

Abstract
In this review, we highlight several studies indicating that modulation of intrinsic neuronal excitability is a key for successful memory formation. Specifically, we will focus our discussion on our hypothesis that the postburst afterhyperpolarization (AHP: a key regulator of intrinsic excitability) is an essential cellular mechanism used by both principle and inhibitory neurons to change their neuronal activity as memory is formed. In addition, we propose that these intrinsic excitability changes occur first in principle neurons, followed by changes in inhibitory neurons; thus maintaining the balance of network activity among neurons for successful encoding and read-out of memory.
Keywords: postburst afterhyperpolarization, CREB, pyramidal neurons, inhibitory interneurons, memory, intrinsic excitability
The hippocampus remains a hot-bed of active research for learning and memory, following the seminal publication by Scoville and Milner (1957) that described case studies of memory deficits in patients who had portions of their medial temporal lobe surgically removed. The most well-known and studied of those patients is H.M. (Henry Gustav Molaison, 1926–2008) who had profound anterograde amnesia after his surgery that removed portions of his medial temporal lobe bilaterally, including the hippocampus (Corkin 2002). Another well studied amnesic is R.B. who suffered memory deficits after ischemic episodes caused specific loss of neurons only in the CA1 region of the hippocampus (Zola-Morgan and others 1986). These and other studies detailing the memory deficits of amnesics have led to many animal studies that demonstrated the importance of hippocampal function for learning various tasks (Alvarez and others 1995; Kim and others 1995; Kim and Fanselow 1992; Morris and others 1982; Moser and others 1993; Moyer and others 1990). Among those tasks, Pavlovian eyeblink conditioning has been an invaluable task used to examine the cellular and network changes that occur within the hippocampus during learning.
Eyeblink conditioning, on the surface, is a simple learning task. Following on the famous experiment by Ivan Pavlov pairing a bell with meat powder in dogs resulting in salivary conditioning, subjects (humans, rabbits, rats, mice) learn over many trials that a neutral stimulus (e.g., a tone: conditioned stimulus, CS) will be followed by a noxious stimulus to the eye (usually a puff of air: unconditioned stimulus, US). When the CS precedes and co-terminates with the US (referred to as delay eyeblink conditioning), the task is readily learned by nearly all subjects of every age group (Christian and Thompson 2003; Disterhoft and Oh 2006). However, when a temporal gap is introduced between the CS and the US (referred to as trace eyeblink conditioning), the task becomes challenging; and many aged subjects fail to reach learning criterion (Disterhoft and Oh 2007). More importantly, learning trace eyeblink conditioning becomes nearly impossible in subjects without the hippocampus (Moyer and others 1990; Solomon and others 1986; Tseng and others 2004; Weiss and others 1999). But recalling the memory for the learned association during trace eyeblink conditioning after memory consolidation does not require the hippocampus, as animals with hippocampal lesions done a month after acquisition are unimpaired during their memory test (Kim and others 1995; Takehara and others 2003). Hence, these studies proved that the hippocampus is an essential brain region for learning, but is not a required permanent storage site for this associative task.
Activity changes within the hippocampus as a subject learns the eyeblink task
Many studies have examined this question using single unit recordings. The seminal in vivo recording studies were reported by Richard Thompson and colleagues (Berger and others 1976; Berger and Thompson 1978) that showed hippocampal principle cells, the pyramidal neurons, increase their firing during the eyeblink conditioning trials in a way that mirrors the behavioral response. Moreover, they discovered that the increased pyramidal neuron firing precedes the expression of the conditioned eyeblink response: i.e., the neural network changes within the hippocampus occurred before the demonstration of the learned response. These findings using the delay (non-hippocampus dependent) eyeblink conditioning task have been extended with studies in trace eyeblink conditioning (Solomon and others 1986). However, unlike delay conditioning, a greater heterogeneity of response patterns were observed within the hippocampus during trace eyeblink conditioning (McEchron and Disterhoft 1997; Weiss and others 1996): i.e., some cells showed increases during the CS, trace, and/or US portion of the training trials; while other cells showed decreases during those portions. Regardless, the greatest increase in hippocampal pyramidal cell activity was during the training trials when the subject first demonstrated learning (i.e., during the training session when the ‘a-ha moment’ occurred) while the activity of presumed interneurons decreased (McEchron and Disterhoft 1997). Furthermore, the activity pattern in the dorsal, but not ventral, CA1 neurons showed learning related changes; resulting in increased baseline firing rate in dorsal CA1 pyramidal neurons with successful learning (Weible and others 2006). Notably, aged subjects that failed to learn the trace eyeblink conditioning task had hippocampal pyramidal neurons with significantly reduced baseline activity as compared to those from aged animals that were able to learn and from young animals (McEchron and others 2001). Together these studies suggest that dynamic hippocampal network changes occur during learning of trace eyeblink conditioning.
What is the cellular mechanism that underlies the learning-related network change?
We have proposed that the postburst afterhyperpolarization (AHP) is a key cellular mechanism recruited by neurons to cause the dynamic learning-related hippocampal network change (Disterhoft and Oh 2006; Disterhoft and Oh 2007). There have been many reviews regarding the characterization of the three distinct phases (fast, medium, slow) of the AHP (e.g.: Faber and Sah 2003; Oh and others 2010; Storm 1990). Here, we will mainly focus on the medium (mediated by apamin-sensitive SK channels) and the slow (mediated by a yet to be discovered channel(s)) AHPs and refer to them as postburst AHP, unless explicitly stated. In brief, the postburst AHP is composed of outward potassium conductances mediated primarily by accumulation of calcium ions within the cytosol following a train of action potentials: i.e., the postburst AHP is not observed without calcium ion influx and without action potentials, even with a train of subthreshold excitatory synaptic inputs (Wu and others 2004). Its main function is to act as a brake to limit or prevent over excitation of cells (i.e., prevent more action potentials). Another way of thinking about it is to imagine the postburst AHP as an energy barrier that must be overcome by successive excitatory inputs onto a cell after initial action potentials have occurred to allow that cell to generate further action potentials. However, the postburst AHP becomes larger with a higher frequency train of action potentials (Wu and others 2004): i.e., the energy barrier gets progressively larger until the postburst AHP decays back to basal levels (Fig. 1). Also, there exists an inverse relationship between the postburst AHP and the well-studied model of synaptic plasticity, long-term potentiation (LTP): a large postburst AHP prevents LTP, whereas a small postburst AHP facilitates LTP (Abraham and Tate 1997; Cohen and others 1999; Kramar and others 2004; Sah and Bekkers 1996). Furthermore, pharmacological compounds that reduce the postburst AHP (and thus, remove the energy barrier) also facilitates long-term synaptic plasticity. Therefore, the postburst AHP plays a significant role in synaptic integration and relay of neural information throughput of neurons.
Figure 1.
