

Abstract
Normal tension glaucoma (NTG) is a complex optic neuropathy characterized by progressive retinal ganglion cell death and glaucomatous visual field loss, despite normal intraocular pressure (IOP). This condition poses a unique clinical challenge due to the absence of elevated IOP, a major risk factor in typical glaucoma. Recent research indicates that up to 21% of NTG patients have a family history of glaucoma, suggesting a genetic predisposition. In this comprehensive review using PubMed studies from January 1990 to December 2023, our focus delves into the genetic basis of autosomal dominant NTG, the only known form of inheritance for glaucoma. Specifically exploring optineurin (OPTN), TANK binding kinase 1 (TBK1), methyltransferase-like 23 (METTL23), and myocilin (MYOC) mutations, we summarize their clinical manifestations, mutant protein behaviors, relevant animal models, and potential therapeutic pathways. This exploration aims to illuminate the intricate pathogenesis of NTG, unraveling the contribution of these genetic components to its complex development.
Keywords: METTL23, MYOC, normal tension glaucoma, OPTN, TBK1
Glaucoma is a major optic neuropathy characterized by optic nerve damage and notable retinal ganglion cell (RGC) loss and is the leading cause of irreversible blindness worldwide.[1] Primary open-angle glaucoma (POAG) represents the most common type of glaucoma.[2] The Baltimore Eye Survey reported that more than half of POAG patients do not exhibit elevated intraocular pressure (IOP), defining the case for normal tension glaucoma (NTG).[3] NTG displays optic nerve head (ONH) damage, progressive retinal nerve fiber layer (RNFL) thinning, distinct visual field defects, open anterior chamber angles upon gonioscopy, and maintains an IOP below 21 mmHg without affecting the rest of the retina.[4]
Epidemiologic studies have identified several risk factors associated with NTG, including age, gender, ethnicity, and family history.[5] First, the average age of NTG patients is in their 60s, generally older than individuals with POAG.[6] Second, some studies have indicated a higher prevalence of NTG among females than males.[7] However, this gender difference may be attributed to the longer life expectancy of females. Third, the incidence of NTG varies among different populations. Notably, research has shown a higher NTG prevalence in Asians (52%–92%), notably Japanese individuals (92%), compared to those of White ethnicity (30%–39%).[8,9,10,11] In addition, Asian Americans have been found to have a 159% increased risk of NTG compared to non-Hispanic White individuals,[10] possibly reflecting an increased demographic association and genetic susceptibility. Moreover, approximately 21% of NTG patients have a family history of glaucoma,[12] further suggesting a genetic predisposition.
While most forms of glaucoma have a hereditary component, they exhibit different patterns of inheritance. Glaucoma can occur at all ages, with Mendelian inheritance typical for the early-onset familial NTG and juvenile open-angle glaucoma (JOAG) and complex traits evident in adult-onset POAG and primary angle-closure glaucoma.[11] Notably, gene mutations associated with early-onset glaucoma have significant biological effects, while variants contributing to adult-onset glaucoma typically have more minor effects.[13]
To date, genome-wide association studies (GWAS) have successfully identified the genomic loci for POAG.[11] A recent multiethnic meta-analysis, encompassing 34,179 cases and 349,321 controls (NTG: 3247 cases, 47,997 controls; High-tension glaucoma (HTG): 5144 cases, 47,997 controls), identified 127 loci.[14] All NTG loci, except rs1812974 near ARHGEF12, showed nominal associations with HTG.[14] In contrast to GWAS, which generally focuses on the common variants with smaller effects (minor allele frequency >1%), NTG pedigree studies have revealed rare mutations in four genes, most of which have been validated through in vitro and animal models.[2,15] Numerous large NTG pedigrees have been described, typically demonstrating an autosomal dominant inheritance pattern, suggesting a Mendelian form of glaucoma caused by mutation in a single gene. This review examines the genetic role in NTG, drawing insights from PubMed studies published between January 1990 and December 2023. It delves into rare mutations identified through NTG pedigree studies, exploring pedigree characteristics, clinical features, protein function, mutation effects, and relevant animal models. The insights gained from these discoveries provide new perspectives on the fundamental molecular mechanisms underlying glaucoma, potentially paving the way for novel gene-based therapies and neuroprotection strategies. Furthermore, gene-based diagnostic testing for mutations responsible for NTG promises to detect individuals in the earliest stages of the disease when therapeutic interventions are more effective.
Optineurin (OPTN)
The human optineurin (OPTN) gene is located on the short arm of chromosome 10 (10p13) and comprises 15 exons, including two noncoding exons and 13 coding exons [Fig. 1].[16] This chromosomal region was previously designated as the GLC1E locus for POAG.[17] Predictions of OPTN promoter sequence reveal the presence of multiple nuclear factor kappa B (NF-kB) binding sites within 600 base pairs (bp) of the transcriptional regulatory region.[16] The OPTN gene encodes a 577-amino acid (aa) protein, with two coiled-coil (CC) domains, highly conserved among species.[18] Furthermore, alternative splicing in the 5’-untranslated region produces at least three distinct isoforms, all sharing the same reading frame.[16]
Figure 1.
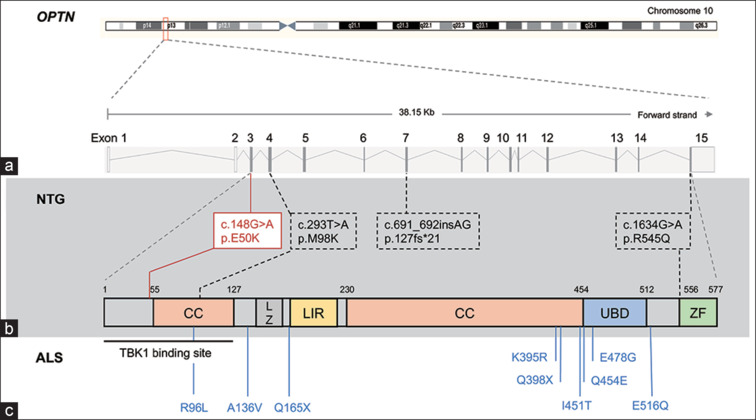
Disease-associated mutations in OPTN gene. (a) OPTN gene encodes a 577-amino acid protein with coiled-coil (CC), leucine zipper (LZ), LC3-interacting region (LIR), ubiquitin-binding (UBD), and zinc finger (ZF) domains. (b) Normal tension glaucoma (NTG)-linked mutations (red box: causative; dotted box: risk variants). (c) Amyotrophic lateral sclerosis (ALS)-associated mutations
OPTN mutations in NTG
In 1998, Sarfarazi et al.[17] reported a four-generation NTG pedigree, comprising 16 affected individuals. Through linkage analysis, a subset of genetic markers within GLC1E locus consistently co-segregated with NTG in the pedigree.[17] Considering the expression in the retina, OPTN was identified as the prime candidate gene for further investigation.[18] Genetic analysis revealed that all family members with NTG carried OPTN (c. 458G > A, p.E50K) mutation, conclusively establishing that OPTNE50K mutation causes NTG.[17] OPTN was marked as the first gene associated with NTG.[18] The OPTNE50K mutation was detected in approximately 1%–2% of NTG patients across various cohorts and was notably absent in matched control populations assembled from clinical settings in the USA, Europe, Japan, and Australia.[19,20,21]
Two additional mutations, OPTN c. 691-692insAG (resulting in a premature stop) and OPTNR545Q (c. 1634G > A), were initially linked to NTG.[18] However, regarding the role of OPTNR545Q in NTG, subsequent analyses have generated conflicting data, as it was frequently found in both NTG patients and controls.[16,19,20,21,22,23,24] In addition, a fourth OPTN variant, OPTNM98K (c.293T>A), was also identified in both NTG-affected cases and controls. Interestingly, the frequency of OPTNM98K is statistically higher in NTG patients compared to controls in certain populations,[21,22,23,24,25] and due to its location within the CC domain, it may potentially contribute as a risk for NTG in specific populations.
