

Abstract
Rationale
Hrd1 is an endoplasmic reticulum (ER)-transmembrane E3 ubiquitin ligase that has been studied in yeast, where it contributes to ER protein quality control by ER-associated degradation (ERAD) of misfolded proteins that accumulate during ER stress. Neither Hrd1 nor ERAD have been studied in the heart, or in cardiac myocytes, where protein quality control is critical for proper heart function.
Objective
The objectives of this study were to elucidate roles for Hrd1 in ER stress, ERAD, and viability in cultured cardiac myocytes and in the mouse heart, in vivo.
Methods and Results
The effects of siRNA-mediated Hrd1 knockdown were examined in cultured neonatal rat ventricular myocytes. The effects of adeno-associated virus (AAV)-mediated Hrd1 knockdown and overexpression were examined in the hearts of mice subjected to pressure-overload induced pathological cardiac hypertrophy, which challenges protein-folding capacity. In cardiac myocytes, the ER stressors, thapsigargin (TG) and tunicamycin (TM) increased ERAD, as well as adaptive ER stress proteins, and minimally affected cell death. However, when Hrd1 was knocked down, TG and TM dramatically decreased ERAD, while increasing maladaptive ER stress proteins and cell death. In vivo, Hrd1 knockdown exacerbated cardiac dysfunction, and increased apoptosis and cardiac hypertrophy, while Hrd1 overexpression preserved cardiac function, and decreased apoptosis and attenuated cardiac hypertrophy in the hearts of mice subjected to pressure-overload.
Conclusions
Hrd1 and ERAD are essential components of the adaptive ER stress response in cardiac myocytes. Hrd1 contributes to preserving heart structure and function in a mouse model of pathological cardiac hypertrophy.
Keywords: Endoplasmic reticulum stress (ER stress), protein folding, HMG-CoA reductase degradation protein 1 (Hrd1), endoplasmic reticulum-associated protein degradation (ERAD), cardiac myocyte, protein degradation
INTRODUCTION
Cellular function depends on protein homeostasis, also known as proteostasis 1. Proteostasis requires the efficient folding of newly synthesized proteins, as well as protein quality control and degradation, which decrease the accumulation of misfolded, potentially toxic proteins 1. At least 1/3 of all proteins, including calcium handling proteins, transmembrane receptors, growth factors, and hormones are synthesized, modified, and folded in the endoplasmic reticulum (ER), then trafficked to various membrane compartments, or secreted 2. Thus, the environment in the ER must be optimal for efficient synthesis and folding of these important proteins 3–5.
A variety of diseases, including many that affect the heart, challenge ER protein folding capacity 6–9. Such challenges can be due to mutations in ER proteins, which can affect their folding or targeting, or to disease-related perturbations of the ER environment 10, which lead to imbalanced proteostasis, in extreme cases causing ER stress. ER stress contributes to pathology by impeding the production of critical ER proteins, and by increasing the accumulation of potentially toxic misfolded proteins.
ER protein misfolding activates the unfolded protein response (UPR), a conserved signaling system that initiates multiple processes to restore proteostasis, including optimization of ER chaperone-assisted protein folding and increased misfolded protein degradation by ER-associated protein degradation (ERAD) 11. ERAD is a four-step quality control process for removing terminally misfolded proteins from the ER by the cytosolic ubiquitin-proteasome system 10, 12, 13. ER-transmembrane and luminal proteins that misfold during ER stress and fail quality control (Figure 1A, Step 1) are transported out of the ER into the cytosol (Step 2), where they are ubiquitylated on the cytosolic side of the ER by ER-transmembrane E3 ubiquitin (Ub) ligases (Step 3), which targets them for degradation by cytosolic proteasomes (Step 4). Accordingly, ERAD is an adaptive process 13. If ERAD, and other aspects of the UPR fail to resolve ER stress, maladaptive features of the UPR, sometimes called maladaptive ER stress, guide cells toward apoptosis, which contributes to the tissue damage and organ dysfunction that are characteristic of pathologies associated with imbalanced proteostasis 14, 15.
Figure 1. Characterization of Hrd1 Expression in Cardiac Myocytes.

A, Diagram of ER-associated protein degradation (ERAD). B, D and E, Cultured cardiac myocytes were treated with AdV-Con, AdV-ATF6, or AdV-XBP1 for 48h. Hrd1 and Gapdh mRNA B, or protein D, were measured by qRT-PCR or immunoblotting, respectively. E, Densitometry of the immunoblot shown in D. C, F and G, Cultured cardiac myocytes were treated with tunicamycin (TM) 10 μg/ml, thapsigargin (TG) 1 μM, or dithiothrietol (DTT) 1mM for 20h. Hrd1 and Gapdh mRNA C, or protein F, were measured by qRT-PCR or immunoblotting, respectively. G, Densitometry of the immunoblot shown in F. * = p ≤ 0.05 different from control.
UPR genes are regulated by several transcription factors including ATF6, an ER-transmembrane protein 16, 17. Although not well studied in cardiac myocytes, or in the heart, in other cultured cell models ER protein misfolding triggers the translocation of ATF6 to the nucleus, where it induces certain ER stress response genes17. Although the causes and consequences of ER stress in the heart remain to be elucidated, previous studies have suggested that ATF6 regulates mainly adaptive ER stress responses 18, 19. Thus, identifying ATF6-regulated genes is required to understand mechanisms that maintain proteostasis and defend against the maladaptive ER stress response and potential cardiac dysfunction.