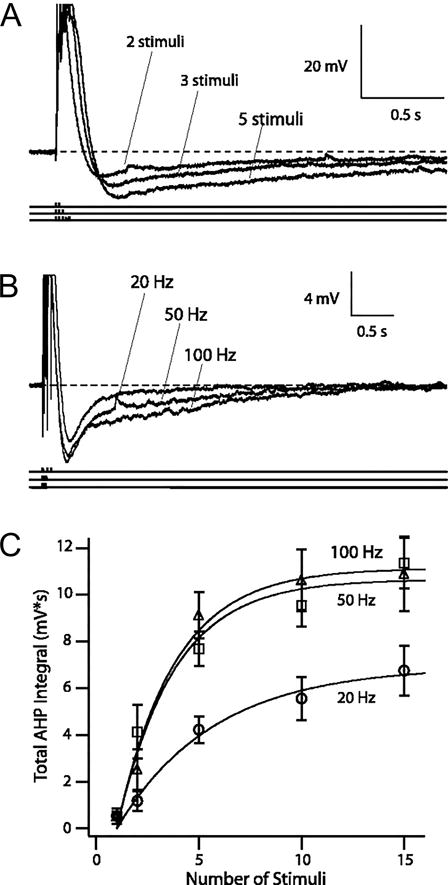
AHPs triggered by different patterns of suprathreshold synaptic stimuli. A: for a given stimulation frequency, AHP increased with each successive suprathreshold stimulus presentation. Representative voltage traces of AHPs triggered by 2, 3, and 5 suprathreshold stimuli at 50 Hz from a resting membrane potential of −68 mV. Action potentials were truncated for clarity. B: for a given number of stimuli, the AHP increased with increasing stimulation frequency. Representative voltage traces of AHPs evoked by 3 suprathreshold stimuli at 20, 50, and 100 Hz. C: frequency- and stimulus-dependence of AHP. Total AHP integral as a function of number of stimuli (1, 2, 5, 10, and 15) is plotted against stimulation frequencies (20 Hz, ○; 50 Hz, □; 100 Hz, ▵). Growths of AHP integrals were fit with monoexponential functions in the following form: Amax × (1 − exp{−[(no. stimuli − 1)/k]}), where Amax is the maximal total AHP integral, and k is the growth constant. Figure modified from Wu, Chan, and Disterhoft, J Neurophysiol. 2004, 92:2346–56.
The experimental support for our postburst AHP hypothesis can be classified into three major groups
First, we have repeatedly demonstrated that the postburst AHP is reduced in hippocampal pyramidal neurons from subjects that learned the eyeblink conditioning task (reviewed in: Oh and others 2010). In contrast, subjects that were trained but failed to learn the task had a similar postburst AHP to that observed in pseudoconditioned and naïve subjects (Matthews and others 2009; Moyer and others 1996; Thompson and others 1996b). Similar learning-related postburst AHP reductions in hippocampal pyramidal neurons have also been found after spatial water maze (Oh and others 2003), inhibitory avoidance (Farmer and Thompson 2012), and fear conditioning (Kaczorowski and Disterhoft 2009; McKay and others 2009; Song and others 2012). Hence, at least in hippocampal pyramidal neurons, postburst AHP reduction is a transient (see below) cellular mechanism recruited within the hippocampus for establishing and maintaining the neural network activity as permanent memory is encoded in neocortical regions.
Second, the postburst AHP must be at a naïve, young-like state for learning to occur. Aged subjects have difficulty learning the trace eyeblink conditioning task, with nearly half of the subjects failing to learn (Knuttinen and others 2001; Thompson and others 1996a). However, for those aged subjects that are able to learn, the postburst AHP of CA1 pyramidal neurons is reduced to nearly identical levels as to that observed in hippocampal neurons from young adults that learned the task (Matthews and others 2009; Moyer and others 2000), and the basal postburst AHP of aged unimpaired animals was significantly reduced as compared to those from aged impaired animals (Tombaugh and others 2005). Furthermore, pharmacological compounds that ameliorated the age-related learning deficits also reduced the postburst AHP of hippocampal neurons from aged animals to young-like levels (Disterhoft and Oh 2007). Notably, pharmacologically increasing the apamin-sensitive medium AHP in dorsal CA1 region of young adult animals caused them to be aged-like and greatly impaired their learning (McKay and others 2012) (Fig. 2). Although these findings with aged animals clearly indicate that reducing the postburst AHP to a young-like levels is beneficial, too little postburst AHP is also detrimental for learning. This could be due to uncontrolled baseline firing in the affected neuron populations. Transgenic mice with reduced postburst AHP in hippocampal pyramidal neurons have difficulty learning various hippocampus-dependent tasks (Gamelli and others 2011; Giese and others 1998; McKinney and Murphy 2006). Mice given high-frequency Schaffer collateral stimulation (HFS) that evokes long-term potentiation of evoked field excitatory postsynaptic potentials (EPSPs) in vivo are also impaired on learning the trace eyeblink conditioning task (Madronal and others 2007); not only due to synaptic modification caused by the HFS, but presumably also due to the reduced postburst AHP evoked with the HFS in the CA1 hippocampal neurons (there is an inverse correlation between LTP and the postburst AHP (Cohen and others 1999)). Furthermore, recent studies demonstrated that pharmacologically increasing the hippocampal neuronal activity of young adults in vivo by blocking BK channels (which partly mediate the fast AHP) significantly impaired learning the trace eyeblink conditioning task (Matthews and Disterhoft 2009; Matthews and others 2008). Hence, these findings suggest that the basal levels of the postburst AHP should be at naïve, young-like levels for learning to be optimized.
Figure 2.