Genetic studies have also linked different sets of OPTN mutations with amyotrophic lateral sclerosis (ALS), a motor neuron disease.[26] Several loss-of-function OPTN mutations, including R96L, A136V, Q165X, K395R, Q398X, I451T, Q454E, E478G, and E516Q, have been associated with ALS [Fig. 1].[26] OPTN c.691-692insAG, initially found in an NTG patient, was later identified as a founder mutation in Moroccan and Ashkenazi Jews, significantly increasing the risk of developing ALS in heterozygous carriers.[27]
Clinical features of OPTN-associated NTG
OPTN-associated NTG patients typically present with early-onset disease following an autosomal dominant inheritance pattern. Aung et al.[28] conducted a comparative analysis, assessing the clinical features of NTG patients with OPTNE50K mutation (n = 11) in contrast to NTG patients without glaucoma-causing mutations (n = 87). It was observed that patients carrying OPTNE50K mutation were diagnosed at a younger age (average of 40.8 ± 15) and exhibited larger cup-to-disc (C/D) ratios (mean of 0.86 ± 0.1) compared to NTG controls.[28] Similar clinical findings have been consistently reported in subsequent studies. For instance, Minegishi et al.[16] described a three-generation Japanese pedigree with six family members carrying OPTNE50K, diagnosed at an early age (mean of 36.8 years) and displayed low IOP (mean of 13 mmHg). In addition, Hauser et al.[19] reported an OPTNE50K NTG patient diagnosed at 38, with normal IOP (15 mmHg in the right eye and 14 mmHg in the left eye) and a C/D ratio of 0.9. Ayala-Lugo et al.[29] also reported an OPTNE50K NTG patient diagnosed at 42 years old with a maximum IOP of 18 mmHg and a C/D ratio of 1.0. More recently, Fox and Fingert[15] documented a four-generation NTG pedigree with six family members carrying OPTNE50K, all exhibiting low IOPs (mean of 14.6 ± 1.2 mmHg).
OPTN protein function in NTG
The human OPTN protein is a 64-kDa scaffold protein consisting of 577 aas, while the mouse Optn gene encodes a 584-aa protein (67 kDa) that shares a 78% identity with human OPTN.[30] OPTN is a highly conserved protein expressed ubiquitously in all human organs, including the retina, heart, brain, liver, kidney, placenta, and pancreas.[18] It exhibits exceptionally high expression in the skeletal muscle.[18] OPTN encompasses several structural domains, including two CC domains, a leucine zipper (LZ) domain, an LC3-interacting region (LIR), a ubiquitin-binding domain (UBD), and a zinc finger (ZF) domain [Fig. 1].[31] Normally, OPTN undergoes posttranslational modifications that significantly affect its function, including phosphorylation and ubiquitination.[26] One of the major kinases capable of phosphorylating OPTN on at least nine serine and two threonine residues is TANK binding kinase-1 (TBK1).[32]
OPTN is a multifunctional protein involved in several cellular processes. First, it acts as an autophagy receptor or adaptor, playing a role in several forms of selective autophagy, including xenophagy,[33] mitophagy,[34] and aggrephagy.[35] Second, it participates in vesicular trafficking,[36] Golgi maintenance,[37] secretion,[38] and receptor recycling.[39,40] Third, OPTN significantly impacts the innate immune response by negatively regulating the NF-kB pathway.[41] This diversity in the functions of OPTN can be attributed to its ability to interact with numerous potential partners and to be involved with multiple pathways.[16]
The effect of OPTNE50K on cell survival and other functions in glaucoma has been investigated using cultured cells and transgenic mouse models, as discussed in the following section. Studies utilizing various cell lines have revealed that the OPTNE50K mutant induces apoptosis-like cell death in retinal cells, such as RGC-5, 661W, and human primary retinal cells.[42,43,44] However, this effect is not observed in several other cell lines tested, including IMR-32, Hela, Cos-1, and NSC34.[42,43,44] These findings suggest that the OPTNE50K mutant may contribute to glaucoma by directly inducing the death of RGCs. Notably, Minegishi et al.[30] have reported an accumulation of insoluble overexpressed OPTNE50K in the endoplasmic reticulum (ER) of HEK293 cells, which aligns with the results obtained from neurons derived from induced pluripotent stem cells from E50K NTG patients (OPTNE50K iPSCs). The OPTNE50K mutant exhibits a strong interaction with TBK1, hindering the proper oligomerization and solubility of OPTN, both of which are crucial for OPTN’s intracellular transition.[30] Treatment with BX795, a TBK1 inhibitor, effectively reverses the abnormal insolubility of the OPTNE50K mutant, which delineates the intracellular dynamics of the endogenous mutant protein, which causes insolubility associated with TBK1, resulting in POAG.[30]
In 2020, VanderWall et al.[45] generated OPTNE50K iPSCs and observed that RGCs differentiated from OPTNE50K iPSCs exhibited numerous neurodegenerative deficits, including neurite retraction, autophagy dysfunction, apoptosis, and increased excitability. Recently, Gomes et al.[46] differentiated RGCs and astrocytes from OPTNE50K iPSCs and identified significant changes in OPTNE50K astrocytes, including autophagy dysfunction. Co-culture experiments demonstrated that astrocytes induce neurodegenerative properties in otherwise healthy RGCs, while healthy astrocytes alleviate some neurodegenerative features in OPTN (E50K) RGCs.[46]
OPTN mouse models
Several mouse models of NTG have been developed to mimic human NTG, primarily caused by the well-characterized OPTNE50K mutation [Table 1]. In 2010, Chi et al.[47] generated the first transgenic mouse that highly overexpressed mouse OptnE50K, equivalent to human OPTNE50K, using the CMV immediate enhancer/β-actin (CAG) promoter. The overexpression of mouse OptnE50K led to progressive retinal degeneration, characterized by loss of RGCs and connecting synapses in the peripheral retina. This resulted in a thinning of RNFL at ONH at normal IOP in 16-month-old mice, thus confirming the association between OPTNE50K and NTG. Furthermore, OptnE50K mice exhibited massive apoptosis and degeneration of the entire retina, leading to approximately 28% reduction in the retina thickness.
Table 1.
Overview of OPTNE50K mouse models for NTG
| Mouse lines | Promotor | Main results | Reference | |||
|---|---|---|---|---|---|---|
| Transgenic mice | ||||||
| Mouse OptnE50K | CAG (CMV immediate enhancer/β-actin) | Progressive retinal degeneration is characterized by loss of RGCs and connecting synapses in the peripheral retina, leading to a thinning of the nerve fiber layer at ONH at normal IOP in 16-month-old mice | [47] | |||
| Human OPTNE50K | c-kit | - | [48] | |||
| BAC-hOPTNE50K | BCA with OPTN endogenous regulatory sequences | More stressed or degenerating optic nerve axons in BAC-hOPTNE50K mice than in 18-month-old control mice at normal IOP | [49] | |||
| Knock-in mice | ||||||
| OptnE50K/E50K | - | The thinning of the RGC fiber layer around ONH from 6 months at normal IOP. At 12 months, the thickness of RGC fiber layer around ONH reduction was 20% compared to WT mice | [16] | |||
| OptnE50K/E50K | - | Aging-related RGC loss and visual dysfunction without elevated IOP | [50] |
BAC=bacterial artificial chromosome, IOP=intraocular pressure, NTG=normal tension glaucoma, ONH=optic nerve head, OPTN=optineurin, RGCs=retinal ganglion cells, WT=wild type
The following year, Meng et al.[48] generated another transgenic mouse that overexpressed human OPTNE50K using the c-kit promoter. However, they only confirmed the expression of human OPTNE50K in the retina of this transgenic mouse model and did not measure IOP or histological abnormalities. In 2015, Tseng et al.[49] created the third OPTN transgenic mouse (BAC-hOPTNE50K) with low expression of OPTNE50K using a bacterial artificial chromosome (BAC) that contained the OPTN promoter region and human OPTN gene. Histological analysis revealed a 40% RGC loss with normal IOP. In addition, the study of optic nerve axons revealed stressed and damaged optic nerve axons in BAC-hOPTNE50K mice compared to 18-month-old control mice.
