jm049515h..
- 23.For designs of AChE inhibitors also able to inhibit ß;-secretase (BACE-1), see: Zhu Y, Xiao K, Ma L, Xiong B, Fu Y, Yu H, Wang W, Wang X, Hu D, Peng H, Li J, Gong Q, Chai Q, Tang X, Zhang H, Li J, Shen J. Bioorg Med Chem. 2009;17:1600. doi: 10.1016/j.bmc.2008.12.067.Camps P, Formosa X, Galdeano C, Muñoz-Torrero D, Ramírez L, Gómez E, Isambert N, Lavilla R, Badia A, Clos MV, Bartolini M, Mancini F, Andrisano V, Arce MP, Rodriguez-Franco MI, Huertas O, Dafni T, Luque J. J Med Chem. 2009;52:5365. doi: 10.1021/jm900859q.Piazzi L, Cavalli A, Colizzi F, Belluti F, Bartolini M, Mancini F, Recanatini M, Andrisano V, Rampa A. Bioorg Med Chem Lett. 2008;18:423. doi: 10.1016/j.bmcl.2007.09.100..
- 24.Cheung J, Gary EN, Shiomi K, Rosenberry TL. ACS Med Chem Lett. 2013;4:1091. doi: 10.1021/ml400304w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For our earliest views on the significance on DE-Ring, see: Douglas CJ, Sklenicka HM, Shen HC, Golding GM, Mathias DS, Degen SJ, Morgan CD, Shih RA, Mueller KL, Seurer LM, Johnson EW, Hsung RP. Tetrahedron. 1999;55:13683.Degen SJ, Mueller KL, Shen HC, Mulder JA, Golding GM, Wei LL, Zificsak CA, Neeno-Eckwall A, Hsung RP. Bioorg Med Chem Lett. 1999;9:973. doi: 10.1016/s0960-894x(99)00115-8..
- 26.Kunitz ID, Chen K, Sharp KA, Kollman PA. Proc Natl Acad Sci USA. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith AJT, Zhang X, Leach AG, Houk KN. J Med Chem. 2009;52:225. doi: 10.1021/jm800498e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.