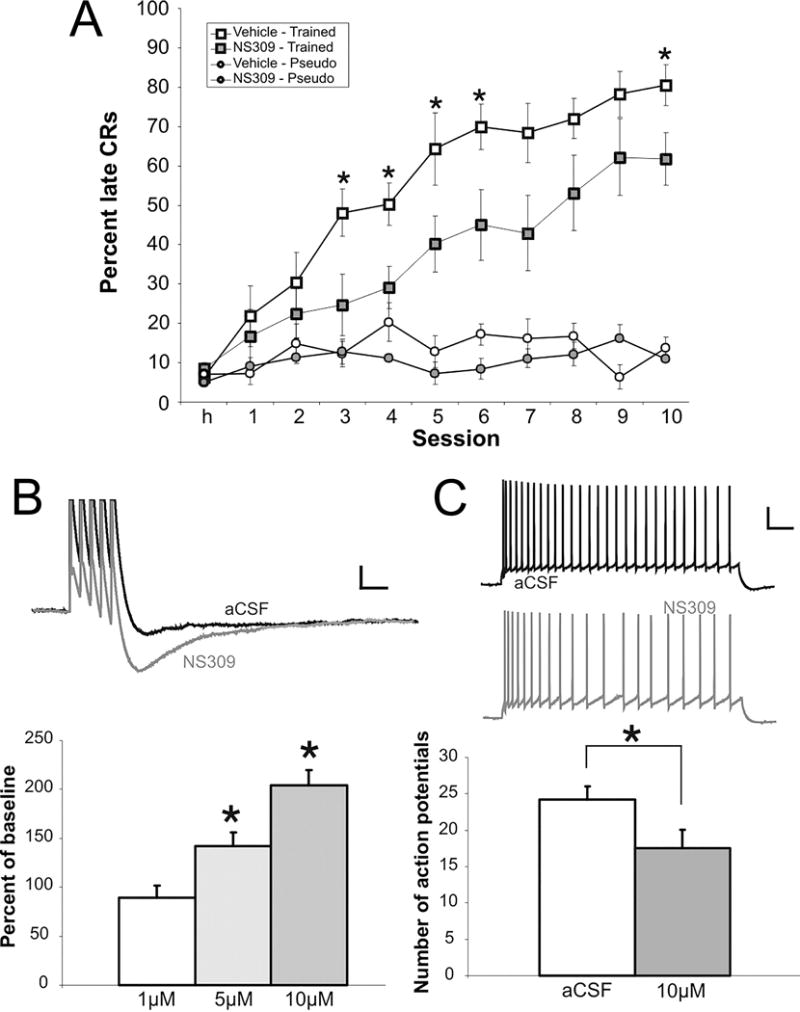
Learning was significantly impaired by pharmacological compound NS309 that increased the apamin-sensitive medium AHP. A: Rats were divided into four groups: vehicle trained (n = 9), NS309 trained (n = 10), vehicle pseudo (n = 5), NS309 pseudo (n = 6). Both control and NS309 (100 μM) animals learned the paradigm, but compared with controls, animals receiving NS309 infusions were significantly impaired on days 3–6 and 10 (*P < 0.05). NS309 had no effect on pseudoconditioning. B,C: NS309 increased the postburst AHP and reduced the excitability of CA1 pyramidal neurons. B: example traces of postburst AHP before and after NS309 bath application are illustrated. The AHP was evoked with five action potentials at 50 Hz. Action potentials are truncated for clarity. Scale bar: 2 mV, 100 ms. C: NS309 (10 μM) significantly increased accommodation. Accommodation was measured as the number of action potentials elicited in 1 s by a depolarizing current step, which evokes five action potentials in the first 100 ms. Scale bar: 20 mV, 100 ms. Data are presented as mean ± SE. *P < 0.05. Figure modified from McKay et al., J Neurophysiol. 2012, 108:863–70.
Lastly, the time course of the learning-related postburst AHP change of hippocampal pyramidal neurons fit well with the proposed function of the hippocampus: essential for learning, but not a required permanent memory storage site. Hippocampal lesions made one day after learning resulted in profound loss of the learned response and inability to relearn the response; whereas lesions made a month after learning did not abolish the learned response (Kim and others 1995). Additional lesion studies demonstrated that hippocampal lesion made a week after successful learning did not impact recall of the learned eyeblink response (Takehara and others 2003); hence further refining the temporal window of hippocampal engagement and recruitment of neocortical regions (e.g., medial prefrontal cortex) for learning and memory. These and other hippocampal lesion studies (e.g., Kim and Fanselow 1992) strongly suggest that neural activity within the hippocampus and/or hippocampal interaction with neocortical memory storage sites are essential during the first few days after reaching learning criterion. It is our hypothesis that this critical temporal window for more permanent memory storage is provided by the transient (up to 5 days) learning-related postburst AHP reductions that we observed in the hippocampal pyramidal neurons (Moyer and others 1996; Thompson and others 1996b) (Fig. 3). This increased excitability allows these neurons to participate in and support the permanent formation of the memory that occurs within permanent storage sites in neocortex (e.g., medial prefrontal cortex) and other brain regions. Notably, an identical transient increase (lasting for up to 5 days) in Schaffer collateral evoked EPSPs measured in CA1 has been observed in vivo after trace eyeblink conditioning (Madronal and others 2007). However, similar learning-related EPSP changes were not observed in our experiments in brain slices. We observed an increased Schaffer collateral evoked EPSP immediately after learning, but not when measured the following day ex vivo (Power and others 1997). Although these findings may appear contradictory, our recent findings regarding increased inhibition during trace eyeblink conditioning mediated by learning-related change in intrinsic excitability of interneurons may shed light to resolve the issue.
Figure 3.
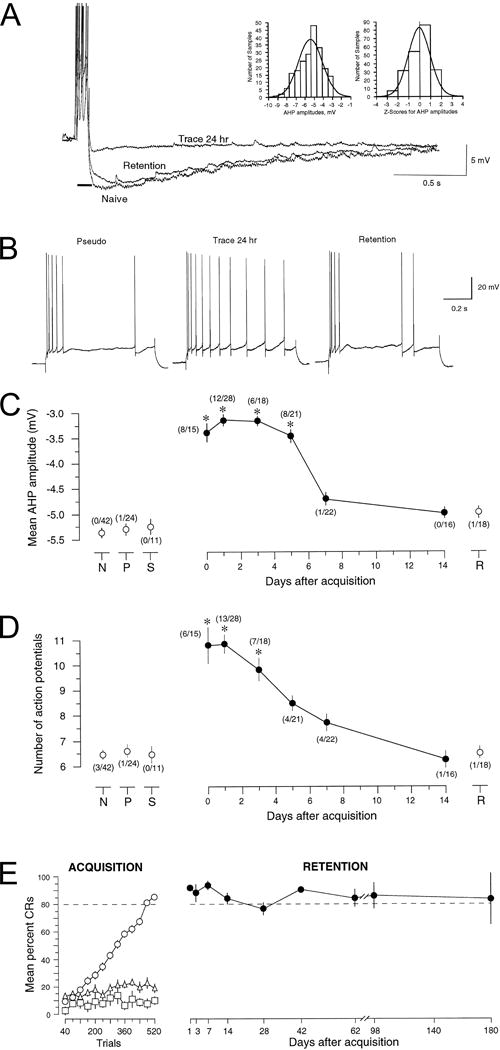
Acquisition of hippocampus-dependent trace eyeblink conditioning increased excitability of hippocampal CA1 pyramidal neurons. A: Voltage trace shows an overlay of recordings of the postburst AHPs in CA1 neurons from a naive (Naive) and from trace-conditioned rabbits studied 24 hr after initial learning (Trace 24 hr) or 24 hr after receiving an additional training session given 14 d after initial learning (Retention). The resting membrane potentials for these cells were approximately −66 mV, with action potentials truncated for visualization of the AHP. The AHP was measured for 5 sec beginning after a 100 msec depolarizing current injection (solid black line), with minimal current (~0.6 nA) required to reliably evoke a burst of four action potentials. The postburst AHPs from experimentally naive rabbits followed a normal distribution as shown in the frequency distribution and z-score graphs (insets). B: Examples of typical accommodation responses in CA1 pyramidal cells from rabbits: 24 hr after pseudoconditioning (Pseudo), 24 hr after acquisition of trace conditioning (Trace 24 hr), and 24 hr after receiving an additional training session 14 d after acquisition (Retention). Notice that although the cell from the trace-conditioned rabbit fired more action potentials, accommodation was certainly not abolished (as evidenced by the increase in interspike interval with time during the 800 msec depolarizing stimulus) but, rather, was significantly and transiently reduced after learning. Increased excitability of CA1 pyramidal neurons after learning was transient while performance remained persistent. C: Learning-related reductions of the AHP amplitude were transient, lasting ~1 week in slices prepared at various times after learning [1 hr (0 d), 1 d, 3 d, 5 d, 7 d, or 14 d]. Such changes were not observed in naive (N), pseudoconditioned (P), or slow-learning (S) control rabbits. Numbers in parentheses indicate the ratio of individual cells with reduced AHPs to number of cells studied in that group. Slow learners (S) were defined as rabbits that did not reach criterion within 15 training sessions, and that exhibited <30% conditioned responses on the last training session. Retention (R) rabbits received an additional 80-trial training session on the 14th day after initial learning. D: Trace eyeblink conditioning also resulted in a transient decrease in spike-frequency adaptation (accommodation) in CA1 neurons. Cells from slow-learning or pseudoconditioned control rabbits showed no changes, nor did cells from retention rabbits that received an additional training session 14 d later. The ratio of individual cells with reduced accommodation versus the number of cells studied for each group is indicated in parentheses. A cell was classified as having reduced accommodation if the number of APs elicited was at least 2 SDs more than the mean for all naive control cells. E: After successful acquisition, rabbits maintained the learned association. The left panel (ACQUISITION) shows the normalized learning curves for trace conditioned compared with pseudoconditioned and slow-learning rabbits. Trace-conditioned rabbits (○, n = 46) required an average of 7.1 ± 0.6 sessions to learn the task. As can be seen clearly, neither the pseudoconditioned (▵, n = 11) nor the slow-learning rabbits (□, n = 3) showed significant improvement across sessions. Thus, the pseudoconditioned and slow-learning rabbits served as excellent controls for nonspecific effects of training unrelated to associative learning. The right panel (RETENTION) shows the percent CRs elicited during 20 paired CS–US trials delivered at various time intervals after acquisition. Notice that when retention rabbits (•, n = 10) received 20 paired CS–US conditioning trials at the indicated times after learning, they maintained their criterion performance. For C and D, asterisks indicate data significantly different from all three control groups: *p < 0.001. Figure modified from Moyer, Thompson, and Disterhoft, J. Neurosci. 1996, 16:5536–5546.