In 2016, Minegishi et al.[16] generated an OptnE50K knock-in (KI) mouse model using CRISPR/Cas9 editing. Thinning of the RGC fiber layer around ONH in OptnE50K KI homozygous (OptnE50K/E50K) mice was observed from 6 months, despite normal IOP. At 12 months, the thickness of the RGC fiber layer around ONH was reduced by 20% compared to wild-type (WT) mice. Furthermore, the optic cup deepened in OptnE50K/E50K mice, similar to the changes seen in NTG patients. The administration of TBK1 inhibitor amlexanox proved effective in treating these OptnE50K KI mice. Presently, amlexanox holds approvals from both the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) and the US Food and Drug Administration (FDA) for clinical trials. In addition, Hou et al.[50] established another OptnE50K KI mouse model and identified aging-related RGCs loss and visual dysfunction in OptnE50K/E50K mice without elevated IOP. Electron microscopy results also demonstrated significant morphological anomalies in the mitochondria of RGCs axons in 3-month-old OptnE50K/E50K mice, with these changes worsening with age, suggesting that the damaged mitochondria-associated dysfunction of RGCs axon may play an etiological role in NTG.
TANK Binding Kinase-1 (TBK1)
TBK1 is located at 12q14.1 and contains 21 exons, of which exon 1, parts of exon 2, and exon 21 are noncoding [Fig. 2].[51] Mutation-associated haploinsufficiency of TBK1 has been implicated as a causal mutation in several types of neuroinflammatory diseases, including ALS, frontotemporal dementia (FTD), Alzheimer’s disease (AD) and related tauopathies, herpes simplex virus-1 (HSV-1) encephalitis (HSE), and atypical parkinsonian syndrome.[52] Copy number gains of TBK1 have been associated with NTG, but TBK1 does not appear to be involved in the pathogenesis of HTG; additional data suggest that TBK1 is not associated with JOAG, pigmentary glaucoma, or steroid-induced glaucoma.[15] In this section, we will focus on the role of TBK1 in NTG.
Figure 2.
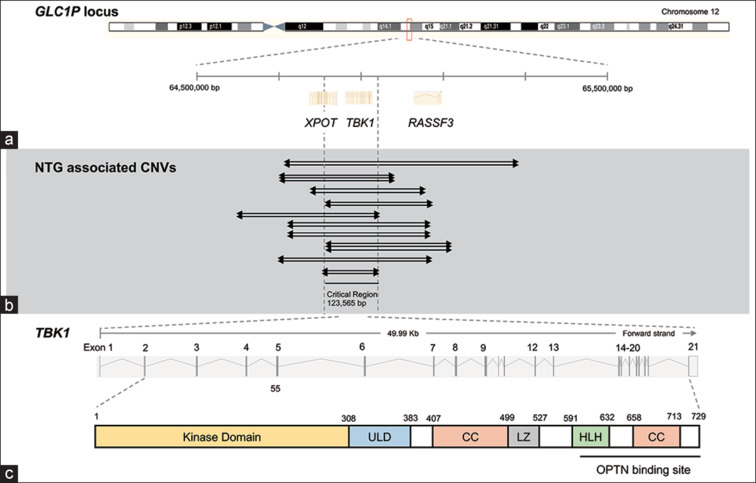
Disease-associated CNVs spanning TBK1 gene. (a) GLC1P locus includes TBK1 with flanking XPOT and RASSF3 genes. (b) CNVs duplicating or triplicating TBK1 in NTG patients (indicated by arrows). (c) TBK1 gene encodes a 729-amino acid protein with kinase, ubiquitin-like (ULD), coiled-coil (CC), leucine zipper (LZ), and helix–loop–helix (HLH) domains. CNV = copy number variation, NTG = normal tension glaucoma
TBK1 mutations in NTG
In 2011, linkage analysis of an African-American NTG pedigree mapped a new glaucoma-causing gene to a 780-kbp duplication within the GLC1P locus on chromosome 12q14.[53] Subsequent copy number variation (CNV) analysis and expression analysis of the GLC1P locus in retina suggested that TBK1 duplication is likely responsible for NTG.[53] Both duplication and triplication of TBK1 have been detected in NTG pedigrees from Europe, Japan, Australia, and Korea [Table 2 and Fig. 2].[53,54,55,56,57] These TBK1 CNVs have been detected and confirmed by various molecular genetic techniques, including quantitative polymerase chain reaction (PCR), chromosomal microarray analysis, and genomic sequencing.[15] The cohort of unrelated NTG patients was also evaluated for TBK1 mutations. TBK1 CNV mutations were detected in two (1.3%) of 152 NTG patients from Iowa,[53] one (0.4%) of 252 Japanese NTG patients,[54] one (1.0%) of 96 NTG patients from New York,[58] four (1.2%) of 334 NTG patients from Australia,[56] two (1.4%) of 143 NTG patients from India,[59] and one (0.2%) of 143 Korean NTG patients.[57]
Table 2.
Summary of TBK1 CNV detected in NTG pedigrees
| Pedigree (ancestry) | CNV (kb) | Diagnosed age (years) | Max IOP (mmHg, OD/OS)/ Cup-to-disc ratio (first examination, OD/OS) | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| GGO-441 (African-American) | Duplication (690–780) | 36±8.2 | 18.2±4.1/16.7±3.6/ 0.95±0.083/0.93±0.10 | [53] | ||||
| GGA-458 (German) | Triplication (646) | 29±6.7 | 19.0±4.3/18.8±3.8/ 0.85±0.071/0.85±0.071 | [55] | ||||
| GGJ-414 (Japanese) | Duplication (296) | 42 (II-4) | 18/17 (II-4)/ 0.75/0.7 (II-4) | [54] | ||||
| AG604 (Australian) | Triplication (347) | 60 (AG604) | 12/12 (AG604)/ 0.95/0.95 (AG604) | [56] | ||||
| AG624 (Australian) | Duplication (406) | 44 (AG624) | 17/17 (AG624)/ 0.95/0.90 (AG624) | [56] | ||||
| AG724 (Australian) | Duplication (406) | 43 (AG724) | 14/14 (AG724)/ 0.80/0.90 (AG724) | [56] | ||||
| GFMC524 (Australian) | Duplication (442) | 32 (GFMC524) | 13/13 (GFMC524)/ 0.85/0.85 (GFMC524) | [56] | ||||
| S227 (Korean) | Duplication (124) | 48±16 (39–68) | 15.0±1.7/15.0±2/ - | [57] |
CNV=copy number variation, IOP=intraocular pressure, NTG=normal tension glaucoma, OD/OS=oculus dexter/oculus sinister, TBK1=TANK binding kinase 1
Clinical features of TBK1-associated NTG
Patients with NTG associated with TBK1 duplication or triplication have typical clinical findings of glaucoma. The presenting features of NTG in seven members of the first TBK1 pedigree from African-Americans (GGO-441) include: 1) an early age at diagnosis with a mean of 36 ± 8.2 years; 2) low IOP with a mean of 18.2 ± 4.1/16.7 ± 3.6 mmHg (oculus dexter/oculus sinister [OD/OS]); and 3) large C/D ratios with a mean of 0.95 ± 0.083/0.93 ± 0.10 (OD/OS).[53] Similar clinical findings were observed in other reported TBK1-associated NTG pedigrees, including GGA-458, GGJ-414, AG624, AG724, and GFMC524.[53,54,55,56,57] These family members also had an early onset of NTG (diagnosed before age 45) and a large C/D ratio, as summarized in Table 2.