Our previous transcriptome analysis showed that in the mouse heart ATF6 induces genes that encode numerous ER-resident proteins predicted to contribute to enhancing ER protein folding through the adaptive ER stress response, including components of the ERAD machinery 20. One of the genes induced by ATF6 in the heart is the ER-transmembrane E3 ubiquitin ligase, HMG-CoA reductase degradation protein 1, or Hrd1 (Figure 1A). Hrd1 was discovered in yeast and named for its ability to ubiquitylate the ER-transmembrane protein, HMG-CoA reductase 21. Since then, Hrd1 has been shown in yeast 22, 23, as well as in several mammalian cell lines 24, 25, to play a key role in ERAD-mediated degradation of a wide spectrum of misfolded proteins. Moreover, Hrd1 has been implicated as being beneficial in several neurodegenerative diseases 26, and maladaptive in other diseases, such as liver cirrhosis 27 and rheumatoid arthritis 28. In addition to Hrd1, other ER-transmembrane E3 ubiquitin ligases, such as gp78, TEB4, and TRC8, also contribute to ERAD, although the range of substrates for these enzymes is more limited than Hrd1 10. Remarkably, amongst nearly 1,000 E3 ubiquitin ligases in the genome 29, Hrd1 was the only ER-transmembrane E3 ubiquitin ligase that was induced by ATF6 in the heart 20. Since ER-transmembrane E3 ubiquitin ligases have not been examined in the cardiac context, we undertook the current study to investigate roles for Hrd1 in cultured cardiac myocytes and in the heart.
METHODS
Further details on the Methods can be found in the Online Supplement.
Laboratory animals
The research reported in this paper has been reviewed and approved by the SDSU Institutional Animal Care and Use Committee and it conforms to the Guide for the Care and Use of Laboratory Animals published by the National Research Council.
Hrd1 Ubiquitylation Assay
The ubiquitin ligase activity of the Hrd1 used in this study was demonstrated as described in the Online Supplement.
ERAD Assay
ER-associated protein degradation (ERAD) was measured using a C-terminally HA-tagged version of the model substrate, TCR-α, essentially as described 30, but using AdV-TCR-α-HA.
Statistics
Unless otherwise stated, values shown are mean ± SEM and statistical treatments are one-way ANOVA followed by Newman-Keuls post hoc analysis.
RESULTS
Hrd1 is induced by ATF6, XBP1, and ER stress in cardiac myocytes
To examine Hrd1 gene expression in response to ER stress in cardiac myocytes, we determined the effects of ATF6 and another ER stress-inducible transcription factor, X-box binding protein 1 (XBP1), on Hrd1 expression in cultured neonatal rat ventricular myocytes. Hrd1 mRNA increased when cardiac myocytes were infected with adenovirus (AdV) encoding activated ATF6, or activated XBP1 (Figure 1B). Hrd1 mRNA also increased when cardiac myocytes were treated with chemical inducers of ER stress, tunicamycin (TM), thapsigargin (TG), or dithiothrietol (DTT) (Figure 1C), which cause ER protein misfolding by inhibiting protein glycosylation 31, decreasing ER calcium 32, or altering ER redox status 33, respectively.
To detect Hrd1 protein, we generated a rabbit antiserum to the C-terminal cytosolic domain of human Hrd1, which is conserved in mouse and rat Hrd1. Using this antiserum, we showed that Hrd1 protein increased when cultured cardiac myocytes were infected with adenovirus (AdV) encoding activated ATF6, or activated XBP1 (Figure 1D and 1E), or when they were treated with TM, TG, or DTT (Figure 1F and 1G). Thus, Hrd1 was upregulated in cultured cardiac myocytes by ER stress and by key transcription factors of the UPR gene program.
Hrd1 knockdown augments ER stress gene expression and decreases cardiac myocyte viability
To examine the function of endogenous Hrd1, we used an siRNA targeted to Hrd1 (siHrd1), which decreased Hrd1 in cultured cardiac myocytes by as much as 75% (Figure 2A and 2B). Hrd1 knockdown increased the ER stress markers, Grp94 and Grp78 in untreated cells, as well as in cells treated with TM or TG for 48h (Figure 2A, 2C and 2D) or 72h (Online Figure IA, IC and ID). These results indicated that a reduction in Hrd1 increased misfolded ER proteins and subsequent ER stress. However, most dramatic was the increase in the ER stress-inducible protein, CHOP, in cells treated with TM or TG (Figure 2A; Online Fig. IA and IE). CHOP is often associated with maladaptive ER stress and cell death. Accordingly, the effects of Hrd1 knockdown on cardiac myocyte viability were examined. We found that Hrd1 knockdown decreased cardiac myocyte viability in cells treated with TM or TG (Figure 2F and 2G; Online Fig. IF and IG). Moreover, ER stress-mediated activation of caspase-12, a marker of maladaptive ER stress and inducer of apoptosis, was also increased by Hrd1 knockdown in cells treated with TM or TG (Figure 2H and 2I). Additionally, Hrd1 knockdown decreased cell number significantly in neonatal rat ventricular myocytes subjected to simulated ischemia/reperfusion (Figure 2J). Hrd1 knockdown was also shown to decrease protein ubiquitylation (Online Fig. IH). These results indicate that endogenous Hrd1 protects cardiac myocytes against cell death due to the maladaptive ER stress response.
Figure 2. Effects of Hrd1 Knockdown on ER Stress and Myocyte Viability.

