

Abstract
The expression efficiency in liver following hydrodynamic delivery of in vitro transcribed mRNA was improved 2000-fold using a codon-optimized mRNA luciferase construct with flanking 3' and 5' human β-globin untranslated regions (UTR mRNA) over an un-optimized mRNA without β-globin UTRs. Nanoparticle UTR mRNA polyplexes were formed using a novel polyacridine PEG-peptide, resulting in an additional 15-fold increase in expression efficiency in the liver. The combined increase in expression for UTR mRNA PEG-peptide polyplexes was 3500-fold over mRNA lacking UTRs and PEG-peptide. The expression efficiency of UTR mRNA polyplex was 10-fold greater than the expression from an equivalent 1 µg dose of pGL3. Maximal expression was maintained from 4 to 24 hours. Serum incubation established the unique ability of the polyacridine PEG-peptide to protect UTR mRNA polyplexes from RNase metabolism by binding to double stranded regions. UTR mRNA PEG-peptide polyplexes are efficient non-viral vectors that circumvent the need for nuclear uptake, representing an advancement toward the development of a targeted gene delivery system to transfect liver hepatocytes.
Introduction
The development of a nonviral gene delivery system that efficiently expresses proteins in the liver has been a long-sought goal for over twenty-five years1. Preclinical studies have demonstrated that protein expression in hepatocytes could lead to curative treatments for liver metabolic diseases as well as diseases in other organs2–4. Much of the effort in developing a non-viral gene delivery system for the liver has focused on packaging and targeting plasmid DNA5–7. Despite much effort, systemic delivery of DNA formulations resulted in either negligible or very low gene transfer efficiency in liver hepatocytes8. In contrast, hydrodynamic delivery of naked plasmid DNA to liver achieves expression efficiency equivalent to Adenovirus or AAV9. While hydrodynamic delivery is highly efficient because it overcomes the rate limiting step of delivery of DNA to the nucleus, it is also an invasive delivery method requiring both high volume and pressure10–13. Alternatively, the delivery of mRNA to the cytosol leading to translation, circumvents the need for delivery to the nucleus. Despite this major advantage, the rapid metabolism of mRNA by ubiquitous RNase remains a significant hurdle to achieving efficient expression of systemically delivered mRNA gene delivery systems14.
Since the earliest report demonstrating in vivo expression following i.m. dosed naked mRNA15, numerous studies have attempted to increase the stability and expression efficiency of mRNA formulations using cationic lipids16–20. Intratracheal high pressure spaying of an mRNA Megafectin™ lipoplex resulted in transfection of the lung21, whereas regeneration following myocardial infarction was achieved by intracardial injection of RNAiMAX™ mRNA22. Stemfect™ mRNA delivered nasally resulted in tumor vaccination23. Alternatively, systemically delivered Stemfect™ mRNA produced low level expression in the spleen17. A mannosylated histidinylated lipoplex dosed systemically resulted in expression in spleen macrophages which primed a tumor vaccine response24. While these studies demonstrate that mRNA lipoplexes possess improved in vivo gene transfer over naked mRNA, their efficiency in vivo is still very low due to relatively weak ionic binding of cationic lipids to mRNA.
In an attempt to further improve mRNA stability, nanoparticle delivery systems have been developed and tested in vitro25–27 and in vivo24,28,29. Systemic delivery of targeted stealth mRNA lipoplexes in vivo led to transfection efficiency similar to DNA formulations in solid tumor28. Intrathecally dosed mRNA polyplex nanomicelles produced measurable expression in the cerebrospinal fluid29. Notably, none of the mRNA cationic lipid or nanoparticle formulations reported to date were able to transfect liver.
There have been only two reports of successful liver transfection with mRNA30,31. The expression of mRNA in the liver was first achieved by McCaffrey et al. in 200230 who measured luciferase expression by bioluminescence imaging (BLI) in mice following hydrodynamic (HD)-dosing of 50 µg of naked mRNA to detect low level expression (106 photons/sec/cm2/steradian). The transient expression in the liver was only detectable at 3 hours and required the co-administration of 30 µg of decoy RNA and 400 units of RNase inhibitor. In an attempt to improve transfection efficiency, in 2006 Wilber et al.31 refined the mRNA construct by inserting 5' and 3' Xenopus laevis β-globin untranslated regions (UTRs) flanking luciferase to increase mRNA cellular half-life20. HD-dosing of 50 µg of UTR mRNA resulted in a 15-fold increase in the expression efficiency at 3 hours relative to mRNA lacking UTRs31, but failed to significantly extend the expression. Co-administration of decoy mRNA and RNase inhibitors significantly improved efficiency but failed to extend peak expression past 12 hours. While these reports demonstrate the feasibility of expressing proteins in the liver when HD-dosing mRNA, the efficiencies reported are far below that achievable with plasmid DNA due to mRNA's susceptibility to metabolism during delivery.
In the present study, mRNA was packaged into polyplexes using a novel PEGylated polyacridine peptide to overcome mRNA instability. This unique peptide has been shown to bind with high affinity to double stranded DNA through polyintercalation, affording protection from metabolism in the circulation of mice for up to 12 hours32–36. mRNA is single stranded, but adopts a complex secondary structure that includes extensive double helices37. We therefore hypothesized that a PEGylated polyacridine peptide would protect mRNA from RNase metabolism relative to PEG-peptides that bind mRNA through ionic interaction34. The results demonstrate synergistic improvements in gene transfer efficiency when dosing UTR mRNA PEGylated polyacridine peptide polyplexes allowing a 1 µg HD-dose to achieve even greater expression than plasmid DNA at 24 hours post-injection.