TBK1 function in NTG
TBK1 is an 82-kDa protein consisting of 729 aa and comprises a kinase domain, a ubiquitin-like domain, and CC domains 1 and 2.[16] It is conserved across eukaryotes, with human paralogs identified in zebrafish, mice, primates, and amphibians. Mouse and human TBK1 exhibit a significant level of sequence similarity, sharing at least 94% homology.[52] TBK1 belongs to the non-canonical IκB kinase (IKK) family and displays 64% homology to IKKε (also known as IKKi), which plays a critical role in regulating the balance between interferon-1 (IFN-1) and IFN-II signaling pathways.[60]
TBK1, also known as NF-kB activating kinase (NAK) and T2K, is a serine/threonine protein kinase that plays a key role in the regulation of various cellular processes, including innate immunity, inflammatory cytokine production, autophagy, mitochondrial metabolism, and cell survival/proliferation.[52] TBK1 can be activated by pathogen-associated molecular patterns, damage-associated molecular patterns, inflammatory cytokines, and oncogenic kinases.[60] Its biological activity was initially recognized in innate defenses against pathogens, where it plays a crucial role in regulating the production of type I IFN, including IFN-α and IFN-β.[52] Recent studies have shown that TBK1 connects the pathogen-stimulated processes of inflammation/immunity, metabolism, and proliferation in diseases, including inflammatory diseases, type II diabetes, obesity, neurovegetative diseases, and some cancers.[60]
TBK1 gene duplications lead to increased TBK1 mRNA transcription in human fibroblast cells.[60] However, the details of how the excess TBK1 can cause RGC death and NTG remain unknown. There are three hypotheses that warrant further investigation: 1) studies of RGCs with a TBK1 duplication derived from induced pluripotent stem cells (iPSCs) have suggested that this mutation is associated with changes in autophagy;[61] 2) stimulation of inflammation and the innate immune system by TBK1 might contribute to RGC death and NTG by production of proinflammatory cytokines and glial cell activation, which have been observed in studies of other neurogenerative diseases;[15] and 3) TBK1 may influence NF-kB signaling upstream of IKK complex.[52] Further research is needed to clarify these potential mechanisms related to excess TBK1 expression in NTG.
TBK1 mouse model
The TBK1 mouse model (Tg-TBK1) was generated using a BAC vector to integrate the natural human TBK1 gene into the mouse genome.[62] This transgene provides the mouse line with an additional copy of TBK1 gene to mimic the TBK1 duplications that cause NTG in humans. Increased TBK1 expression was detected in RGCs of Tg-TBK1 mice. Furthermore, Tg-TBK1 mice developed RGC loss that increased with age at normal IOP.[62] Heterozygous Tg-TBK1 mice exhibited a 13% RGC loss at 18 months of age compared to controls, while homozygous Tg-TBK1 mice had a 20% loss at 6 months.
Methyltransferase-like 23 (METTL23)
Human methyltransferase-like 23 (METTL23) is located at 17q25.1 and comprises five exons, with exon 1 and a part of exon 2 being noncoding [Fig. 3]. METTL23 functions as a histone arginine methyltransferase. Several pathogenic variants of METTL23 are associated with familial NTG and familial intellectual disability [Fig. 3],[2,63] suggesting potential etiologic links among arginine methylation, nervous system disorders, and glaucoma.[1]
Figure 3.
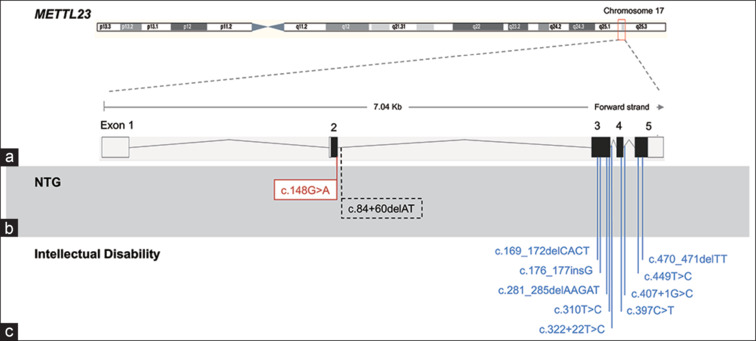
Disease-associated variants in METTL23 gene. (a) METTL23 consists of five exons; exon 1 and part 2 are noncoding. Several pathogenic variants in METTL23 have been reported to be associated with normal tension glaucoma (NTG) (b) and familial intellectual disability (c). Red box: causal mutation; dotted box: risk variant
METTL23 mutations
Recently, in a whole exon data analysis involving 11 samples from three-generation Japanese NTG pedigree (comprising six affected and five unaffected), METTL23 and CEP290 were identified as the putative NTG-causing genes.[2] The in silico predictive tool (Human splicing Finder) indicated that METTL23 c.A83G causes abnormal splicing, resulting in functionally null proteins, whereas the CEP290 variant was excluded due to its low pathogenicity score. The METTL23 c.A83G mutation was heterozygous in all NTG patients. This allele was transmitted vertically from the affected parents to the affected children, indicating an autosomal dominant inheritance pattern.[2] No additional exon-spanning duplications or deletions were detected in any of the patients, and no TBK1 CNVs were detected.[2] In addition, the METTL23 c.A83G mutation is not found in any other glaucoma patients enrolled in the Japan Whole Exome Project.[2]
A second METTL23 variant, specifically METTL23 c.84+60delAT, was identified in 14 individuals from 1029 unrelated Japanese NTG cases and found in eight of 1402 age-matched Japanese controls.[2] No TBK1 CNVs or OPTNE50K mutations were detected in any NTG patient with the METTL23 c.84+60delAT variant. The distribution of this allele between the patients and controls suggests a potential contribution to NTG, as indicated by statistical tests (P = 0.03, Fisher’s exact test or c2 with Yates’ correction; P = 0.038, logistic regression adjusting for age and sex).[2] However, further work, including analyses of additional data sets, will be necessary to confirm and further explore this finding.
Clinical features of METTL23-associated NTG
Patients carrying METT23 c.A83G have typical clinical characteristics of NTG, including 1) early age at diagnosis, with a mean age of 47 ± 9.6 years; 2) low IOP with a mean of 15.4 ± 3.5 in the right eye and 14.6 ± 2.9 mmHg in the left eye; and 3) large C/D ratios with a mean of 0.88 ± 0.05 in the right eye and 0.88 ± 0.04 in the left eye at the first examination.[2] None of the affected individuals with METT23 c.A83G had a history of developmental delay or retinal disease.
NTG patients carrying METTL23 c.84+60delAT had distinct clinical characteristics, including 1) a mean age at diagnosis of 62.3 ± 12.0 years, 2) maximum IOP with a mean of 15.0 ± 2.8/15.0 ± 2.6 (OD/OS), and 3) C/D ratio of 0.76 ± 0.07/0.82 ± 0.11 (OD/OS).[2] Interestingly, NTG patients with METT23 c.A83G had an earlier onset of disease and a larger C/D ratio at the initial examination than those with METTL23 c.84+60delAT. Though the sample size is limited, these data suggest that METT23 c.A83G mutation may lead to more severe NTG than METTL23 c.84+60delAT.
METTL23 function in NTG
METTL23 is expressed at low levels, but is found ubiquitously in human tissues.[2] It belongs to a methyltransferase-like (METTL) family and shares homology with the conserved domains related to protein arginine methyltransferases (PRMT) activity, particularly PRMT1–8.[64,65] In 2017, Hatanaka et al.[64] demonstrated that METTL23 possesses PRMT activity. It catalyzes the dimethylation of histone H3 at arginine 17 (H3R17). This enzymatic activity suggests that METTL23 plays a role in histone arginine methylation, which may affect gene expression regulation and cellular processes.
The aberrant splicing by METT23 c.A83G mutation at the end of METTL23 exon 2 was confirmed in various experimental models, including exon-trapping vector transfected HEK293T cells, iPSCs derived from NTG patients, and the retinas of Mettl23 KI mice. NF-κB transcription factors play an important role in regulating the immune response, while PS2 (small secreted protein that belongs to the trefoil factor family) has been shown to suppress the NF-κB–activated expression of proinflammatory cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-1β.[66,67] Elevated expression of TNF-α and IL-1β by activated microglial cells in hypoxic neonatal retina has been linked to RGC apoptosis.[68,69] METTL23 was expressed in RGCs and was found to be associated with NF-κB–mediated transcription of TNF-α and IL-1β through dimethylation of histone 3 arginine 17 (H3R17).[2]
Both METTL23 c.A83G mutation and METTL23 deficiency were associated with RGC loss and axonal degeneration at ONH in Mettl23-KI and -KO mice, similar to the findings in NTG patients. Similarly, METTL23 c.84+60delAT mutation led to partially aberrant splicing in exon-trapping vector-transfected HEK293T cells, consistent with the milder clinical features observed in NTG patients with this mutation. These findings suggest a direct link among METTL23, histone methylation, immune response regulation, and RGC degeneration in NTG.