Cultured cardiac myocytes were treated with siCon or siHrd1 for 48h, then vehicle, TM (10 μg/ml) or TG (1 μM) for 48h. A, Hrd1, Grp94, Grp78, Gapdh, and CHOP were measured by immunoblotting. B–E, Densitometry of the blots shown in A normalized to vehicle-treated siCon, except for CHOP, which was normalized to TM-treated siCon. * = p ≤ 0.05 different from Con. # = p ≤ 0.05 different from Con/siCon. F and G, Cultured cardiac myocytes were treated with siCon or siHrd1 for 48h, then with various doses of TM or TG for 48h, after which cell viability was determined by MTT assay. * = p ≤ 0.05 different from siCon at the same dose and time of TM or TG treatment. Note: at 10 μM TG there was no MTT value in either siCon or siHrd1. H, Cultured cardiac myocytes were treated with siCon or siHrd1 and then with vehicle, TM, or TG for 48h. Extracts were then immunoblotted for caspase-12. I, Densitometry of the 40 kD active version of caspase-12, normalized to vehicle-treated siCon cells. * = p ≤ 0.05 different from treatment-matched siCon. J, Cultured cardiac myocytes were treated with siCon or siHrd1, then subjected to simulated ischemia/reperfusion (I/R), after which cell numbers were determined by microscopy. * = p ≤ 0.05 different from Con/siHrd1, as determined by t-test.
The effect of Hrd1 knockdown on ERAD
Since Hrd1 is best known for its roles in ERAD, but functional roles for ERAD have not been investigated in cardiac myocytes, we examined the effects of Hrd1 knockdown on ERAD in cardiac myocytes using an HA epitope-tagged version of the T-cell antigen receptor α-chain (TCR-α-HA) as a model misfolded ER protein 30. When expressed in cells that do not normally express the other TCR subunits, TCR-α-HA, an ER-transmembrane protein, misfolds and is degraded by ERAD 30,34. Accordingly, cultured cardiac myocytes that had been treated with siCon or siHrd1, were infected with an adenovirus that expresses TCR-α-HA. The rate of TCRα degradation, which is a measure of ERAD, was then assessed by examining TCR-α-HA levels by anti-HA immunoblotting after various times of blocking protein synthesis with cycloheximide (CHX). In siCon-treated cells, TM and TG increased the rate of ERAD, as evidenced by a decrease in TCR-α-HA levels after each time of CHX treatment (Figure 3A siCon; Figure 3B and 3D). This finding suggests that ER stress-mediated upregulation of proteins that comprise the ERAD machinery augments the degradation of misfolded ER proteins. In contrast, in siHrd1-treated cells, TM and TG decreased ERAD (Fig. 3A siHrd1; Figure 3C and 3E). Moreover, the rate of ERAD was decreased by Hrd1 knockdown even in the absence of ER stress. These results demonstrate that endogenous Hrd1 plays a key role in the adaptation of cardiac myocytes to ER protein misfolding.
Figure 3. Effects of Hrd1 Knockdown, Tunicamycin, and Thapsigargin on ERAD.

A, Cultured cardiac myocytes were treated with AdV-TCR-α-HA and siCon or siHrd1 for 48h, then with vehicle, TM (10 μg/ml) or TG (1μM) for 24h. Cultures were then treated with cycloheximide (CHX) for the times shown (hours), then immunoblotted for TCR-α-HA and Gapdh. The mass of TCR-α-HA decreased upon TM treatment because TM inhibits its glycosylation. B and C, Densitometry of the TCR-α-HA blots shown in A. * = p ≤ 0.05 different from Con at the same CHX treatment time. D and E, ERAD is displayed here as the relative rates of TCR-α-HA degradation at the 1h CHX treatment time. * = p ≤ 0.05 different from Con.
Hrd1 knockdown is maladaptive in a mouse model of pathological cardiac hypertrophy
To determine roles for endogenous Hrd1 during cardiac pathology, mice were injected with 1011 genome-containing units of AAV9-sh-Con or AAV9-sh-Hrd1 and then subjected to sham or trans-aortic constriction (TAC) surgery to produce pressure overload-induced pathological cardiac hypertrophy (Figure 4A). Because of its effects on increasing cardiac myocyte protein synthesis, pathological cardiac hypertrophy can potentially challenge cellular protein folding and quality control machinery. Immunoblots of the mouse hearts showed that AAV9-sh-Hrd1 decreased Hrd1 levels of sham and TAC mouse hearts by about 50% (Figure 4B). Echocardiography showed that, compared to control, Hrd1 knockdown did not significantly affect fractional shortening (Figure 4C, blue vs green) or ejection fraction (Online Table I). However, in mice subjected to TAC surgery, Hrd1 knockdown exacerbated the functional impairment, as evidenced by further significant decreases in fractional shortening (Figure 4C, red vs brown) and ejection fraction (Online Table I).
Figure 4. Hrd1 Knockdown Impairs Cardiac Function.

A, Experimental protocol for AAV9 administration, TAC, and termination of the experiment. n = number of mice used for each treatment. B, Immunoblots of AAV9-sh-Con or AAV9-sh-Hrd1 mouse heart extracts. C–E, Echocardiography of AAV9-sh-Con and AAV9-sh-Hrd1 treated mice subjected to sham or TAC surgery. Echocardiography was done just prior to animal sacrifice. C, Fractional shortening (%). D and E, LVEDV and LVESV = left ventricular end diastolic or systolic volumes, respectively. F, Heart weights normalized to tibia lengths (HW/TL). G, mRNA levels in mouse heart extracts were determined by qRT-PCR. *, # = p ≤ 0.05 different from all other values. Additional echocardiography data and statistical analyses can be found in Online Table I.
In terms of LV structure, in mice subjected to TAC, Hrd1 knockdown increased LV end diastolic and systolic volumes (LVEDV and LVESV) (Figure 4D and 4E), as well as LV diastolic and systolic inner diameters (Online Table I), all of which are indicators of pathological LV dilation in response to pressure overload. These results indicated that Hrd1 knockdown accelerated disease progression in response to pressure overload, as evidenced by worsened systolic dysfunction, as well as increased systolic and diastolic LV dilation.