Results
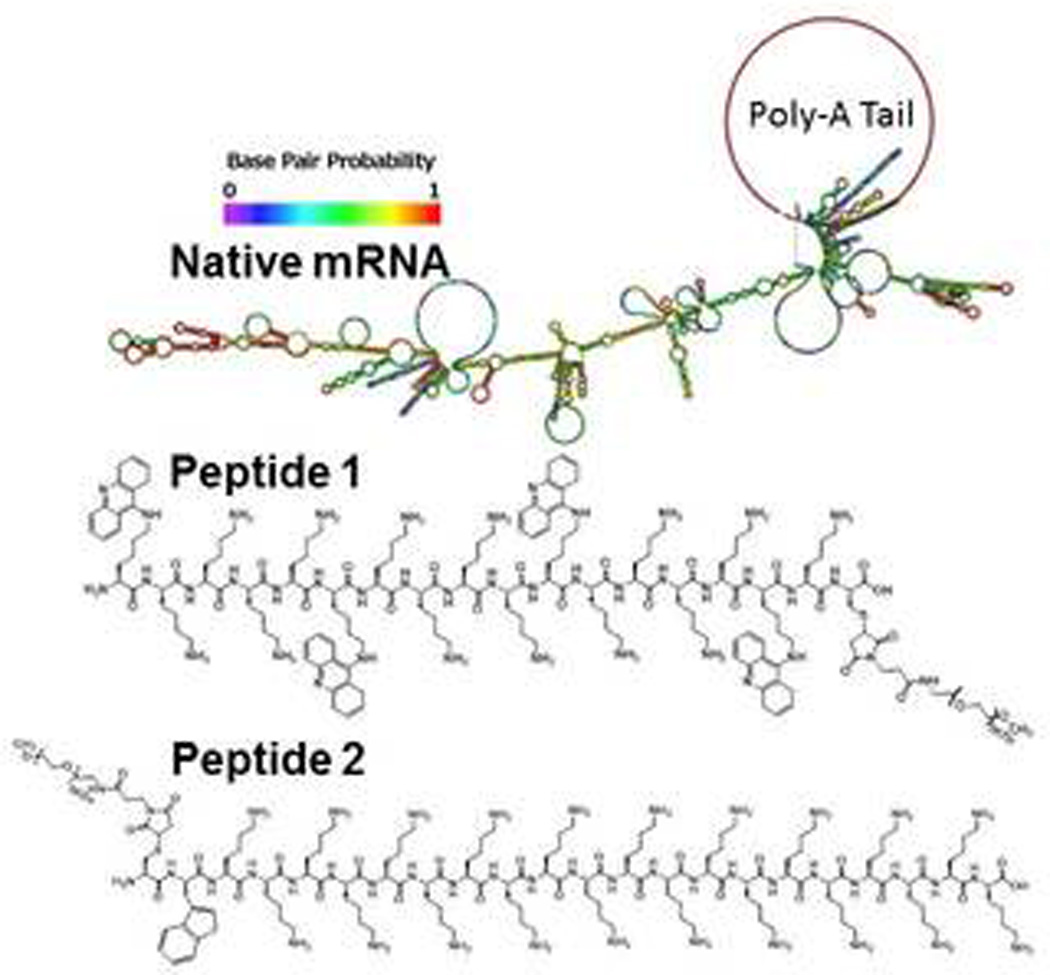
Two earlier studies established the ability to hydrodynamically dose 50 µg of mRNA resulting in transient low level (106 photons/sec/cm2/steradian) luciferase expression in the liver at 3 hours post-mRNA delivery30,31. A major goal of the present study was to significantly improve the efficiency of mRNA expression as a first step toward further development of a targeted mRNA delivery system for liver. To achieve this, a codon optimized mRNA construct containing 3' and 5' β-globin UTRs was generated to extend mRNA half-life in the cytoplasm as first described by Malone et al20. β-globin untranslated regions have been used to improve the translation of several proteins19,32,33 including erythropoietin, urokinase receptor, and β-globin, and may be generally applicable. In addition, mRNA was packaged into a polyplex by combining it with a PEG-peptide to improve its metabolic stability (Scheme 1). We have previously shown that short polylysine PEG-peptides that possess multiple acridine residues incorporated onto Lys side chains (Acr) possess significantly higher binding affinity for double stranded plasmid DNA affording PEG-peptide DNA polyplexes with much greater metabolic stability in the circulation34. Consequently, the predicted folded stem-loop structures of mRNA should also provide significant double stranded character to support high affinity binding by peptide 1 through a combination of polyintercalation and ionic binding (Scheme 1). Conversely, the ionic binding of peptide 2 to mRNA was predicted to result in mRNA polyplexes that were less metabolically stable (Scheme 1).
Scheme 1. Predicted Folded Structure of UTR mRNA.
Panel A illustrates the predicted stem-loop structure of tailed UTR mRNA. The structure of (Acr-Lys4)3-Acr-Lys-Cys-PEG5kDa (peptide 1) and PEG5kDa-Cys-Trp-Lys20 (peptide 2) used to form mRNA polyplexes are shown. Acr refers to a Lys residue modified with acridine on the ε-amine.