Much attention and focus have been on principle cells, the pyramidal neurons, to identify the cellular mechanisms that underlie successful learning. A potential reason for the emphasis on the pyramidal neurons may be due to the diversity of GABAergic cells, as there are more than 20 different functional and/or anatomically classified interneurons within CA1 region alone (Freund and Buzsaki 1996; Somogyi and others 2014), that comprise about 10–30 percent of the neurons in the cortex (DeFelipe and others 2013; Markram and others 2004). This makes it difficult to carry out systematic studies to attribute specific learning and memory related changes to a specific interneuron subtype. However, numerous studies have demonstrated that proper function of interneurons is important for successful learning (Cui and others 2008; Fuchs and others 2007; Lovett-Barron and others 2014; Ruediger and others 2011) and that interneuron activity changes with learning (Brosh and Barkai 2009; Saar and others 2012; Urban-Ciecko and others 2010). Recently it has been demonstrated using various genetic manipulations, including use of optogentics, that contextual fear learning is severely impaired without proper function of somatostatin positive interneurons in CA1 region (Lovett-Barron and others 2014). Therefore, proper modulation of interneuronal function is essential for successful learning. But how is the function of interneuron(s) changed during learning and what is the cellular mechanism used to support these alterations?
We have serendipitously discovered that reduction of calcium-activated potassium currents as measured by the postburst AHP is also a cellular mechanism recruited by interneurons to alter their activity with learning (McKay and others 2013). Most studies investigating the learning-related postburst AHP changes in hippocampal pyramidal neurons have used direct current injections into the soma to evoke bursts of action potentials. The outward potassium currents mediating the afterhyperpolarization followed these action potentials. To measure afterhyperpolarizations following synaptic activation, we conducted a series of experiments using suprathreshold Schaffer collateral stimulations to verify that the learning-related postburst AHP reductions in hippocampal CA1 pyramidal neurons are observed using a naturalistic stimulation pattern to evoke action potentials in these neurons (McKay and others 2013). Contrary to expectations, we initially found no learning-related changes in the postburst AHP evoked with a train of synaptically evoked action potentials in ACSF (Fig. 4A); yet, we still observed the learning-related postburst AHP reductions with trains of action potentials evoked using direct somatic current injections in this same bathing solution. This dichotomy prompted us to pharmacologically block GABA receptors and reexamine the synaptically evoked postburst AHP. In a series of occlusion experiments, we showed that the synaptically evoked postburst AHP is reduced while the synaptically evoked inhibition is increased in CA1 pyramidal neurons from animals that learned the trace eyeblink conditioning task (Fig. 4A,B). Specifically, we observed learning-related increased inhibition following a synaptically evoked action potential in CA1 pyramidal neuron (Fig. 4C,D), but no learning-related changes were observed with subthreshold synaptic stimulation – even with a train of subthreshold EPSPs (Fig. 4E,F). We also discovered learning-related increases in the frequency of inhibitory postsynaptic currents onto CA1 pyramidal neurons without a change in their amplitude. Parenthetically, this increased inhibition onto CA1 pyramidal neurons after learning may be the reason for the lack of field EPSP increase we observed in the earlier study (Power and others 1997). Together these findings strongly suggested that learning changed not only the excitability of principle neurons but also altered the activity of feedback inhibitory interneurons, and prompted us to systematically examine intrinsic membrane properties of inhibitory CA1 neurons following learning.
Figure 4.