In terms of cardiac hypertrophy, compared to control, Hrd1 knockdown increased heart weights in mice subjected to sham surgery (Figure 4F, blue vs green), suggesting that endogenous Hrd1 is a regulator of cardiac growth and knocking down Hrd1 may prime the heart for a hypertrophic growth response. This trend was also seen when LV mass was calculated from the echocardiography data, although it did not reach statistical significance (Online Table I). In control mice, heart weights were increased upon TAC (Figure 4F, red), consistent with the expected pressure overload-induced cardiac hypertrophy. Hrd1 knockdown resulted in a further increase in heart weight upon TAC (Figure 4F, brown) but, as determined gravimetrically, this increase did not reach significance. However, when calculated from the echocardiography data, the increase in LV mass in mice subjected to TAC was significantly increased by Hrd1 knockdown, compared to control (Online Table I).
With regards to genetic markers of hypertrophy, compared to AAV9-sh-Con, AAV9-sh-Hrd1 increased the levels of atrial natriuretic factor (ANF), B-type natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) in the hearts of mice subjected to TAC (Figure 4G, Group 3 vs 4), consistent with exacerbated hypertrophy upon Hrd1 knockdown. The level of collagen 1A1 (Col1A1) was also increased by Hrd1 knockdown (Figure 4G), suggesting an increase in fibrosis, which was supported by histological examination (Figure 5A through 5D). Finally, examination of apoptosis in mouse heart sections using a TUNEL stain also showed that Hrd1 knockdown increased the number of TUNEL positive nuclei (Figure 5E). Thus, by most measures, Hrd1 knockdown exacerbated cardiac pathology in the hearts of mice subjected to pressure overload.
Figure 5. Histology and TUNEL Analyses of Mouse Hearts Treated with AAV9-sh-Con and AAV9-sh-Hrd1.
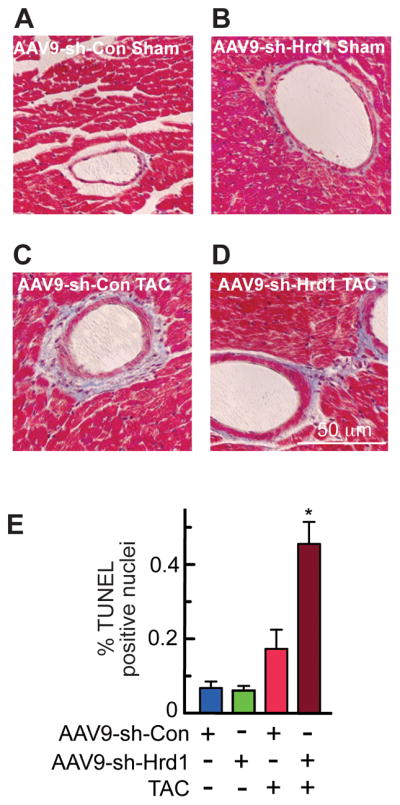
A–D, Sections of hearts from mice treated with either AAV9-sh-Con or AAV9-sh-Hrd1, and then subjected to sham or TAC surgery, as described in the Experimental Protocol shown in Fig. 4A, were stained with Masson’s trichrome to examine fibrosis (blue). E, Sections of hearts from mice treated with either AAV9-sh-Con or AAV9-sh-Hrd1, and then subjected to TAC were analyzed for apoptosis by TUNEL staining, then quantified to determine % of nuclei that were TUNEL-positive. * = p ≤ 0.05 different from all other values.
Hrd1 overexpression is adaptive in a mouse model of pathological cardiac hypertrophy
Since Hrd1 loss-of-function was maladaptive, we examined whether Hrd1 gain-of-function would be adaptive in the mouse heart. Accordingly, we generated a recombinant AAV9 encoding an untagged version of mouse Hrd1. Expression of Hrd1 was directed to ventricular myocytes using a previously described form of the myosin regulatory light chain 2 promoter 35, 36, which we previously showed to support increased protein expression in >80% of mouse cardiac myocytes, in vivo 37, 38. In the present study, 1011 genome-containing units of AAV9-Con or AAV9-Hrd1 were administered to mice by tail vein injection. Immunocytofluorescence (ICF) showed that, compared to AAV9-Con, AAV9-Hrd1 increased Hrd1 expression in most cardiac myocytes in mouse hearts, and that some myocytes expressed more Hrd1 than others (Online Figure IIA and IIB). Additional ICF showed that Hrd1 co-localized with cardiac troponin T and, to some extent, with SERCA2a (Online Figure IIC and IID), consistent with Hrd1 localization to the longitudinal SR. Since Hrd1 appeared to localize to the SR, the effects of Hrd1 overexpression on Ca2+ transients in adult mouse cardiac myocytes were examined. Overexpression of Hrd1 did not change the Ca2+ transient amplitude, the rate of Ca2+ removal from the cytosol by SERCA2, SR Ca2+ concentration, or sarcolemmal sodium-calcium exchanger (NCX) activity (Online Figure IIIA through IIID). Moreover, Hrd1 overexpression did not affect Ca2+ spark properties (Online Figure IIIE through IIIH). Therefore, Hrd1 overexpression had no adverse effects on contractile Ca2+ handling in the heart.