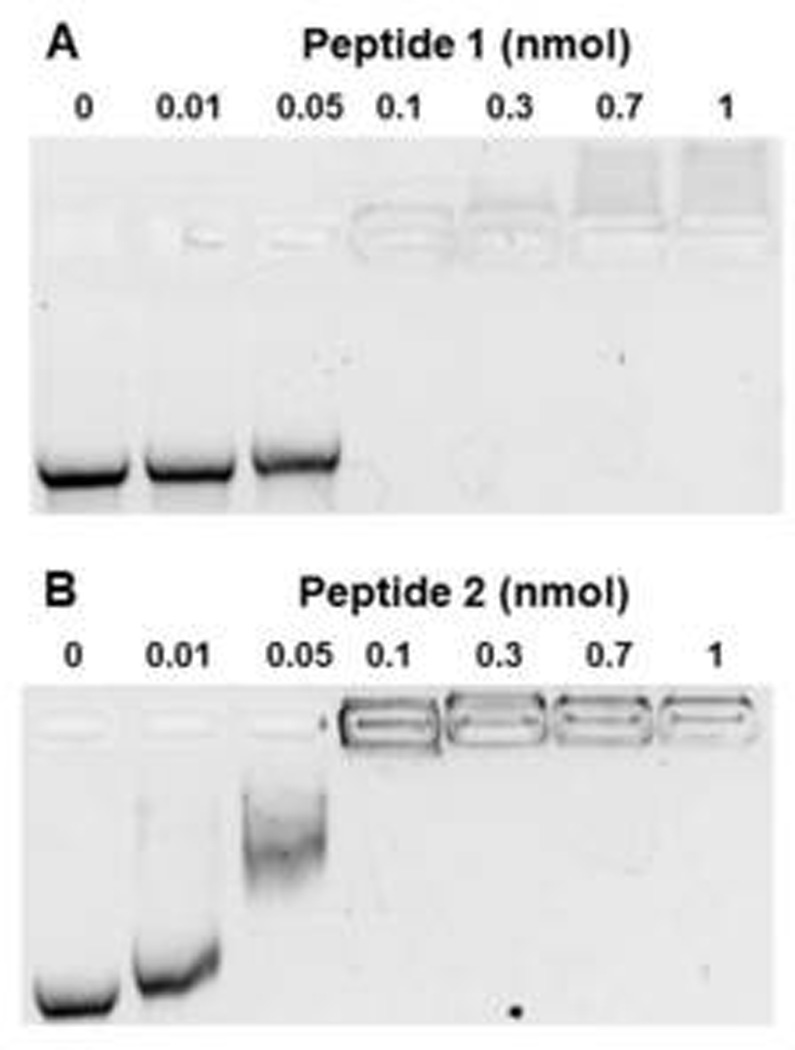
Comparison of the binding of peptide 1 and peptide 2 to mRNA demonstrated that both peptides formed mRNA polyplexes that inhibited migration on gel electrophoresis at 0.1 nmol of PEG-peptide per µg mRNA (Fig. 1). PEG-peptide polyplexes used for physical studies and in vivo experiments were formed at 0.8 nmol of peptide per µg of mRNA to fully protect the mRNA from RNase metabolism. Dynamic light scattering revealed that mRNA polyplexes possessed an average diameter of 104 nm and an average zeta potential of +15 mV. By comparison, pGL3 PEG-peptide polyplexes possessed a diameter of 170 nm and a zeta potential of +15 mV34. The smaller size of an mRNA polyplex is consistent with the shorter length of mRNA (1.6 kb) versus plasmid DNA (5.3 kbp).
Figure 1. Formation of PEG-Peptide mRNA Polyplexes.
UTR mRNA (1 µg) was combined with 0, 0.01, 0.05, 0.1, 0.3, 0.7, or 1 nmol of peptide 1 (panel A) or peptide 2 (panel B) and electrophoresed on 1% agarose gel then stained with ethidium bromide. mRNA migration was prevented at 0.1 nmol of Peptide 1 and 2, indicating both peptides bind ionically to mRNA to form polyplexes.
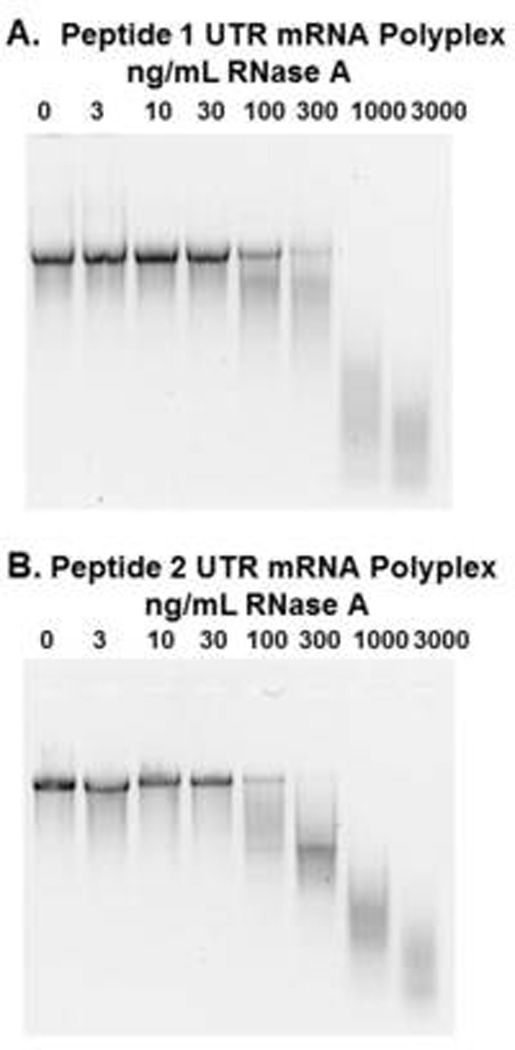
The ability of mRNA polyplexes to resist direct digestion with RNase was evaluated using gel electrophoresis (Fig. 2). Both PEG-peptides were found to stabilize mRNA to RNase digestion, as demonstrated by the recovery of intact mRNA when PEG-peptide polyplexes were digested with up to 30 ng/mL RNase A (Fig. 2). By comparison, naked mRNA was completely digested by 3 ng/ml of RNase. However, incubation of mRNA polyplexes with 100 ng/mL RNase or higher resulted in either partial or complete digestion of mRNA. mRNA polyplexes prepared with PEG-peptides 1 and 2 were equally stabilized to direct RNase digestion (Fig. 2).