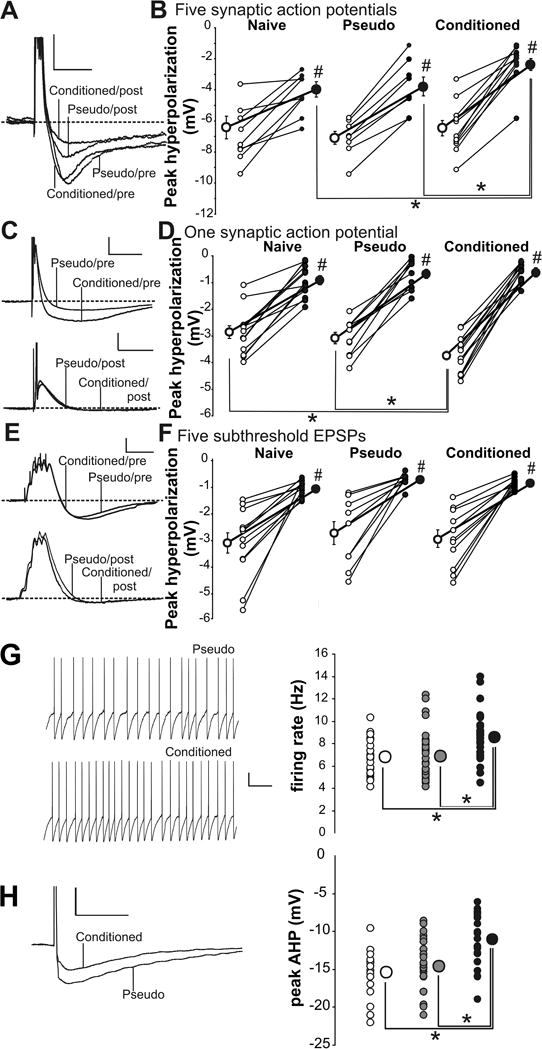
Learning resulted in postburst AHP reduction and IPSP increase in CA1 pyramidal neurons. A: Example postburst hyperpolarization traces evoked with 50 Hz train of five suprathreshold Schaffer collateral stimulation from a CA1 neuron from a conditioned and a pseudoconditioned rat before (Pre) and after (Post) GABA blockers were added to the perfusate. The action potentials have been truncated to focus on the hyperpolarization. Calibration: 4 mV, 500 ms. B: GABA blockers significantly reduced the hyperpolarization in CA1 neurons from all rats (#paired t test p < 0.05) and unveiled the learning-related postburst AHP reduction in neurons from conditioned rats. Open circles, Pre-GABA blockers; closed circles, post-GABA blockers. C: Example traces of hyperpolarization evoked with single synaptically evoked action potential from CA1 neurons from conditioned and pseudoconditioned rats before (Pre) and after (Post) GABA blockers. Calibration: 5 mV, 100 ms. D: GABA blockers significantly reduced the IPSP in CA1 neurons from all behavioral groups (#paired t test p < 0.05) and abolished the learning-related IPSP enlargement observed in normal aCSF. E: Illustrated are example traces evoked with 50 Hz trains of five subthreshold Schaffer collateral stimulation in CA1 neurons from conditioned and pseudoconditioned rats before (Pre) and after (Post) GABA blockers. Calibration: 3 mV, 100 ms. F: GABA blockers significantly reduced the IPSP in CA1 neurons from all behavioral groups (#paired t test p < 0.05). However, no significant IPSP differences were observed between groups either before or after GABA blockers. G: Intrinsic excitability is increased in SOMs from trained mice compared with controls. Learning resulted in significantly increased spontaneous firing rate at resting membrane potential in SOM neurons. Conditioned, black circle; pseudoconditioned, gray circle; naive, open circle. Calibration: 10 mV, 500 ms. H: The apamin-sensitive AHP following a single action potential is significantly reduced in SOMs from conditioned mice compared with controls. Calibration: 10 mV, 50 ms. Note: *Fischer’s PLSD p < 0.05. Smaller circles are the data points from individual neurons. Larger circles are the group means. In some instances, the SEM of each group is smaller than the size of the circle that represents the group mean data. Figure modified from McKay, Oh, and Disterhoft, J Neurosci. 2013, 33:5499–506.
As discussed above, there are many different types of interneurons within the CA1 region (Somogyi and others 2014). Among them, the O-LM interneurons (soma located within the oriens/alveus and axons innervating the lacunosum-moleculare) have been demonstrated to express somatostatin (Freund and Buzsaki 1996) and provide feedback inhibition onto CA1 pyramidal neurons (Maccaferri and McBain 1995). Therefore, we decided to train transgenic mice that were created to express the enhanced green fluorescent protein (GFP: to visually identify them) specifically in somatostatin expressing cells (Oliva and others 2000), and record from those GFP expressing cells with somas located within the oriens of the CA1 region. Remarkably, we discovered that the baseline firing was increased and the AHP was reduced in somatostatin expressing interneurons from mice that learned the trace eyeblink conditioning task (McKay and others 2013) (Fig. 4G,F). Importantly, a calcium-dependent, apamin-sensitive AHP (mediated by SK channels) is present in interneurons (Zhang and McBain 1995), as well as pyramidal neurons (Oh and others 2000; Stocker and others 1999). Therefore, we systematically determined if the learning-related AHP reduction in these somatostatin-positive interneurons was also mediated by a reduction in the apamin-sensitive AHP. We found that indeed the learning-related AHP in these somatostatin interneurons was mediated by a reduction in the apamin-sensitive AHP. This is the first demonstration that the calcium-activated postburst AHP is used by both excitatory and inhibitory neurons to modulate their intrinsic excitability following successful learning. Although it is yet to be determined if intrinsic excitability is also altered by learning in other interneurons, we hypothesize that the learning-related intrinsic excitability change will be due to modulation of the postburst AHP in other interneuron classes that demonstrate learning-associated increases in baseline firing.
Thus far, we have mainly focused the discussion to the learning-related postburst AHP changes in hippocampal neurons. However, the learning-related postburst AHP modulation appears to be a mechanism used throughout the brain. In the piriform cortex, odor discrimination learning leads to a transient postburst AHP reduction in the piriform cortical neurons (Saar and others 1998) similar to that observed in hippocampal pyramidal neurons following trace eyeblink conditioning (Moyer and others 1996; Thompson and others 1996b). The postburst AHP reduction in the piriform cortical neurons is mediated by extracellular signal-regulated kinase activity (Cohen-Matsliah and others 2007). In the lateral amygdala, learning fear conditioning leads to postburst AHP reductions in the lateral amygdala pyramidal neurons (Sehgal and others 2014). In the prefrontal cortex, infralimbic pyramidal neurons increase, not decrease, their postburst AHP during fear conditioning (Santini and others 2008); but the converse (i.e., postburst AHP reduction) via activation of PKA is necessary for extinction of the fear response (Mueller and others 2008). Hence, the postburst AHP appears to be a universal cellular mechanism used by neurons (both excitatory and inhibitory) to alter their intrinsic excitability during learning and memory.
Together the learning-related changes reported by in vivo and ex vivo studies paint an intriguing picture. We have hypothesized that the postburst AHP modulation (specifically the AHP reduction in hippocampal pyramidal neurons) must precede learning (Disterhoft and Oh 2006; Disterhoft and Oh 2007). This is based on the in vivo recordings of pyramidal neurons and interneurons in the CA1 hippocampal region showing maximal change on the training session when the association is first demonstrated (McEchron and Disterhoft 1997) and on the numerous studies with aged animals that demonstrated rescued learning by treatments with pharmacological compounds that also reduced the postburst AHP in CA1 pyramidal neurons (Disterhoft and Oh 2007). However, our recent discovery of learning-related changes in intrinsic excitability of somatostatin expressing interneurons adds another important layer to our hypothesis. We propose that the postburst AHP modulation in principle neurons allows the neural network to form the learned memories, and this change in the excitatory circuitry must be kept in check after learning by modulating the postburst AHP of interneurons (Fig. 5). Hence, in the hippocampus, the postburst AHP in hippocampal pyramidal neurons will be reduced to allow increased communication between hippocampal and neocortical neurons, and thus allow neural network formation supporting learning and memory to occur. But once this network is established and learning has occurred, the increase in the network must be dampened to prevent over-excitation and allow selective information flow through the network (that is, increase the signal-to-noise selection bias). This latter event would be mediated by increasing the output of appropriate inhibitory neurons via reducing their postburst AHP.
Figure 5.