METTL23 mouse model
The mouse models of METTL23 c.A83G-KI and METTL23-knockout (KO) generated by CRISPR/Cas9 demonstrated key pathological features relevant to glaucoma: 1) Ganglion cell complex (GCC) thickness reduction. In B-circular scans focused on ONH, both homozygous Mettl23-KI and -KO mice exhibited a reduction in GCC thickness starting at 2 months of age with normal IOP. This reduction was observed to progress in all Mettl23-KI and -KO mice by 6 months of age. 2) Optic nerve injury. Optic nerve fiber injury was evident in optic nerve cross sections, with statistically significant differences observed in both Mettl23-KI and -KO mice compared to control mice at 2 and 6 months. 3) RGC deficiency. Slight reductions in photopic negative scotopic threshold response (pSTR) amplitudes were noted in Mettl23-KI and -KO mice at 2 months of age. These reductions became more significant at 6 months of age, indicating a substantial loss of RGCs. 4) RGC loss. Examination of retinal whole mounts revealed a slight, though insignificant, decrease in RGCs in both Mettl23-KI and -KO mice compared to controls at 2 months, which developed significant RGC loss at 6 months. These findings support the conclusion that RGC degeneration in the Mettl23-KI and -KO mouse models is a progressive process, beginning with the axonal injury that precedes the loss of RGCs. This pattern of disease progression in the mouse models resembles the pathophysiological changes observed in NTG patients. Therefore, the Mettl23-KI and -KO mouse models provide valuable insights for studying specific aspects of glaucoma, particularly in cases with normal IOP.
Myocilin (MYOC)
The human myocilin (MYOC) gene is located on chromosome 1q24 and spans approximately 17 kb. It comprises three exons and two intros.[70] Initially, MYOC was referred to as “the trabecular meshwork inducible glucocorticoid response” (TIGR) gene, but it was later renamed MYOC.[71] This gene was the first to be associated with POAG.[72]
MYOC mutations in POAG
More than 100 POAG-associated mutations have been identified in MYOC gene, accounting for 3%–5% of POAG cases.[73] Specific mutations in MYOC gene can cause POAG with varying ages of onset and degrees of severity. One of the significant associations is with JOAG, characterized by autosomal dominant inheritance and highly penetrant severe phenotype with markedly elevated IOP (greater than 50 mmHg).[15] Another notable variant is the MYOC p.Q368T (rs74315329) variant, which is linked to a less-severe form (IOP <30 mmHg). This variant is more commonly observed in adult-onset POAG, the most prevalent form (approximately 1.6% of POAG patients).[15]
MYOC mutations in NTG
Mutations in MYOC, including p.W286R and p.Q368T, have also been detected in NTG patients.[74,75,76] However, the presence of these mutations in NTG patients has been subjected to some controversy and variability in different studies. In 2019, a case–control study identified the p.Q368T mutation in 11 (0.8%) of 1333 NTG patients and 17 (0.4%) of 4758 controls. This finding suggested a potential association, with P = 0.04 and an odds ratio of 2.3.[77] However, this association was not replicated in a more extensive study of 218,792 Finnish citizens.[78] In addition, the prevalence of the MTOC p.Q368T mutation is higher in POAG patients (1.6%), who typically have elevated IOP, compared to NTG patients (0.8%), indicating that this mutation is more strongly associated with POAG with elevated IOP.[77,79]
MYOC function
In humans and mice, MYOC is expressed in numerous ocular tissues, with its most prominent presence being in trabecular meshwork (TM).[80] This expression is known to be induced in cultured human TM cells by various factors, including glucocorticoids, oxidative stress, cell stress signaling pathways, and potentially unidentified factors in the aqueous humor.[81,82] Normally, TM cells secrete MYOC; however, mutations in MYOC can disrupt the secretion of both mutant and WT MYOC.[83] Notably, TM plays an important role in regulating IOP, with elevated IOP being the primary risk factor associated with glaucoma, but not for NTG. Despite extensive research, the precise function of MYOC in the retina remains unknown, but it appears to affect TM and the outflow of aqueous humor.[70] The genetic association and functional relevance to NTG are still under investigation.
MYOC animal models
Several groups have used mouse genetics to gain insights into the normal and pathological functions of MYOC in retina. In 2001, Kim et al.[84] generated null mutations in mice, not resulting in a glaucoma phenotype. These findings helped elucidate genetic studies of glaucoma in human patients; a likely null mutation was identified as unrelated to glaucoma in humans.[85] Furthermore, in 2004, Gould et al.[86] observed that mice overexpressing mouse Myoc also did not develop elevated IOP or glaucoma. Conversely, more than 90% of glaucoma-related mutations affect the highly conserved functional domain of MYOC, known as the olfactomedin domain, likely affecting the structure.[87] The results from genetic models suggested that MYOC does not have a significant role in regulating IOP, and mutated MYOC with abnormal function (gain of function) was likely responsible for ocular hypertension.
Subsequently, several groups began testing disease-specific MYOCY437H mutation in mice, a mutation responsible for a severe form of JOAG. Expression of human MYOCY437H and mouse Myoc with a mutation orthologous to Y437H did not lead to RGC loss.[80] An important discovery was made when Shepard et al.[88] used the adenovirus to express human MYOCY437H in the iridocorneal angle and observed elevated IOP. It is worth noting that human MYOC contains a targeting signal for peroxisomes that does not exist in mouse Myoc, which appears to be critical for the toxicity induced by mutant MYOC in TM cells.[88] Subsequently, three other groups have reported IOP elevation using different strategies to express human MYOCY437H in mouse eyes.[80] Other MYOC mutations are also believed to involve alternative pathogenic mechanisms.[80] However, to date, there are still no animal models specifically linked between the MYOC gene and NTG.
Conclusion
To date, only three genes (OPTN, TBK1, and METTL23) have been linked to familial NTG. Both OPTN and TBK1 proteins play essential roles in NF-κB signaling and autophagy. Furthermore, they share common upstream signals and regulators and can mutually influence each other through positive or negative feedback loops.[11,31,32,60,67] Recently, METTL23 has been shown to catalyze the dimethylation of H3R17 and upregulate SP2 transcription both in vitro and in vivo, along with TNF-α and IL-1β in vivo.[2] TNF-α and IL-1β are particularly significant for RGCs and optic nerve fibers. The loss of SP2 is associated with NF-κB–mediated activation of TNF-α and IL-1β in vivo.[66] Consequently, similar to OPTN and TBK1, METTL23 may also negatively regulate NF-κB–mediated TNF-α and IL-1β expression. These findings suggest that the other proteins involved in NF-κB signaling may also contribute to the development of NTG, and the modulation of NF-κB signaling could be a potential direction for the development of therapeutics.
Research into the role of histone arginine methylation in the development of neuronal diseases is rapidly advancing. The METTL23 gene has recently been linked to histone arginine methylation in NTG.[2] In addition to NTG, mutations in OPTN and TBK1 genes have been identified in patients with ALS.[15] Another gene associated with ALS, ataxin 2 (ATXN2), has also been linked to POAG.[89,90] Furthermore, ALS-linked mutations in the RNA-binding protein fused in sarcoma (FUS) result in the formation of insoluble intracytoplasmic protein aggregates.[91] This process was found to be alleviated by the depletion of PRMT1, which serves as a transcriptional coactivator by depositing dimethylarginines on histone H4R3.[92] Specifically, METTL23 c.A83G mutation was shown to hinder the dimethylation of histone H3R17, leading to the onset of NTG.[2] Notably, several pathogenic variants of METTL23 are also associated with familial intellectual disability,[2,63] indicating a possible connection among arginine methylation, nervous system disorders, and glaucoma.