To examine the effects of Hrd1 overexpression on cardiac pathology, mice that had been injected with either AAV9-Con or AAV9-Hrd1 were subjected to sham or TAC surgery (Figure 6A). Immunoblots showed that AAV9-Hrd1 increased Hrd1 expression in both sham and TAC treated mouse hearts (Figure 6B). TAC resulted in a slight increase in Hrd1 protein (Figure 6B), as well as Hrd1 mRNA (Online Figure IVA). Echocardiography showed that Hrd1 overexpression did not affect fractional shortening (Figure 6C, blue vs green) or ejection fraction (Online Table II) in mice subjected to sham surgery. However, Hrd1 overexpression improved fractional shortening and ejection fraction in mice subjected to TAC (Figure 6C, red vs brown; Online Table II). In terms of LV structure, Hrd1 overexpression also diminished TAC-mediated increases LVEDV and LVESV (Figure 6D and 6E, red vs brown), as well as LV diameter (Online Table II). In terms of cardiac hypertrophy, AAV9-Con-treated mice subjected to TAC exhibited increased heart weights, which were diminished in AAV9-Hrd1-treated mice (Figure 6F, red vs brown). Hrd1 overexpression decreased the levels of ANF, BNP, and β-MHC in mice subjected to TAC (Figure 6G, Group 3 vs 4). Moreover, the level of collagen 1A1 (Col1A1) in mice subjected to TAC was also decreased by Hrd1 overexpression (Figure 6G), suggesting a decrease in fibrosis, which was supported by histological examination (Figure 7A through 7D). Finally, examination of apoptosis in mouse heart sections using a TUNEL stain also showed that Hrd1 knockdown increased the number of TUNEL positive nuclei (Figure 7E). Thus, by most measures, Hrd1 overexpression decreased cardiac pathology in the hearts of mice subjected to pressure overload.
Figure 6. Hrd1 Overexpression Preserves Cardiac Function.

A, Experimental protocol for AAV9 administration, TAC, and termination of the experiment. n = number of mice used for each treatment. B, Immunoblots of AAV9-Con or AAV9-Hrd1 mouse heart extracts. C–E, Echocardiography of AAV9-Con and AAV9-Hrd1 treated mice subjected to sham or TAC surgery. Echocardiography was done just prior to animal sacrifice. C, Fractional shortening (%). D and E, LVEDV and LVESV = left ventricular end diastolic or systolic volumes, respectively. F, Heart weights normalized to tibia lengths (HW/TL). G, mRNA levels in mouse heart extracts were determined by qRT-PCR. * = p ≤ 0.05 different from all other values. Additional echocardiography data and statistical analyses can be found in Supplemental Table II.
Figure 7. Histology and TUNEL Analyses of Mouse Hearts Treated with AAV9-Con and AAV9-Hrd1.
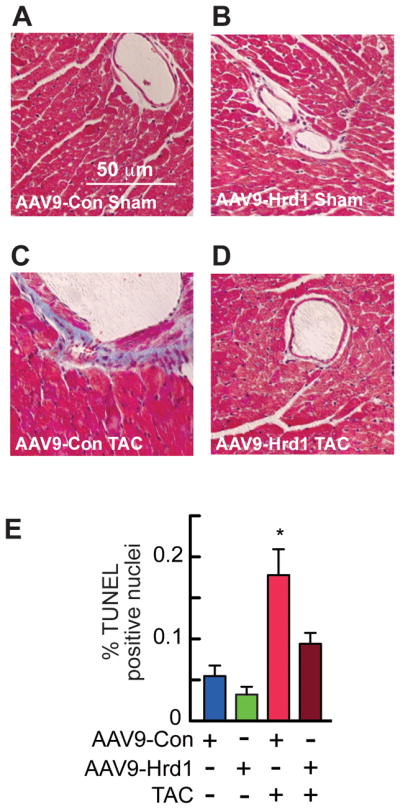
A–D, Sections of hearts from mice treated with either AAV9-Con or AAV9-Hrd1, and then subjected to sham or TAC surgery, as described in the Experimental Protocol shown in Fig. 6A, were stained with Masson’s trichrome to examine fibrosis (blue). E, Sections of hearts from mice treated with either AAV9- Con or AAV9-Hrd1, and then subjected to TAC were analyzed for apoptosis by TUNEL staining, then quantified to determine % of nuclei that were TUNEL-positive. * = p ≤ 0.05 different from all other values.
To examine the effects of Hrd1 overexpression on cardiac myocyte growth directly, we generated an adenovirus encoding Hrd1 (AdV-Hrd1), which resulted in overexpression of Hrd1 in cultured cardiac myocytes by about 3-fold over control (Online Figure VA). Immunocytofluorescence showed that, compared to AdV-Con, AdV-Hrd1 increased the levels of Hrd1, which co-localized with ER proteins (Online Figure VI). Overexpression of Hrd1 decreased myocyte surface area and protein synthesis, as well as expression of ANF and BNP, two markers of hypertrophic cardiac myocyte growth (Online Figure VB through VE). These results are consistent with the ability of Hrd1 to diminish hypertrophy, in vivo, and indicate that Hrd1 exhibits cardiac myocyte growth moderating effects.
DISCUSSION
ER stress and heart disease
Many diseases, including heart disease, are associated with protein misfolding, which contributes to organ dysfunction 2, 39–42. Such protein misfolding can take place in the ER, as well as elsewhere in cardiac myocytes. Protein quality control outside the ER in cardiac myocytes has been addressed in a number of relatively recent studies 42; however, less is known about the effects of protein misfolding in the ER of cardiac myocytes. While protein misfolding can have dire consequences, regardless of where it takes place, disease-related ER protein misfolding is particularly problematic in cardiac myocytes, since it could affect the levels of key secreted and membrane proteins, such as calcium-handling proteins and adrenergic receptors, which can impair cardiac myocyte function. Moreover, some forms of cardiac disease, such as those associated with hypertrophy, can increase protein synthesis, which potentially challenges an already disease-impaired ER protein-folding environment 19, 39, 43. Although not studied extensively in cardiac myocytes, in other model cell types ER protein misfolding activates the UPR, which is designed to restore ER protein folding and degrade misfolded proteins, two processes that are essential for the adaptive ER stress response. Therefore, throughout essentially all of a cellular lifetime, ER stress is met with adaptive responses 11. However, when such adaptive responses are not sufficient to restore ER proteostasis, continued ER stress can impair cardiac myocyte function and, ultimately, it can lead to cell death and organ damage 15.