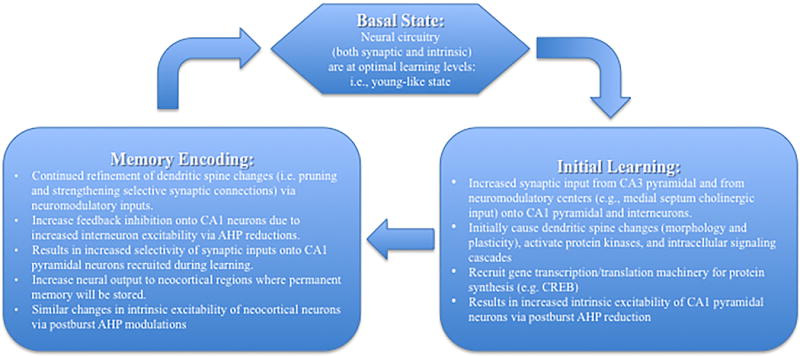
Schematic model of events that occur within CA1 region during acquisition and encoding of memory. We hypothesize that the cellular and neural network changes during Initial Learning lead to the events that occur during Memory Encoding. The temporal time window for the events that occur between the two events may be hours to days, depending on the difficulty of the task being learned and encoded. Importantly the events during the Memory Encoding phase are transient, and will last for few days as memory related cellular changes (e.g., postburst AHP modulation) occur within the neocortical neurons. Once the memory trace has been established in the permanent memory storage sites, the hippocampal circuit returns to the Basal State.
Although the learning-related postburst AHP reduction in hippocampal neurons has been repeatedly demonstrated, it is still unclear how the reduction is induced. One potential cellular signaling cascade involves activation of a cellular transcription factor CREB (cAMP response element-binding protein) via protein kinase A (PKA). The PKA-CREB signaling cascade has been well studied as a mechanism for long-term synaptic modifications and for learning in numerous brain regions and species (Kandel 2001; Kandel 2012). PKA activation also has been shown to reduce the postburst AHP in hippocampal neurons (Pedarzani and others 1998; Pedarzani and Storm 1993). More importantly, PKA activity mediates the learning-related postburst AHP reduction in hippocampal CA1 pyramidal neurons (Oh and others 2009) and primes (by phosphorylation) the cellular machinery necessary for fear conditioning in the amygdala (Parsons and Davis 2012). In addition to PKA, numerous other cascades can activate CREB; all of which involve activation of various protein kinases (Alberini 2009; Benito and Barco 2010). Notably, genetically increasing CREB levels has been shown to reduce the postburst AHP (Lopez de Armentia and others 2007; Zhou and others 2009) and facilitate learning (Han and others 2007; Han and others 2009). Furthermore, the genetically modified neurons that have more CREB (and thus, reduced postburst AHP) are also preferentially recruited in the neural network during learning and for recall of fear memory (Han and others 2009; Zhou and others 2009). Hence these findings suggest that the learning-related postburst AHP modulation and its maintenance most likely involve the activation of the CREB signaling cascade.
Undoubtedly future work will refine our hypothesis. Advent of optogentics and specific cre-line transgenic mouse models has led to identifying the role of general classes of neurons for learning. For example, recent work by Losonczy and colleagues demonstrated that inactivation of dorsal CA1 somatostatin interneurons during US presentation period was sufficient to prevent contextual fear conditioning in mice (Lovett-Barron and others 2014). With better genetic targeting to specific cell types, similar optogentic approaches can be used to further narrow the role of specific neurons (e.g., somatostatin-positive O-LM interneurons) and interactions of the neurons within and across the neural networks during encoding and recall of memory. Continued refinement of viral mediated gene transfer and silencing will also allow specific examination of cellular signaling cascades that are essential for learning (Han and others 2007; Han and others 2009; Zhou and others 2009). Although CREB activation has been shown to be a critical factor for successful learning and for reducing the postburst AHP, much work remains to clarify if there is a direct relationship between the two cellular events, or if they are two independent events that coincidentally occur as by-products of common protein kinase(s) activation. If there is a cause-and-effect linkage between the postburst AHP reduction and CREB activation during learning, then identifying the signaling pathway that underlies it will prove to be an invaluable reference tool for finding cures for learning and memory deficits caused by to numerous genetic disorder and those that occur with normal aging. Furthermore, new tools (genetic and technological innovations) will also allow us to examine the temporal interaction of connected brain regions, and the neurons that allow the connection, during learning and during recall of the learned memory. For example, Deisseroth and colleagues recently demonstrated that recall of a learned fear response is severely impaired when the anterior cingulate cortex (ACC) is optogenetically inactivated during testing for the fear response nearly a month (but not one day) after training (Goshen and others 2011). We have recently reported using single unit recordings in vivo that a temporal transfer of neural activity occurs from the caudal ACC to prelimbic subregions of the medial prefrontal cortex as rabbits learn (initial few days of training; caudal ACC) and remember (a month after training; prelimbic) the trace eyeblink response (Hattori and others 2014). Together, these recent in vivo data strongly support the hypothesis that a transfer of memory from one brain region to another (e.g., from hippocampus to medial prefrontal cortex) occurs over a temporal window that is evidenced by changes in neural firing within the brain regions. Furthermore, the cellular mechanisms used by the cells (both pyramidal neurons and interneurons) within the permanent memory storage sites as memory is encoded have yet to be determined. We hypothesize that the postburst AHP modulation will be one of the cellular mechanisms recruited by neurons within the network that permanently stores the memory after consolidation has occurred.