NTG is an irreversible blinding disease, and its diagnosis is more complex compared to high-pressure forms of glaucoma. Understanding the role of NF-κB–mediated inflammation in NTG could serve as a catalyst for the development of novel diagnostic and therapeutic approaches. Considering that the optic nerve is part of the central nervous system, these findings may also have broader implications for understanding the pathogenesis of other neurodegenerative diseases.
Financial support and sponsorship
This work has been supported by a research grant to TI by the Japan Agency for Medical Research and Development (AMED, 15ek0109072h0003, 16ek0109072h0003, 17ek0109282s0001, 18ek0109282h0002, 19ek0109282h0003); the Japanese Ministry of Health, Labor, and Welfare (H22-Kankaku-Ippan-002); and the National Hospital Organization (R2-NHO (Kankaku)-02). This work was also supported by grants to YP by the Japan Society for the Promotion of Science (JSPS KAKENHI Grant Numbers 20K18366 and 23K15923).
Conflicts of interest
There are no conflicts of interest.
References
- 1.Liu WW, Sun Y. Epigenetics in glaucoma: A link between histone methylation and neurodegeneration. J Clin Invest. 2022;132:e163670. doi: 10.1172/JCI163670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan Y, Suga A, Kimura I, Kimura C, Minegishi Y, Nakayama M, et al. METTL23 mutation alters histone H3R17 methylation in normal-tension glaucoma. J Clin Invest. 2022;132:e153589. doi: 10.1172/JCI153589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sommer A, Tielsch JM, Katz J, Quigley HA, Gottsch JD, Javitt J, et al. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans. The Baltimore Eye Survey. Arch Ophthalmol. 1991;109:1090–5. doi: 10.1001/archopht.1991.01080080050026. [DOI] [PubMed] [Google Scholar]
- 4.Chen MJ. Normal tension glaucoma in Asia: Epidemiology, pathogenesis, diagnosis, and management. Taiwan J Ophthalmol. 2020;10:250–4. doi: 10.4103/tjo.tjo_30_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahrlich KG, De Moraes CG V, Teng CC, Prata TS, Tello C, Ritch R, et al. Visual field progression differences between normal-tension and exfoliative high-tension glaucoma. Invest Ophthalmol Vis Sci. 2010;51:1458–63. doi: 10.1167/iovs.09-3806. [DOI] [PubMed] [Google Scholar]
- 6.Bonomi L, Marchini G, Marraffa M, Bernardi P, De Franco I, Perfetti S, et al. Prevalence of glaucoma and intraocular pressure distribution in a defined population. The Egna-Neumarkt Study. Ophthalmology. 1998;105:209–15. doi: 10.1016/s0161-6420(98)92665-3. [DOI] [PubMed] [Google Scholar]
- 7.Dielemans I, Vingerling JR, Wolfs RC, Hofman A, Grobbee DE, de Jong PT. The prevalence of primary open-angle glaucoma in a population-based study in The Netherlands. The Rotterdam study. Ophthalmology. 1994;101:1851–5. doi: 10.1016/s0161-6420(94)31090-6. [DOI] [PubMed] [Google Scholar]
- 8.Klein BE, Klein R, Sponsel WE, Franke T, Cantor LB, Martone J, et al. Prevalence of glaucoma. The Beaver Dam eye study. Ophthalmology. 1992;99:1499–504. doi: 10.1016/s0161-6420(92)31774-9. [DOI] [PubMed] [Google Scholar]
- 9.Cho HK, Kee C. Population-based glaucoma prevalence studies in Asians. Surv Ophthalmol. 2014;59:434–47. doi: 10.1016/j.survophthal.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Stein JD, Kim DS, Niziol LM, Talwar N, Nan B, Musch DC, et al. Differences in rates of glaucoma among Asian Americans and other racial groups, and among various Asian ethnic groups. Ophthalmology. 2011;118:1031–7. doi: 10.1016/j.ophtha.2010.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiggs JL, Pasquale LR. Genetics of glaucoma. Hum Mol Genet. 2017;26:R21–7. doi: 10.1093/hmg/ddx184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trivli A, Koliarakis I, Terzidou C, Goulielmos GN, Siganos CS, Spandidos DA, et al. Normal-tension glaucoma: Pathogenesis and genetics. Exp Ther Med. 2019;17:563–74. doi: 10.3892/etm.2018.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan BJ, Wiggs JL. Glaucoma: Genes, phenotypes, and new directions for therapy. J Clin Invest. 2010;120:3064–72. doi: 10.1172/JCI43085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gharahkhani P, Jorgenson E, Hysi P, Khawaja AP, Pendergrass S, Han X, et al. Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat Commun. 2021;12:1258. doi: 10.1038/s41467-020-20851-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox AR, Fingert JH. Familial normal tension glaucoma genetics. Prog Retin Eye Res. 2023;96:101191. doi: 10.1016/j.preteyeres.2023.101191. [DOI] [PubMed] [Google Scholar]
- 16.Minegishi Y, Nakayama M, Iejima D, Kawase K, Iwata T. Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res. 2016;55:149–81. doi: 10.1016/j.preteyeres.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Sarfarazi M, Child A, Stoilova D, Brice G, Desai T, Trifan OC, et al. Localization of the fourth locus (GLC1E) for adult-onset primary open-angle glaucoma to the 10p15-p14 region. Am J Hum Genet. 1998;62:641–52. doi: 10.1086/301767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002;295:1077–9. doi: 10.1126/science.1066901. [DOI] [PubMed] [Google Scholar]
- 19.Hauser MA, Sena DF, Flor J, Walter J, Auguste J, Larocque-Abramson K, et al. Distribution of optineurin sequence variations in an ethnically diverse population of low-tension glaucoma patients from the United States. J Glaucoma. 2006;15:358–63. doi: 10.1097/01.ijg.0000212255.17950.42. [DOI] [PubMed] [Google Scholar]
- 20.Aung T, Ebenezer ND, Brice G, Child AH, Prescott Q, Lehmann OJ, et al. Prevalence of optineurin sequence variants in adult primary open angle glaucoma: Implications for diagnostic testing. J Med Genet. 2003;40:e101. doi: 10.1136/jmg.40.8.e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alward WLM, Kwon YH, Kawase K, Craig JE, Hayreh SS, Johnson AT, et al. Evaluation of optineurin sequence variations in 1,048 patients with open-angle glaucoma. Am J Ophthalmol. 2003;136:904–10. doi: 10.1016/s0002-9394(03)00577-4. [DOI] [PubMed] [Google Scholar]
- 22.Funayama T, Ishikawa K, Ohtake Y, Tanino T, Kurosaka D, Kimura I, et al. Variants in optineurin gene and their association with tumor necrosis factor-alpha polymorphisms in Japanese patients with glaucoma. Invest Ophthalmol Vis Sci. 2004;45:4359–67. doi: 10.1167/iovs.03-1403. [DOI] [PubMed] [Google Scholar]
- 23.Tang S, Toda Y, Kashiwagi K, Mabuchi F, Iijima H, Tsukahara S, et al. The association between Japanese primary open-angle glaucoma and normal tension glaucoma patients and the optineurin gene. Hum Genet. 2003;113:276–9. doi: 10.1007/s00439-003-0964-y. [DOI] [PubMed] [Google Scholar]
- 24.Toda Y, Tang S, Kashiwagi K, Mabuchi F, Iijima H, Tsukahara S, et al. Mutations in the optineurin gene in Japanese patients with primary open-angle glaucoma and normal tension glaucoma. Am J Med Genet A. 2004;125A:1–4. doi: 10.1002/ajmg.a.20439. [DOI] [PubMed] [Google Scholar]
- 25.Fuse N, Takahashi K, Akiyama H, Nakazawa T, Seimiya M, Kuwahara S, et al. Molecular genetic analysis of optineurin gene for primary open-angle and normal tension glaucoma in the Japanese population. J Glaucoma. 2004;13:299–303. doi: 10.1097/00061198-200408000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Toth RP, Atkin JD. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front Immunol. 2018;9:1017. doi: 10.3389/fimmu.2018.01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldstein O, Nayshool O, Nefussy B, Traynor BJ, Renton AE, Gana-Weisz M, et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology. 2016;86:446–53. doi: 10.1212/WNL.0000000000002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aung T, Rezaie T, Okada K, Viswanathan AC, Child AH, Brice G, et al. Clinical features and course of patients with glaucoma with the E50K mutation in the optineurin gene. Invest Ophthalmol Vis Sci. 2005;46:2816–22. doi: 10.1167/iovs.04-1133. [DOI] [PubMed] [Google Scholar]