The adaptive ER stress response and cardiac development
In contrast to the neonatal heart, we found that the adult heart expresses low levels of Hrd1, as well as Grp78 and ATF6 (Online Fig. IVb). Previous studies of a variety of tissues, including liver, kidney, brain, and heart, have shown that other proteins that are part of the ER protein synthesis and quality control machinery are also expressed at higher levels early in development compared to the adult 44–46. Moreover, targeted deletion of the genes encoding several of these proteins, such as Grp78, Grp94, calreticulin, and Hrd1, is embryonic-lethal in mice, and in many of these cases, this lethality is associated with impaired cardiac development 47–50, underscoring the essential nature of these genes in the embryonic heart. The relatively high levels of ER proteins in early development may be required to support cellular differentiation and the high rate of production of secreted and membrane proteins. Moreover, in the developing heart, cardiac myocytes proliferate and grow in size, placing demands on the ER protein synthesis and folding machinery. Additionally, the sarcoplasmic reticulum (SR) must grow dramatically to meet the needs of the developing excitation-contraction coupling machinery in growing cardiac myocytes. This SR expansion requires active lipid synthesis in the ER, as well as synthesis, proper folding, and trafficking of the proteins that participate in contractile calcium handling. Therefore, while it is not surprising that levels of Hrd1, as well as other ER protein quality control proteins are higher in the developing heart than in the adult heart, it has been previously unappreciated that in the adult heart, the adaptive ER stress response may be less dynamic, and may therefore exhibit a narrower range of responsiveness to misfolded protein accumulation, than in the developing heart.
Conclusions
Previous studies have shown the importance of the transcription factors, ATF6 and XBP1, in the adaptive ER stress response in ischemic and hypertrophic heart disease in mice 19, 51–53. The present study demonstrates that a gene that can be induced by these transcription factors, Hrd1, contributes to the adaptive ER stress response in cultured cardiac myocytes and that it preserves cardiac function in a mouse model of pathological cardiac hypertrophy. Based on this study, we hypothesize that under certain conditions, sufficient levels of Hrd1 facilitate the degradation of misfolded proteins in the ER, which adaptively enhances myocyte viability. However, when Hrd1 is not sufficient, such as in the adult heart, pathology-driven maladaptive accumulation of misfolded proteins threatens myocyte viability and cardiac function. This is the first study to report on roles for any ER-transmembrane E3 ubiquitin ligase and ERAD in cardiac myocytes and in the mouse heart, in vivo. Moreover, this is the first study to show that ER stress accelerates ERAD in a Hrd1-dependend manner in cardiac myocytes.
Supplementary Material
Novelty and Significance.
What Is Known?
The ER is the location of the synthesis and folding of secreted and membrane proteins.
A decrease in protein folding and accumulation of unfolded proteins in the ER may contribute to the pathology of several disease states.
ER-associated protein degradation, ERAD, removes misfolded ER proteins, and reduces and contributes to the adaptive rejuvenation of protein folding; however, the role of ERAD in cardiac myocytes and in heart disease has not been examined.
What New Information Does This Article Contribute?
The ER-transmembrane protein, Hrd1, is expressed in the SR/ER of cardiac myocytes, where it ubiquitylates and targets misfolded proteins for removal, thus enhancing cell survival during stress.
Increasing Hrd1 expression using an AAV9-based gene therapy approach preserved cardiac function and decreased cardiac hypertrophy in a model of pressure overload-induced cardiac pathology.
Hrd1 plays a central role in ERAD and contributes to the ability of cardiac myocytes to adapt to pathological conditions that threaten myocyte viability by impairing ER protein folding.
The endoplasmic reticulum (ER) is the site of secreted and membrane protein synthesis and folding. In the diseased heart, impaired ER protein folding could contribute to pathology. The ER-transmembrane protein, Hrd1, is a ubiquitin ligase that has been shown in yeast to ubiquitylate misfolded, potentially toxic proteins, targeting them for degradation by ER-associated degradation, ERAD, which improves cell viability. However, neither Hrd1 nor ERAD have been studied in the heart. Accordingly, we examined the functions of Hrd1 and ERAD in cardiac myocytes and determined whether they play adaptive roles in the pathological heart. We found that cardiac myocytes express Hrd1 in the SR and that Hrd1 is required for ERAD. Knocking down Hrd1 decreased ERAD, increased misfolded ER protein accumulation, and decreased cardiac myocyte viability. When we knocked down Hrd1 in mouse hearts, in vivo, cardiac function was impaired in mice subjected to a surgery that causes pathological cardiac hypertrophy; in contrast, overexpression of Hrd1 preserved cardiac function. These findings suggest that overexpressing Hrd1 and improving ERAD in the heart has potential for the treatment of heart failure.
Acknowledgments
The authors thank Dr. Roger Hajjar (Mount Sinai Hospital) for help with AAV9 generation, Dr. Ron Kopito (Stanford University) for the TCR-α-HA plasmid.
SOURCES OF FUNDING
SD was supported by grants from the Rees-Stealy Research Foundation, the SDSU Heart Institute, the San Diego Chapter of the Achievement Rewards for College Scientists (ARCS) Foundation, the American Heart Association (Predoctoral Fellowship 10PRE3410005), and the Inamori Foundation.
MV was supported by Deutsche Forschungsgemeinschaft (DFG) 1659/1-1.