Acknowledgments
Supported by: NIH grant R37 AG008796
References
- Abraham WC, Tate WP. Metaplasticity: a new vista across the field of synaptic plasticity. Prog Neurobiol. 1997;52(4):303–23. doi: 10.1016/s0301-0082(97)00018-x. [DOI] [PubMed] [Google Scholar]
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89(1):121–45. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Zola-Morgan S, Squire LR. Damage limited to the hippocampal region produces long-lasting memory impairment in monkeys. J Neurosci. 1995;15(5 Pt 2):3796–807. doi: 10.1523/JNEUROSCI.15-05-03796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33(5):230–40. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Berger TW, Alger B, Thompson RF. Neuronal substrate of classical conditioning in the hippocampus. Science. 1976;192(4238):483–5. doi: 10.1126/science.1257783. [DOI] [PubMed] [Google Scholar]
- Berger TW, Thompson RF. Identification of pyramidal cells as the critical elements in hippocampal neuronal plasticity during learning. Proc Natl Acad Sci U S A. 1978;75(3):1572–6. doi: 10.1073/pnas.75.3.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosh I, Barkai E. Learning-induced enhancement of feedback inhibitory synaptic transmission. Learn Mem. 2009;16(7):413–6. doi: 10.1101/lm.1430809. [DOI] [PubMed] [Google Scholar]
- Christian KM, Thompson RF. Neural substrates of eyeblink conditioning: acquisition and retention. Learn Mem. 2003;10(6):427–55. doi: 10.1101/lm.59603. [DOI] [PubMed] [Google Scholar]
- Cohen-Matsliah SI, Brosh I, Rosenblum K, Barkai E. A novel role for extracellular signal-regulated kinase in maintaining long-term memory-relevant excitability changes. J Neurosci. 2007;27(46):12584–9. doi: 10.1523/JNEUROSCI.3728-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Coussens CM, Raymond CR, Abraham WC. Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J Neurophysiol. 1999;82(6):3139–3148. doi: 10.1152/jn.1999.82.6.3139. [DOI] [PubMed] [Google Scholar]
- Corkin S. What’s new with the amnesic patient H.M.? Nat Rev Neurosci. 2002;3(2):153–60. doi: 10.1038/nrn726. [DOI] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135(3):549–60. doi: 10.1016/j.cell.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, Lopez-Cruz PL, Benavides-Piccione R, Bielza C, Larranaga P, Anderson S, et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat Rev Neurosci. 2013;14(3):202–16. doi: 10.1038/nrn3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29(10):587–99. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Alterations in intrinsic neuronal excitability during normal aging. Aging Cell. 2007;6(3):327–36. doi: 10.1111/j.1474-9726.2007.00297.x. [DOI] [PubMed] [Google Scholar]
- Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9(3):181–94. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- Farmer GE, Thompson LT. Learning-dependent plasticity of hippocampal CA1 pyramidal neuron postburst afterhyperpolarizations and increased excitability after inhibitory avoidance learning depend upon basolateral amygdala inputs. Hippocampus. 2012;22(8):1703–19. doi: 10.1002/hipo.22005. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6(4):347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53(4):591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- Gamelli AE, McKinney BC, White JA, Murphy GG. Deletion of the L-type calcium channel Ca(V) 1.3 but not Ca(V) 1.2 results in a diminished sAHP in mouse CA1 pyramidal neurons. Hippocampus. 2011;21(2):133–41. doi: 10.1002/hipo.20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KP, Storm JF, Reuter D, Fedorov NB, Shao LR, Leicher T, et al. Reduced K+ channel inactivation, spike broadening, and after-hyperpolarization in Kvbeta1.1-deficient mice with impaired learning. Learn Mem. 1998;5:4–5. 257–273. [PMC free article] [PubMed] [Google Scholar]
- Goshen I, Brodsky M, Prakash R, Wallace J, Gradinaru V, Ramakrishnan C, et al. Dynamics of retrieval strategies for remote memories. Cell. 2011;147(3):678–89. doi: 10.1016/j.cell.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, et al. Neuronal competition and selection during memory formation. Science. 2007;316(5823):457–60. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Han JH, Kushner SA, Yiu AP, Hsiang HL, Buch T, Waisman A, et al. Selective erasure of a fear memory. Science. 2009;323(5920):1492–6. doi: 10.1126/science.1164139. [DOI] [PubMed] [Google Scholar]
- Hattori S, Yoon T, Disterhoft JF, Weiss C. Functional reorganization of a prefrontal cortical network mediating consolidation of trace eyeblink conditioning. J Neurosci. 2014;34(4):1432–45. doi: 10.1523/JNEUROSCI.4428-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft JF. Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice. Learn Mem. 2009;16(6):362–6. doi: 10.1101/lm.1365609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294(5544):1030–8. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain. 2012;5:14. doi: 10.1186/1756-6606-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Clark RE, Thompson RF. Hippocampectomy impairs the memory of recently, but not remotely, acquired trace eyeblink conditioned responses. Behav Neurosci. 1995;109(2):195–203. doi: 10.1037//0735-7044.109.2.195. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256(5057):675–7. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Knuttinen MG, Gamelli AE, Weiss C, Power JM, Disterhoft JF. Age-related effects on eyeblink conditioning in the F344 × BN F1 hybrid rat. Neurobiol Aging. 2001;22(1):1–8. doi: 10.1016/s0197-4580(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24(22):5151–5161. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Armentia M, Jancic D, Olivares R, Alarcon JM, Kandel ER, Barco A. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27(50):13909–18. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, et al. Dendritic inhibition in the hippocampus supports fear learning. Science. 2014;343(6173):857–63. doi: 10.1126/science.1247485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. Passive propagation of LTD to stratum oriens-alveus inhibitory neurons modulates the temporoammonic input to the hippocampal CA1 region. Neuron. 1995;15(1):137–45. doi: 10.1016/0896-6273(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Madronal N, Delgado-Garcia JM, Gruart A. Differential effects of long-term potentiation evoked at the CA3 CA1 synapse before, during, and after the acquisition of classical eyeblink conditioning in behaving mice. J Neurosci. 2007;27(45):12139–46. doi: 10.1523/JNEUROSCI.3397-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5(10):793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- Matthews EA, Disterhoft JF. Blocking the BK channel impedes acquisition of trace eyeblink conditioning. Learn Mem. 2009;16(2):106–9. doi: 10.1101/lm.1289809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EA, Linardakis JM, Disterhoft JF. The fast and slow afterhyperpolarizations are differentially modulated in hippocampal neurons by aging and learning. J Neurosci. 2009;29(15):4750–5. doi: 10.1523/JNEUROSCI.0384-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EA, Weible AP, Shah S, Disterhoft JF. The BK-mediated fAHP is modulated by learning a hippocampus-dependent task. Proc Natl Acad Sci USA. 2008;105(39):15154–9. doi: 10.1073/pnas.0805855105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEchron MD, Disterhoft JF. Sequence of single neuron changes in CA1 hippocampus of rabbits during acquisition of trace eyeblink conditioned responses. J Neurophysiol. 1997;78(2):1030–44. doi: 10.1152/jn.1997.78.2.1030. [DOI] [PubMed] [Google Scholar]
- McEchron MD, Weible AP, Disterhoft JF. Aging and learning-specific changes in single-neuron activity in CA1 hippocampus during rabbit trace eyeblink conditioning. J Neurophysiol. 2001;86(4):1839–57. doi: 10.1152/jn.2001.86.4.1839. [DOI] [PubMed] [Google Scholar]