Figure 2. Metabolic Stability of mRNA Polyplexes.
UTR mRNA (2 µg) was complexed with 1.6 nmol of peptide 1 (panel A) or peptide 2 (panel B) to form mRNA polyplexes that were digested with 0, 3, 10, 30, 100, 300, 1000, or 3000 ng/ml of RNase A in 20 µL 5 mM HEPES buffer, pH 7.4 at 37°C for 10 min. mRNA polyplexes were digested with proteinase K to remove PEG-peptides. Following phenol:chloroform:isoamyl alcohol extraction, mRNA was electrophoresed on 1% agarose gel then stained with ethidium bromide. Both PEG-peptides were found to protect mRNA from RNase digestion up to 100 ng/mL, whereas naked mRNA was completely digested with 3 ng/ml.
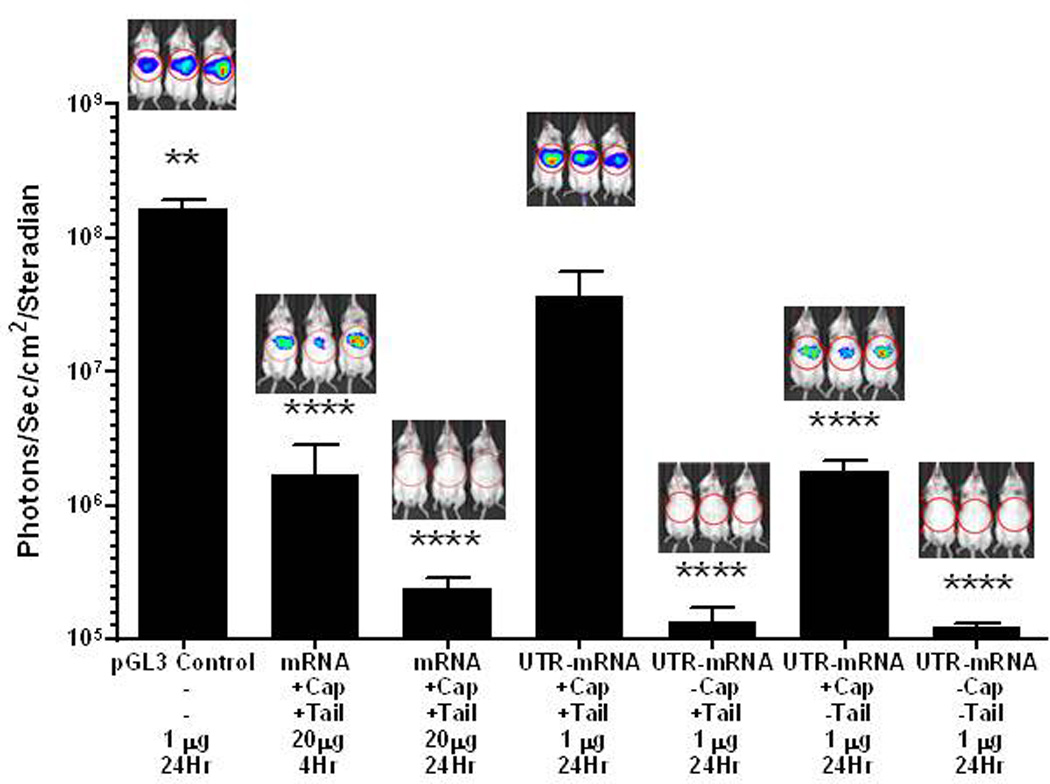
Direct HD-dosing of 20 µg of an un-optimized mRNA encoding Luc (Fig. 1 Supplemental) resulted in transient expression as determined by BLI detection of 106 photons/sec/cm2/steradian in the liver at 4 hours, returning to background signal at 24 hours (Fig. 3), consistent with the results from McCaffrey et al.30. To improve the expression efficiency of mRNA, a codon optimized luciferase gene was synthesized possessing 3' and 5' human β-globin UTRs flanking the Luc gene (Fig 2, supplemental). Direct HD-dosing of 1 µg of UTR mRNA resulted in BLI detection of 3.7×107 photons/sec/cm2/steradian in the liver at 24 hours post-mRNA delivery (Fig. 3). The expression efficiency of UTR mRNA was approximately 200-fold greater than mRNA lacking UTRs, and was only 10-fold lower than the efficiency afforded from HD-delivery of 1 µg of pGL3 (Fig. 3). Control experiments suggested that the HD-delivery of UTR mRNA to the cytosol of hepatocytes was sufficient to mediate efficient expression. HD-dosing of tailed, but uncapped UTR mRNA, or uncapped and untailed UTR mRNA produced background BLI signal (Fig. 3). However, HD-dosing of capped, but untailed UTR mRNA, produced approximately 1.8×106 photons/sec/cm2/steradian in liver at 24 hours, which was only 10-fold less than fully capped and tailed UTR mRNA (Fig. 3). This result suggests that HD-dosing delivers capped UTR mRNA to the nucleus where some tailing occurs, resulting in 10-fold less expression compared to fully capped and tailed UTR mRNA.