JLR was supported by NIH grant T32 HL007676-23.
OJM was supported by the DZHK (German Centre for Cardiovascular Research) and by the BMBF (German Ministry of Education and Research).
XHTW was supported by National Institutes of Health (NIH) grants RO1 HL089598 and R01 HL091947, and American Heart Association Grant 13EIA14560061.
MAS was supported by NIH grants R37 HL091102, R01 HL105759, R01 HL067245, R01 HL113656, R01 HL117163, R01 HL113647, and P01 HL 08577.
CCG was supported by National Institutes of Health (NIH) grants R01 HL75573, R01 HL104535, R03 EB011698, and P01 HL085577.
Nonstandard Abbreviations and Acronyms
- AAV
adeno-associated virus
- AdV
adenovirus
- ANF
atrial natriuretic factor
- ANOVA
analysis of variance
- ATF6
activating transcription factor 6 alpha
- β-MHC
β-myosin heavy chain
- BNP
b-type natriuretic peptide
- CHOP
C/EBP-homologous protein
- CHX
cycloheximide
- Col1A1
collagen 1A1
- DTT
dithiothrietol
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum associated degradation
- gp78
E3 ubiquitin-protein ligase AMFR
- Grp78
78 kilodalton glucose-regulated protein
- HA
hemaglutinin
- HMG-CoA
hydroxymethyl glutaryl-coenzyme A
- HR
heart rate
- Hrd1
HMG-CoA reductase degradation protein 1
- HW
heart weight
- ICF
0 immunocytofluorescence
- LV
left ventricle
- LVEDV
left ventricular end diastolic volume
- LVESV
left ventricular end systolic volume
- LVIDD
left ventricular inner diameter in diastole
- LVIDS
left ventricular inner diameter in systole
- MLC2
myosin regulatory light chain 2
- PWTD
left ventricular posterior wall thickness in diastole
- PWTS
left ventricular posterior wall thickness in systole
- SERCA2a
sarcoplasmic/endoplasmic reticulum calcium ATPase 2a
- SR
sarcoplasmic reticulum
- TAC
trans-aortic constriction
- TCR-α
T-cell antigen receptor α-chain
- TEB4
E3 ubiquitin-protein ligase MARCH6
- TRC8
E3 ubiquitin-protein ligase RNF139
- TG
thapsigargin
- TL
tibia length
- TM
tunicamycin
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- Ub
ubiquitin
- UPR
unfolded protein response
- XBP1
X-box-binding protein 1
Footnotes
DISCLOSURES
None.
References
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 2.Glembotski CC. Roles for the sarco-/endoplasmic reticulum in cardiac myocyte contraction, protein synthesis, and protein quality control. Physiology (Bethesda) 2012;27:343–350. doi: 10.1152/physiol.00034.2012. [DOI] [PubMed] [Google Scholar]
- 3.Gidalevitz T, Stevens F, Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta. 2013;1833:2410–2424. doi: 10.1016/j.bbamcr.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta. 2014;1843:182–196. doi: 10.1016/j.bbamcr.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 5.Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- 7.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res. 2010;107:1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 8.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: Programmed to conserve. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Millott R, Dudek E, Michalak M. The endoplasmic reticulum in cardiovascular health and disease. Can J Physiol Pharmacol. 2012;90:1209–1217. doi: 10.1139/y2012-058. [DOI] [PubMed] [Google Scholar]
- 10.Olzmann JA, Kopito RR, Christianson JC. The Mammalian Endoplasmic Reticulum-Associated Degradation System. Cold Spring Harb Perspect Biol. 2012 doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 12.Brodsky JL. Cleaning Up: ER-Associated Degradation to the Rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claessen JH, Kundrat L, Ploegh HL. Protein quality control in the ER: balancing the ubiquitin checkbook. Trends Cell Biol. 2012;22:22–32. doi: 10.1016/j.tcb.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu C, Johansen FE, Prywes R. Interaction of ATF6 and serum response factor. Mol Cell Biol. 1997;17:4957–4966. doi: 10.1128/mcb.17.9.4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glembotski CC. Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J Mol Cell Cardiol. 2014;71:11–15. doi: 10.1016/j.yjmcc.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, Lynch KA, Okada T, Aronow BJ, Osinska H, Prywes R, Lorenz JN, Mori K, Lawler J, Robbins J, Molkentin JD. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149:1257–1268. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, Gude N, Sussman MA, Glembotski CC. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–14021. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampton RY, Gardner RG, Rine J. Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell. 1996;7:2029–2044. doi: 10.1091/mbc.7.12.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bordallo J, Plemper RK, Finger A, Wolf DH. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell. 1998;9:209–222. doi: 10.1091/mbc.9.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151:69–82. doi: 10.1083/jcb.151.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadav E, Shmueli A, Barr H, Gonen H, Ciechanover A, Reiss Y. A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast Hrd1. Biochem Biophys Res Commun. 2003;303:91–97. doi: 10.1016/s0006-291x(03)00279-1. [DOI] [PubMed] [Google Scholar]
- 25.Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem. 2004;279:3525–3534. doi: 10.1074/jbc.M307453200. [DOI] [PubMed] [Google Scholar]