- 29.Ayala-Lugo RM, Pawar H, Reed DM, Lichter PR, Moroi SE, Page M, et al. Variation in optineurin (OPTN) allele frequencies between and within populations. Mol Vis. 2007;13:151–63. [PMC free article] [PubMed] [Google Scholar]
- 30.Minegishi Y, Iejima D, Kobayashi H, Chi ZL, Kawase K, Yamamoto T, et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet. 2013;22:3559–67. doi: 10.1093/hmg/ddt210. [DOI] [PubMed] [Google Scholar]
- 31.Qiu Y, Wang J, Li H, Yang B, Wang J, He Q, et al. Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy. 2022;18:73–85. doi: 10.1080/15548627.2021.1908722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113:4039–44. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–33. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore AS, Holzbaur ELF. Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy. 2016;12:1956–7. doi: 10.1080/15548627.2016.1212788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126:580–92. doi: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundaramoorthy V, Walker AK, Tan V, Fifita JA, Mccann EP, Williams KL, et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:3830–46. doi: 10.1093/hmg/ddv126. [DOI] [PubMed] [Google Scholar]
- 37.Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol. 2005;169:285–95. doi: 10.1083/jcb.200501162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sippl C, Bosserhoff AK, Fischer D, Tamm ER. Depletion of optineurin in RGC-5 cells derived from retinal neurons causes apoptosis and reduces the secretion of neurotrophins. Exp Eye Res. 2011;93:669–80. doi: 10.1016/j.exer.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 39.Nagabhushana A, Chalasani ML, Jain N, Radha V, Rangaraj N, Balasubramanian D, et al. Regulation of endocytic trafficking of transferrin receptor by optineurin and its impairment by a glaucoma-associated mutant. BMC Cell Biol. 2010;11:4. doi: 10.1186/1471-2121-11-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park B, Ying H, Shen X, Park JS, Qiu Y, Shyam R, et al. Impairment of protein trafficking upon overexpression and mutation of optineurin. PLoS One. 2010;5:e11547. doi: 10.1371/journal.pone.0011547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Kang J, Horwitz MS. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol Cell Biol. 1998;18:1601–10. doi: 10.1128/mcb.18.3.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sayyad Z, Sirohi K, Radha V, Swarup G. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci Rep. 2017;7:16855. doi: 10.1038/s41598-017-17241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chalasani ML, Radha V, Gupta V, Agarwal N, Balasubramanian D, Swarup G. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest Ophthalmol Vis Sci. 2007;48:1607–14. doi: 10.1167/iovs.06-0834. [DOI] [PubMed] [Google Scholar]
- 44.Sayyad Z, Vishwakarma S, Dave TV, Naik MN, Radha V, Kaur I, et al. Human primary retinal cells as an in-vitro model for investigating defective signalling caused by OPTN mutants associated with glaucoma. Neurochem Int. 2021;148:105075. doi: 10.1016/j.neuint.2021.105075. [DOI] [PubMed] [Google Scholar]
- 45.VanderWall KB, Huang KC, Pan Y, Lavekar SS, Fligor CM, Allsop AR, et al. Retinal ganglion cells with a glaucoma OPTN(E50K) mutation exhibit neurodegenerative phenotypes when derived from three-dimensional retinal organoids. Stem Cell Reports. 2020;15:52–66. doi: 10.1016/j.stemcr.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomes C, VanderWall KB, Pan Y, Lu X, Lavekar SS, Huang KC, et al. Astrocytes modulate neurodegenerative phenotypes associated with glaucoma in OPTN(E50K) human stem cell-derived retinal ganglion cells. Stem Cell Reports. 2022;17:1636–49. doi: 10.1016/j.stemcr.2022.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chi ZL, Akahori M, Obazawa M, Minami M, Noda T, Nakaya N, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet. 2010;19:2606–15. doi: 10.1093/hmg/ddq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meng Q, Xiao Z, Yuan H, Xue F, Zhu Y, Zhou X, et al. Transgenic mice with overexpression of mutated human optineurin(E50K) in the retina. Mol Biol Rep. 2012;39:1119–24. doi: 10.1007/s11033-011-0840-0. [DOI] [PubMed] [Google Scholar]
- 49.Tseng HC, Riday TT, McKee C, Braine CE, Bomze H, Barak I, et al. Visual impairment in an optineurin mouse model of primary open-angle glaucoma. Neurobiol Aging. 2015;36:2201–12. doi: 10.1016/j.neurobiolaging.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou M, Shao Z, Zhang S, Liu X, Fan P, Jiang M, et al. Age-related visual impairments and retinal ganglion cells axonal degeneration in a mouse model harboring OPTN (E50K) mutation. Cell Death Dis. 2022;13:362. doi: 10.1038/s41419-022-04836-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Revach OY, Liu S, Jenkins RW. Targeting TANK-binding kinase 1 (TBK1) in cancer. Expert Opin Ther Targets. 2020;24:1065–78. doi: 10.1080/14728222.2020.1826929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Runde AP, Mack R, S J PB, Zhang J. The role of TBK1 in cancer pathogenesis and anticancer immunity. J Exp Clin Cancer Res. 2022;41:135. doi: 10.1186/s13046-022-02352-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fingert JH, Robin AL, Stone JL, Roos BR, Davis LK, Scheetz TE, et al. Copy number variations on chromosome 12q14 in patients with normal tension glaucoma. Hum Mol Genet. 2011;20:2482–94. doi: 10.1093/hmg/ddr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kawase K, Allingham RR, Meguro A, Mizuki N, Roos B, Solivan-Timpe FM, et al. Confirmation of TBK1 duplication in normal tension glaucoma. Exp Eye Res. 2012;96:178–80. doi: 10.1016/j.exer.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeLuca AP, Alward WLM, Liebmann J, Ritch R, Kawase K, Kwon YH, et al. Genomic organization of TBK1 copy number variations in glaucoma patients. J Glaucoma. 2017;26:1063–7. doi: 10.1097/IJG.0000000000000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Awadalla MS, Fingert JH, Roos BE, Chen S, Holmes R, Graham SL, et al. Copy number variations of TBK1 in Australian patients with primary open-angle glaucoma. Am J Ophthalmol. 2015;159:124–30.e1. doi: 10.1016/j.ajo.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim MJ, Kim YW, Jeoung JW, Seong MW, Lee JS, Kim DM, et al. Genomic characterization of TBK1 duplication in Korean normal-tension glaucoma patients. J Glaucoma. 2020;29:331–6. doi: 10.1097/IJG.0000000000001466. [DOI] [PubMed] [Google Scholar]
- 58.Ritch R, Darbro B, Menon G, Khanna CL, Solivan-Timpe F, Roos BR, et al. TBK1 gene duplication and normal-tension glaucoma. JAMA Ophthalmol. 2014;132:544–8. doi: 10.1001/jamaophthalmol.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaurani L, Vishal M, Ray J, Sen A, Ray K, Mukhopadhyay A. TBK1 duplication is found in normal tension and not in high tension glaucoma patients of Indian origin. J Genet. 2016;95:459–61. doi: 10.1007/s12041-016-0637-y. [DOI] [PubMed] [Google Scholar]