Figure 3. HD-dosing of Luc mRNA and DNA.
Hydrodynamic delivery of pGL3 (1 µg) was used as a benchmark to compare the magnitude of expression in liver determined by bioluminescence imaging. The hydrodynamic delivery of mRNA (20 µg) resulted in low-level detectable expression at 4 hours, that diminished to background by 24 hours. Alternatively, the hydrodynamic delivery of UTR mRNA (1 µg) resulted in expression comparable to pGL3 at 24 hours and 500-fold greater than mRNA assayed at 4 hours when normalized for dose. Omission of the 5’ cap or 3’ Poly A tail, resulted in complete loss of expression, whereas omission of only the 3’ Poly A tail resulted in expression approximately 20-fold higher than un-optimized Luc mRNA assayed at 4 hours when normalized for dose. Statistically significant differences relative to 1 µg dose of capped and tailed UTR mRNA at 24 hours are marked, with ** p ≤ 0.01, **** p ≤ 0.0001.
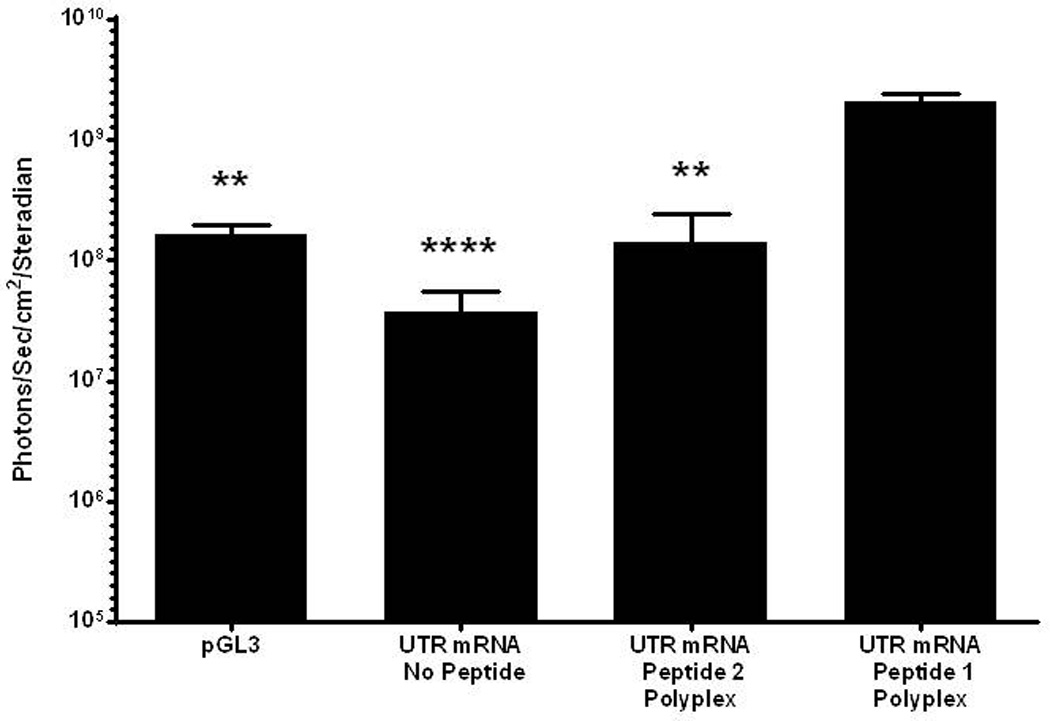
PEG-peptide UTR mRNA polyplexes were partially protected from the action of RNase (Fig. 2). Consequently, HD-dosing of peptide 1 UTR mRNA polyplex produced 1.4×109 photons/sec/cm2/steradian in liver at 24 hours, which is significantly higher than naked UTR mRNA and peptide 2 UTR mRNA polyplex, supporting the hypothesis that peptide 1 polyintercalation affords greater mRNA polyplex stability in vivo (Fig. 4). Additionally, the peptide 1 UTR mRNA polyplex produced significantly higher expression than pGL3 plasmid DNA.
Figure 4. HD-dosing of UTR mRNA Polyplexes.
UTR mRNA (1 µg) was combined with 0.8 nmol of either peptide 1 or 2 to form polyplexes, delivered hydrodynamically to triplicate mice, and assayed for luciferase expression in liver by bioluminescence imaging at 24 hours. Peptide 1 UTR mRNA polyplexes produced 10–15 fold higher luciferase expression compared to pGL3 (1 µg), UTR mRNA alone, or peptide 2 UTR mRNA polyplex. Statistically significant differences relative to UTR mRNA peptide 1 polyplex dose are marked, with ** p ≤ 0.01, *** p ≤ 0.001.