- 26.Omura T, Kaneko M, Okuma Y, Matsubara K, Nomura Y. Endoplasmic reticulum stress and Parkinson’s disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxid Med Cell Longev. 2013;2013:239854. doi: 10.1155/2013/239854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasegawa D, Fujii R, Yagishita N, Matsumoto N, Aratani S, Izumi T, Azakami K, Nakazawa M, Fujita H, Sato T, Araya N, Koike J, Tadokoro M, Suzuki N, Nagata K, Senoo H, Friedman SL, Nishioka K, Yamano Y, Itoh F, Nakajima T. E3 ubiquitin ligase synoviolin is involved in liver fibrogenesis. PLoS One. 2010;5:e13590. doi: 10.1371/journal.pone.0013590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamasaki S, Yagishita N, Nishioka K, Nakajima T. The roles of synoviolin in crosstalk between endoplasmic reticulum stress-induced apoptosis and p53 pathway. Cell Cycle. 2007;6:1319–1323. doi: 10.4161/cc.6.11.4277. [DOI] [PubMed] [Google Scholar]
- 29.Neutzner A, Neutzner M, Benischke AS, Ryu SW, Frank S, Youle RJ, Karbowski M. A systematic search for endoplasmic reticulum (ER) membrane-associated RING finger proteins identifies Nixin/ZNRF4 as a regulator of calnexin stability and ER homeostasis. J Biol Chem. 2011;286:8633–8643. doi: 10.1074/jbc.M110.197459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu H, Kaung G, Kobayashi S, Kopito RR. Cytosolic degradation of T-cell receptor alpha chains by the proteasome. J Biol Chem. 1997;272:20800–20804. doi: 10.1074/jbc.272.33.20800. [DOI] [PubMed] [Google Scholar]
- 31.Olden K, Pratt RM, Jaworski C, Yamada KM. Evidence for role of glycoprotein carbohydrates in membrane transport: specific inhibition by tunicamycin. Proc Natl Acad Sci U S A. 1979;76:791–795. doi: 10.1073/pnas.76.2.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jamsa E, Simonen M, Makarow M. Selective retention of secretory proteins in the yeast endoplasmic reticulum by treatment of cells with a reducing agent. Yeast. 1994;10:355–370. doi: 10.1002/yea.320100308. [DOI] [PubMed] [Google Scholar]
- 34.Ishikura S, Weissman AM, Bonifacino JS. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J Biol Chem. 2010;285:23916–23924. doi: 10.1074/jbc.M110.127936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boecker W, Bernecker OY, Wu JC, Zhu X, Sawa T, Grazette L, Rosenzweig A, del Monte F, Schmidt U, Hajjar RJ. Cardiac-specific gene expression facilitated by an enhanced myosin light chain promoter. Mol Imaging. 2004;3:69–75. doi: 10.1162/15353500200404103. [DOI] [PubMed] [Google Scholar]
- 36.Muller OJ, Leuchs B, Pleger ST, Grimm D, Franz WM, Katus HA, Kleinschmidt JA. Improved cardiac gene transfer by transcriptional and transductional targeting of adeno-associated viral vectors. Cardiovasc Res. 2006;70:70–78. doi: 10.1016/j.cardiores.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 37.Volkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S, Joyo A, Ornelas L, Samse K, Thuerauf DJ, Gude N, Glembotski CC, Sussman MA. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128:2132–2144. doi: 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, Ornelas L, Joyo E, Thuerauf DJ, Konstandin MH, Gude N, Glembotski CC, Sussman MA. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci U S A. 2013;110:12661–12666. doi: 10.1073/pnas.1301455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–1110. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 40.Christians ES, Mustafi SB, Benjamin IJ. Chaperones and cardiac misfolding protein diseases. Curr Protein Pept Sci. 2014;15:189–204. doi: 10.2174/1389203715666140331111518. [DOI] [PubMed] [Google Scholar]
- 41.Sandri M, Robbins J. Proteotoxicity: an underappreciated pathology in cardiac disease. J Mol Cell Cardiol. 2014;71:3–10. doi: 10.1016/j.yjmcc.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol. 2014;71:16–24. doi: 10.1016/j.yjmcc.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 44.Kim SK, Kim YK, Lee AS. Expression of the glucose-regulated proteins (GRP94 and GRP78) in differentiated and undifferentiated mouse embryonic cells and the use of the GRP78 promoter as an expression system in embryonic cells. Differentiation. 1990;42:153–159. doi: 10.1111/j.1432-0436.1990.tb00756.x. [DOI] [PubMed] [Google Scholar]
- 45.Barnes JA, Smoak IW. Immunolocalization and heart levels of GRP94 in the mouse during post-implantation development. Anat Embryol (Berl) 1997;196:335–341. doi: 10.1007/s004290050102. [DOI] [PubMed] [Google Scholar]
- 46.Barnes JA, Smoak IW. Glucose-regulated protein 78 (GRP78) is elevated in embryonic mouse heart and induced following hypoglycemic stress. Anat Embryol (Berl) 2000;202:67–74. doi: 10.1007/s004290000090. [DOI] [PubMed] [Google Scholar]
- 47.Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, Opas M, MacLennan DH, Michalak M. Calreticulin is essential for cardiac development. J Cell Biol. 1999;144:857–868. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amano T, Yamasaki S, Yagishita N, Tsuchimochi K, Shin H, Kawahara K, Aratani S, Fujita H, Zhang L, Ikeda R, Fujii R, Miura N, Komiya S, Nishioka K, Maruyama I, Fukamizu A, Nakajima T. Synoviolin/Hrd1, an E3 ubiquitin ligase, as a novel pathogenic factor for arthropathy. Genes Dev. 2003;17:2436–2449. doi: 10.1101/gad.1096603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, Glembotski CC. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–1193. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 52.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 53.Wang ZV, Deng Y, Gao N, Pedrozo Z, Li DL, Morales CR, Criollo A, Luo X, Tan W, Jiang N, Lehrman MA, Rothermel BA, Lee AH, Lavandero S, Mammen PP, Ferdous A, Gillette TG, Scherer PE, Hill JA. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell. 2014;156:1179–1192. doi: 10.1016/j.cell.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.