- 60.Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: A gatekeeper of neuroinflammation. Trends Mol Med. 2016;22:511–27. doi: 10.1016/j.molmed.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tucker BA, Solivan-Timpe F, Roos BR, Anfinson KR, Robin AL, Wiley LA, et al. Duplication of TBK1 stimulates autophagy in iPSC-derived retinal cells from a patient with normal tension glaucoma. J Stem Cell Res Ther. 2014;3:161. doi: 10.4172/2157-7633.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fingert JH, Miller K, Hedberg-Buenz A, Roos BR, Lewis CJ, Mullins RF, et al. Transgenic TBK1 mice have features of normal tension glaucoma. Hum Mol Genet. 2017;26:124–32. doi: 10.1093/hmg/ddw372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smaili W, Elalaoui SC, Zrhidri A, Raymond L, Egéa G, Taoudi M, et al. Exome sequencing revealed a novel homozygous METTL23 gene mutation leading to familial mild intellectual disability with dysmorphic features. Eur J Med Genet. 2020;63:103951. doi: 10.1016/j.ejmg.2020.103951. [DOI] [PubMed] [Google Scholar]
- 64.Hatanaka Y, Tsusaka T, Shimizu N, Morita K, Suzuki T, Machida S, et al. Histone H3 methylated at arginine 17 is essential for reprogramming the paternal genome in zygotes. Cell Rep. 2017;20:2756–65. doi: 10.1016/j.celrep.2017.08.088. [DOI] [PubMed] [Google Scholar]
- 65.Feng L, Wang C, Zhang C, Zhang W, Song W. Role of epigenetic regulation in glaucoma. Biomed Pharmacother. 2023;168:115633. doi: 10.1016/j.biopha.2023.115633. [DOI] [PubMed] [Google Scholar]
- 66.Soutto M, Belkhiri A, Piazuelo MB, Schneider BG, Peng D, Jiang A, et al. Loss of TFF1 is associated with activation of NF-κB-mediated inflammation and gastric neoplasia in mice and humans. J Clin Invest. 2011;121:1753–67. doi: 10.1172/JCI43922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc Natl Acad Sci U S A. 2014;111:11181–6. doi: 10.1073/pnas.1402819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sivakumar V, Foulds WS, Luu CD, Ling EA, Kaur C. Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J Pathol. 2011;224:245–60. doi: 10.1002/path.2858. [DOI] [PubMed] [Google Scholar]
- 70.Sharma R, Grover A. Myocilin-associated glaucoma: A historical perspective and recent research progress. Mol Vis. 2021;27:480–93. [PMC free article] [PubMed] [Google Scholar]
- 71.Stone EM, Fingert JH, Alward WL, Nguyen TD, Polansky JR, Sunden SL, et al. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–70. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 72.Liu Y, Allingham RR. Major review: Molecular genetics of primary open-angle glaucoma. Exp Eye Res. 2017;160:62–84. doi: 10.1016/j.exer.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trivli A, Zervou MI, Goulielmos GN, Spandidos DA, Detorakis ET. Primary open angle glaucoma genetics: The common variants and their clinical associations (Review) Mol Med Rep. 2020;22:1103–10. doi: 10.3892/mmr.2020.11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mardin CY, Velten I, Ozbey S, Rautenstrauss B, Michels-Rautenstrauss K. A GLC1A gene Gln368Stop mutation in a patient with normal-tension open-angle glaucoma. J Glaucoma. 1999;8:154–6. doi: 10.1097/00061198-199904000-00012. [DOI] [PubMed] [Google Scholar]
- 75.Michels-Rautenstrauss K, Mardin C, Wakili N, Jünemann AM, Villalobos L, Mejia C, et al. Novel mutations in the MYOC/GLC1A gene in a large group of glaucoma patients. Hum Mutat. 2002;20:479–80. doi: 10.1002/humu.9092. [DOI] [PubMed] [Google Scholar]
- 76.Alward WLM, Kwon YH, Khanna CL, Johnson AT, Hayreh SS, Zimmerman MB, et al. Variations in the myocilin gene in patients with open-angle glaucoma. Arch Ophthalmol. 2002;120:1189–97. doi: 10.1001/archopht.120.9.1189. [DOI] [PubMed] [Google Scholar]
- 77.Alward WLM, van der Heide C, Khanna CL, Roos BR, Sivaprasad S, Kam J, et al. Myocilin mutations in patients with normal-tension glaucoma. JAMA Ophthalmol. 2019;137:559–63. doi: 10.1001/jamaophthalmol.2019.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liuska PJ, Lemmelä S, Havulinna AS, Kaarniranta K, Uusitalo H, Laivuori H, et al. Association of the MYOC p.(Gln368Ter) variant with glaucoma in a Finnish population. JAMA Ophthalmol. 2021;139:762–8. doi: 10.1001/jamaophthalmol.2021.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fingert JH, Héon E, Liebmann JM, Yamamoto T, Craig JE, Rait J, et al. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 1999;8:899–905. doi: 10.1093/hmg/8.5.899. [DOI] [PubMed] [Google Scholar]
- 80.Fernandes KA, Harder JM, Williams PA, Rausch RL, Kiernan AE, Nair KS, et al. Using genetic mouse models to gain insight into glaucoma: Past results and future possibilities. Exp Eye Res. 2015;141:42–56. doi: 10.1016/j.exer.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tamm ER. Myocilin and glaucoma: Facts and ideas. Prog Retin Eye Res. 2002;21:395–428. doi: 10.1016/s1350-9462(02)00010-1. [DOI] [PubMed] [Google Scholar]
- 82.Polansky JR, Fauss DJ, Chen P, Chen H, Lütjen-Drecoll E, Johnson D, et al. Cellular pharmacology and molecular biology of the trabecular meshwork inducible glucocorticoid response gene product. Ophthalmologica. 1997;211:126–39. doi: 10.1159/000310780. [DOI] [PubMed] [Google Scholar]
- 83.Jacobson N, Andrews M, Shepard AR, Nishimura D, Searby C, Fingert JH, et al. Non-secretion of mutant proteins of the glaucoma gene myocilin in cultured trabecular meshwork cells and in aqueous humor. Hum Mol Genet. 2001;10:117–25. doi: 10.1093/hmg/10.2.117. [DOI] [PubMed] [Google Scholar]
- 84.Kim BS, Savinova OV, Reedy MV, Martin J, Lun Y, Gan L, et al. Targeted disruption of the myocilin gene (Myoc) suggests that human glaucoma-causing mutations are gain of function. Mol Cell Biol. 2001;21:7707–13. doi: 10.1128/MCB.21.22.7707-7713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pang CP, Lam DSC. Differential occurrence of mutations causative of eye diseases in the Chinese population. Hum Mutat. 2002;19:189–208. doi: 10.1002/humu.10053. [DOI] [PubMed] [Google Scholar]
- 86.Gould DB, Miceli-Libby L, Savinova OV, Torrado M, Tomarev SI, Smith RS, et al. Genetically increasing Myoc expression supports a necessary pathologic role of abnormal proteins in glaucoma. Mol Cell Biol. 2004;24:9019–25. doi: 10.1128/MCB.24.20.9019-9025.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Resch ZT, Fautsch MP. Glaucoma-associated myocilin: A better understanding but much more to learn. Exp Eye Res. 2009;88:704–12. doi: 10.1016/j.exer.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shepard AR, Jacobson N, Millar JC, Pang IH, Steely HT, Searby CC, et al. Glaucoma-causing myocilin mutants require the Peroxisomal targeting signal-1 receptor (PTS1R) to elevate intraocular pressure. Hum Mol Genet. 2007;16:609–17. doi: 10.1093/hmg/ddm001. [DOI] [PubMed] [Google Scholar]
- 89.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bailey JNC, Loomis SJ, Kang JH, Allingham RR, Gharahkhani P, Khor CC, et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat Genet. 2016;48:189–94. doi: 10.1038/ng.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blanc RS, Richard S. Arginine methylation: The coming of age. Mol Cell. 2017;65:8–24. doi: 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Tradewell ML, Yu Z, Tibshirani M, Boulanger MC, Durham HD, Richard S. Arginine methylation by PRMT1 regulates nuclear-cytoplasmic localization and toxicity of FUS/TLS harbouring ALS-linked mutations. Hum Mol Genet. 2012;21:136–49. doi: 10.1093/hmg/ddr448. [DOI] [PubMed] [Google Scholar]