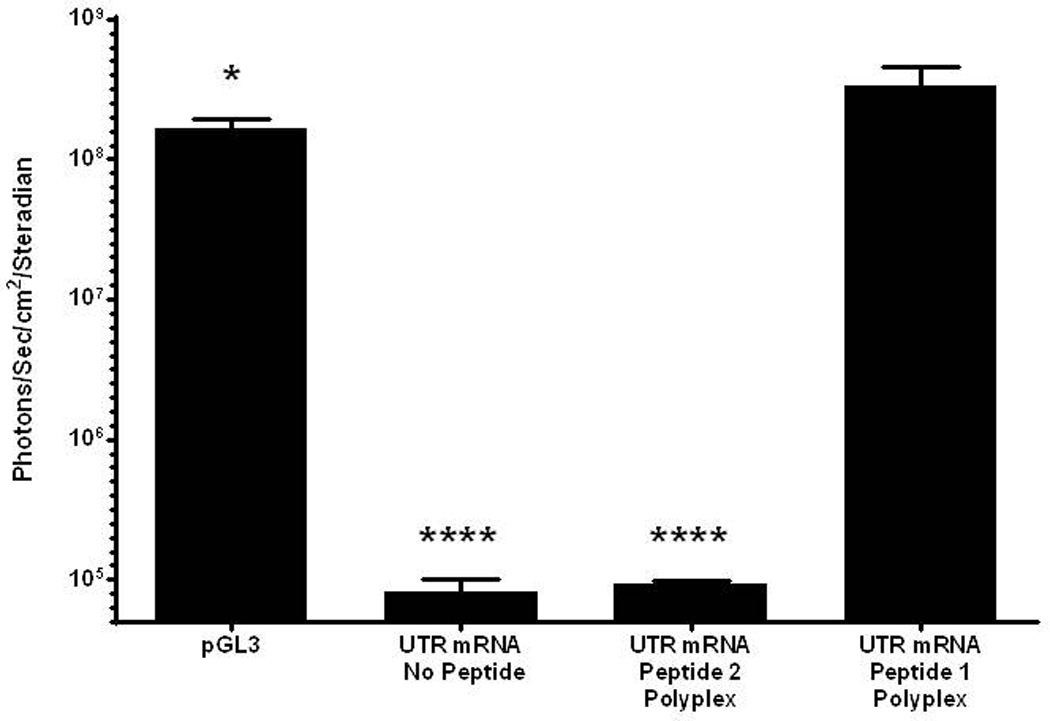
Further evidence of the enhanced stability of peptide 1 UTR mRNA polyplex resulted from incubation in mouse serum (Fig. 5). Naked UTR mRNA and peptide 2 UTR mRNA polyplex were unable to protect mRNA from metabolism, resulting in the complete loss of luciferase expression. However, incubation of peptide 1 UTR mRNA polyplex in mouse serum followed by HD-dosing produce 1×108 photons/sec/cm2/steradian in liver at 24 hours post-mRNA delivery, indicating increased RNase protection resulting from PEG-peptide binding to double stranded regions of mRNA (Fig. 5). Serial BLI analysis was used to examine the time course of luciferase expression mediated by HD-delivery of a 1 µg dose of PEG-peptide UTR mRNA polyplex. The expression determined at 4 hours remained constant for 24 hours, after which it declined 10-fold per 24 hours to reach background in 84 hours (Fig 6).
Figure 5. Serum Incubation of mRNA Polyplexes.
UTR mRNA (1 µg) was combined with 0.8 nmol peptide 1 or 2 and incubated in mouse serum (20 µl) for 30 min. mRNA polyplexes were diluted with saline, dosed hydrodynamically in mice, and assayed for luciferase expression by bioluminescence imaging at 24 hours. Naked UTR mRNA and peptide 2 UTR mRNA polyplexes produced no expression, whereas peptide 1 polyplexes produced luciferase expression comparable to pGL3. Statistically significant differences relative to UTR mRNA peptide 1 polyplex dose are marked, with * p ≤ 0.05, **** p ≤ 0.0001.
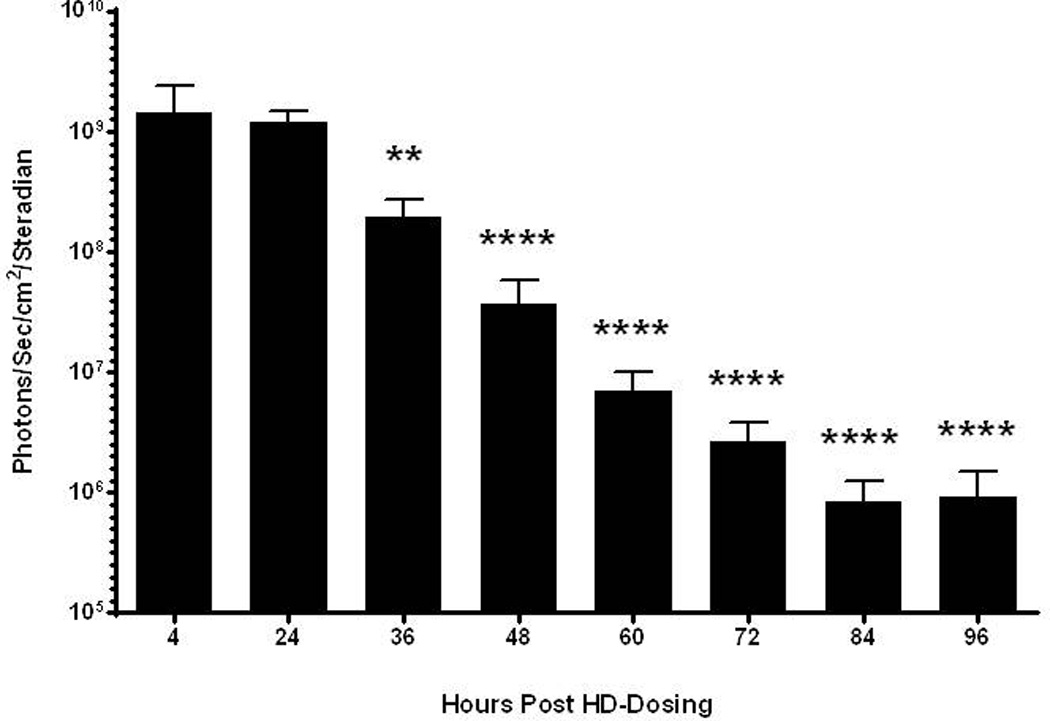
Figure 6. Kinetics of mRNA Polyplex Expression Following Hydrodynamic Delivery.