- McKay BM, Matthews EA, Oliveira FA, Disterhoft JF. Intrinsic neuronal excitability is reversibly altered by a single experience in fear conditioning. J Neurophysiol. 2009;102(5):2763–70. doi: 10.1152/jn.00347.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BM, Oh MM, Disterhoft JF. Learning increases intrinsic excitability of hippocampal interneurons. J Neurosci. 2013;33(13):5499–506. doi: 10.1523/JNEUROSCI.4068-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BM, Oh MM, Galvez R, Burgdorf J, Kroes RA, Weiss C, et al. Increasing SK2 channel activity impairs associative learning. J Neurophysiol. 2012;108(3):863–70. doi: 10.1152/jn.00025.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney BC, Murphy GG. The L-Type voltage-gated calcium channel Cav1.3 mediates consolidation, but not extinction, of contextually conditioned fear in mice. Learn Mem. 2006;13(5):584–9. doi: 10.1101/lm.279006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O’Keefe J. Place navigation impaired in rats with hippocampal lesions. Nature. 1982;297(5868):681–3. doi: 10.1038/297681a0. [DOI] [PubMed] [Google Scholar]
- Moser E, Moser MB, Andersen P. Spatial learning impairment parallels the magnitude of dorsal hippocampal lesions, but is hardly present following ventral lesions. J Neurosci. 1993;13(9):3916–25. doi: 10.1523/JNEUROSCI.13-09-03916.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Deyo RA, Disterhoft JF. Hippocampectomy disrupts trace eye-blink conditioning in rabbits. Behav Neurosci. 1990;104(2):243–252. doi: 10.1037//0735-7044.104.2.243. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Power JM, Thompson LT, Disterhoft JF. Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. J Neurosci. 2000;20(14):5476–5482. doi: 10.1523/JNEUROSCI.20-14-05476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Thompson LT, Disterhoft JF. Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci. 1996;16(17):5536–5546. doi: 10.1523/JNEUROSCI.16-17-05536.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28(2):369–75. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol. 2003;90(4):2171–2179. doi: 10.1152/jn.01177.2002. [DOI] [PubMed] [Google Scholar]
- Oh MM, McKay BM, Power JM, Disterhoft JF. Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A. Proc Natl Acad Sci U S A. 2009;106(5):1620–5. doi: 10.1073/pnas.0807708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Oliveira FA, Disterhoft JF. Learning and aging related changes in intrinsic neuronal excitability. Front Aging Neurosci. 2010;2:2. doi: 10.3389/neuro.24.002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Power JM, Thompson LT, Disterhoft JF. Apamin increases excitability of CA1 hippocampal pyramidal neurons. Neuroscience Research Communications. 2000;27(2):135–142. [Google Scholar]
- Oliva AA, Jr, Jiang M, Lam T, Smith KL, Swann JW. Novel hippocampal interneuronal subtypes identified using transgenic mice that express green fluorescent protein in GABAergic interneurons. J Neurosci. 2000;20(9):3354–68. doi: 10.1523/JNEUROSCI.20-09-03354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Davis M. A metaplasticity-like mechanism supports the selection of fear memories: role of protein kinase a in the amygdala. J Neurosci. 2012;32(23):7843–51. doi: 10.1523/JNEUROSCI.0939-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Krause M, Haug T, Storm JF, Stuhmer W. Modulation of the Ca2+-activated K+ current sIAHP by a phosphatase-kinase balance under basal conditions in rat CA1 pyramidal neurons. J Neurophysiol. 1998;79(6):3252–3256. doi: 10.1152/jn.1998.79.6.3252. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11(6):1023–35. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Power JM, Thompson LT, Moyer JR, Jr, Disterhoft JF. Enhanced synaptic transmission in CA1 hippocampus after eyeblink conditioning. J Neurophysiol. 1997;78(2):1184–7. doi: 10.1152/jn.1997.78.2.1184. [DOI] [PubMed] [Google Scholar]
- Ruediger S, Vittori C, Bednarek E, Genoud C, Strata P, Sacchetti B, et al. Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature. 2011;473(7348):514–8. doi: 10.1038/nature09946. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci. 1998;10(4):1518–23. doi: 10.1046/j.1460-9568.1998.00149.x. [DOI] [PubMed] [Google Scholar]
- Saar D, Reuveni I, Barkai E. Mechanisms underlying rule learning-induced enhancement of excitatory and inhibitory synaptic transmission. J Neurophysiol. 2012;107(4):1222–9. doi: 10.1152/jn.00356.2011. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16(15):4537–42. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28(15):4028–36. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20(1):11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal M, Ehlers VL, Moyer JR., Jr Learning enhances intrinsic excitability in a subset of lateral amygdala neurons. Learn Mem. 2014;21(3):161–70. doi: 10.1101/lm.032730.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon PR, Vander Schaaf ER, Thompson RF, Weisz DJ. Hippocampus and trace conditioning of the rabbit’s classically conditioned nictitating membrane response. Behav Neurosci. 1986;100(5):729–744. doi: 10.1037//0735-7044.100.5.729. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Katona L, Klausberger T, Lasztoczi B, Viney TJ. Temporal redistribution of inhibition over neuronal subcellular domains underlies state-dependent rhythmic change of excitability in the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2014;369(1635):20120518. doi: 10.1098/rstb.2012.0518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Detert JA, Sehgal M, Moyer JR., Jr Trace fear conditioning enhances synaptic and intrinsic plasticity in rat hippocampus. J Neurophysiol. 2012;107(12):3397–408. doi: 10.1152/jn.00692.2011. [DOI] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 1999;96(8):4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Takehara K, Kawahara S, Kirino Y. Time-dependent reorganization of the brain components underlying memory retention in trace eyeblink conditioning. J Neurosci. 2003;23(30):9897–905. doi: 10.1523/JNEUROSCI.23-30-09897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR, Disterhoft JF. Trace eyeblink conditioning in rabbits demonstrates heterogeneity of learning ability both between and within age groups. Neurobiol Aging. 1996a;17(4):619–629. doi: 10.1016/0197-4580(96)00026-7. [DOI] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR, Disterhoft JF. Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J Neurophysiol. 1996b;76(3):1836–1849. doi: 10.1152/jn.1996.76.3.1836. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM. The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25(10):2609–2616. doi: 10.1523/JNEUROSCI.5023-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng W, Guan R, Disterhoft JF, Weiss C. Trace eyeblink conditioning is hippocampally dependent in mice. Hippocampus. 2004;14(1):58–65. doi: 10.1002/hipo.10157. [DOI] [PubMed] [Google Scholar]
- Urban-Ciecko J, Kossut M, Mozrzymas JW. Sensory learning differentially affects GABAergic tonic currents in excitatory neurons and fast spiking interneurons in layer 4 of mouse barrel cortex. J Neurophysiol. 2010;104(2):746–54. doi: 10.1152/jn.00988.2009. [DOI] [PubMed] [Google Scholar]
- Weible AP, O’Reilly J, Weiss C, Disterhoft JF. Comparisons of dorsal and ventral hippocampal CA1 pyramidal neuron activity during trace eye-blink conditioning in the rabbit. Neuroscience. 2006;141(3):1123–1137. doi: 10.1016/j.neuroscience.2006.04.065. [DOI] [PubMed] [Google Scholar]
- Weiss C, Bouwmeester H, Power JM, Disterhoft JF. Hippocampal lesions prevent trace eyeblink conditioning in the freely moving rat. Behav Brain Res. 1999;99(2):123–132. doi: 10.1016/s0166-4328(98)00096-5. [DOI] [PubMed] [Google Scholar]
- Weiss C, Kronforst-Collins MA, Disterhoft JF. Activity of hippocampal pyramidal neurons during trace eyeblink conditioning. Hippocampus. 1996;6(2):192–209. doi: 10.1002/(SICI)1098-1063(1996)6:2<192::AID-HIPO9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Disterhoft JF. Slow afterhyperpolarization governs the development of NMDA receptor-dependent afterdepolarization in CA1 pyramidal neurons during synaptic stimulation. J Neurophysiol. 2004;92(4):2346–56. doi: 10.1152/jn.00977.2003. [DOI] [PubMed] [Google Scholar]
- Zhang L, McBain CJ. Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol. 1995;488(Pt 3):661–72. doi: 10.1113/jphysiol.1995.sp020998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nature Neuroscience. 2009;12(11):1438–43. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG. Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci. 1986;6(10):2950–67. doi: 10.1523/JNEUROSCI.06-10-02950.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]