UTR mRNA (1 µg) peptide 1 polyplexes were hydrodynamically dosed in triplicate mice. Serial bioluminescent imaging was performed at 4, 24, 36, 48, 60, 72, 84, and 96 hours to monitor luciferase expression. The luciferase expression reached a maximum at 4 hours and remained above background for 72 hours. Statistically significant differences relative to the 24 hour measurement are marked, with ** p ≤ 0.01, **** p ≤ 0.0001.
Discussion
The ease of plasmid DNA production along with its successful packaging into polyplexes and lipoplexes has led to the development of efficient in vitro transfection agents38,39. More sophisticated blood compatible polyplexes and liposomes possessing targeting ligands have been delivered systemically to tumors in vivo40,41. However, the systemic delivery of plasmid DNA to non-dividing quiescent liver hepatocytes remains a significant challenge42. Once inside hepatocytes, the endosomal escape of DNA nanoparticles remains inefficient and nuclear delivery has yet to be convincingly demonstrated. The nuclear envelope presents a major barrier to DNA delivery. Although the membrane is perforated by many nuclear pore complexes, the maximum diameter allowed through the pore is approximately 39 nm43, smaller than most DNA nanoparticles. When plasmid DNA was microinjected into non-dividing cells, only 0.1% of plasmids were detected in the nucleus44.
The successful delivery of siRNA to the cytosol of hepatocytes in animals45 has prompted this and other investigations into the delivery of mRNA to the cytosol of hepatocytes, as a first step toward by-passing nuclear uptake as the major impediment to the development of i.v. dosed targeted gene delivery systems. As with the development of siRNA delivery systems, optimization of the oligonucleotide construct for potency is a prerequisite to packaging and delivery46. HD-dosing lends itself well to the optimization of DNA constructs that are designed for hepatocyte expression. The rapid delivery in a large volume of saline displaces serum nucleases and results in the nuclear delivery of naked plasmid DNA. However, prior HD-dosing studies demonstrated that luciferase expressing mRNA was not efficiently expressed30,31, pointing to the need to improve the mRNA construct and further stabilize mRNA to RNase.
Compared to an un-optimized mRNA construct, codon optimization combined with installing a 5' and 3' human β-globin UTR into the mRNA produced a 2000-fold improvement in luciferase expression efficiency following HD-dosing (Fig. 3). These changes increased the expression so that a 1 µg dose of UTR mRNA was equivalent to 1 µg of plasmid DNA (pGL3) when assayed at 24 hours following hydrodynamic delivery (Fig. 3) in addition to producing a transient expression profile (Fig. 6) equivalent to pGL347. The 2000-fold enhancement is significantly greater than the 15-fold increase reported by Wilber et al. who included 5' and 3'-UTRs from Xenopus laevis31. While many elements of an mRNA's design impact stability and expression efficiency, such as configuration of 3' UTRs and the length of the poly-A tail48, the improvement in efficiency reported here most likely result from individual or combined contributions from the use of a β-globin 3' and 5' UTRs from human origin instead of Xenopus laevis31, codon optimization of luciferase, and the length of the poly-A resulting from tailing pre-mRNA using polyadenosine polymerase.
One of the greatest challenges in developing an i.v. dosed mRNA delivery system is protecting the oligonucleotide from metabolism by endogenous RNase14. Non-viral gene delivery systems that rely solely upon ionic binding with oligonucleotides either possess insufficient affinity to remain stable in the circulation or are highly charged making them incompatible with blood49,50. The present study used a polyacridine PEG-peptide with proven ability to protect plasmid DNA in the circulation35 to stabilize mRNA through intercalative binding to double stranded regions of the stem-loop structures (Scheme 1). Two experimental results establish that a PEGylated polyacridine peptide provides superior protection to mRNA relative to a comparable polylysine PEG-peptide that lacks the ability to intercalate mRNA. Polyacridine PEG-peptide mRNA polyplexes provided a significant 15-fold enhanced luciferase expression upon HD-dosing relative to mRNA polyplexes prepared with polylysine PEG-peptide (Fig. 4). Furthermore, the transfection of an mRNA polyplex is preserved following pre-incubation in mouse serum only when protected by a polyacridine PEG-peptide (Fig. 5).
This study demonstrates that in vitro transcribed mRNA possessing natural nucleotides is of comparable efficiency as plasmid DNA (pGL3) when delivered by HD-dosing. It is also possible that mRNA mediated expression may be further enhanced through the use of modified nucleotides such as pseudouridine and/or 5-methylcytidine, which have been shown to improve protein expression and reduce immune response19,51,52. These findings show that mRNA has the potential to replace DNA in some systemically delivered gene delivery applications. The transient expression of mRNA may be desirable in certain applications such as genome editing using Zinc Finger Nucleases, TALEN nucleases, or the CRISPR/Cas9 system53, or integration through transposases31 where short term mRNA expression is needed to avoid potential genotoxicity. PEGylated polyacridine peptide protects mRNA from RNase activity in vitro, and enhances luciferase expression in liver by approximately 15-fold, suggesting that mRNA stability may be improved even further by optimizing peptides for higher affinity binding to mRNA. While HD-dosing may not be directly applicable to delivery of mRNA to humans, it remains a useful research tool to allow optimization of mRNA constructs and polyplex design to increase transfection efficiency, which is prerequisite to the development of efficient targeted mRNA gene delivery systems.
Materials and Methods
DNA and mRNA Preparation
pGL3 control vector (Promega, Madison, WI, USA), a 5.3 kbp plasmid encoding the firefly luciferase gene controlled by an SV40 promoter, was grown in DH5α E. coli and purified by Qiagen Gigaprep kit (Qiagen, Germantown, MD, USA). Template DNA for in vitro transcription was prepared by synthesizing the firefly luciferase gene with flanking 5' and 3' untranslated regions (UTRs) derived from human β-globin and codon optimization for expression in mice (GenScript, Piscataway, NJ, USA). The synthetic gene (Luc-UTR) was inserted into the pcDNA3.1(−) vector (Life Technologies, Grand Island, NY, USA) between the XbaI and BamHI sites, downstream from the T7 promoter site. Luc-UTR pcDNA3.1(−) was grown in DH5α E. coli and isolated using a Qiagen Miniprep kit (Qiagen, Germantown, MD, USA).
Purified Luc-UTR pcDNA3.1(−) plasmid was linearized with HindIII-HF (New England Biolabs, Ipswich, MA, USA) at 37°C for 60 min. Residual RNase A from the miniprep was removed by digestion with 1.2 U of proteinase K (Thermo Fisher Scientific, Pittsburgh, PA, USA) in 0.5% SDS (Research Products International, Mt. Prospect, IL, USA). Linearized template DNA was purified by phenol:chloroform:isoamyl alcohol extraction and isopropanol precipitation. Pre-mRNA was produced by in vitro transcription using the Ambion MEGAscript™ T7 Kit (Life Technologies, Grand Island, NY, USA) according to manufacturer's instructions. Briefly, 1 µg of linearized template Luc-UTR pcDNA3.1(−) DNA was combined with 7.5 mM ATP, GTP, CTP, UTP and 10X reaction buffer and T7 RNA Polymerase in a total volume of 20 µL, then reacted at 37°C for 4 hours. After transcription, 2 units of TURBO DNAse was added and allowed to digest template DNA at 37°C for 15 min followed by the addition of 100 mM EDTA in 5 M ammonium acetate to terminate the digestion. The resulting pre-mRNA was purified by phenol:chloroform:isoamyl alcohol extraction and isopropanol precipitation, then quantified by absorbance.
A 3' PolyA tail was added to the pre-mRNA using the Ambion PolyA Tailing Kit (Life Technologies, Grand Island, NY, USA) adapted from the manufacturer's instructions. Briefly, 200 µg of pre-mRNA was prepared in 100 µL of 50 mM Tris-HCl pH 7.9 containing 1 mM ATP, 250 mM NaCl, 10 mM MgCl2, and 8 units of E. coli polyadenosine polymerase (E.C. 2.7.7.19). The reaction was incubated at 37°C for 1 hour, then terminated by the addition of 5 M ammonium acetate. Tailed mRNA was purified by phenol:chloroform:isoamyl alcohol extraction and isopropanol precipitation and quantified by absorbance.
The 5' cap was added to tailed mRNA using Vaccinia Capping System and mRNA Cap 2'O-methyltransferase (New England Biolabs, Ipswich, MA, USA) according to the manufacturer's instructions. Tailed mRNA (150 µg) was heat denatured at 65°C for 5 min, then chilled on ice for 5 min, prior to combining it with 0.5 mM GTP, 0.2 mM SAM, 150 units of Vaccinia Capping Enzyme, and 750 units mRNA cap 2'O-methyltransferase in a total volume of 300 µL. The reaction was incubated at 37°C for 1 hour, then purified by phenol:chloroform:isoamyl alcohol extraction and isopropanol precipitation, quantified by absorbance at 260 nm and stored at −70°C. Typical yields for a 20 µL reaction were approximately 200 µg of pre-mRNA. After tailing and capping, the final yield ranged from 100 to 150 µg. Pre-mRNA produced a 260/280 nm absorbance ratio of approximately 2.0, as did purified tailed and capped mRNA.
The size and purity of mRNA was determined using agarose native gel electrophoresis. Prior to electrophoresis, mRNA (1 µg) was denatured at 65°C for 5 min and chilled on ice for 5 min then combined with 5X LB Loading Medium (Faster Better Media, Baltimore, MD, USA) and loaded onto a 1% agarose gel prepared with 50 mL of 0.5 X lithium boric acid electrophoresis buffer containing 2 µL of 50 µg/mL ethidium bromide (Faster Better Media, Baltimore, MD, USA), then electrophoresed at 145 V for 30 min. Gels were imaged using a UVP BioSpectrum Imaging System and VisionWorks®LS software (UVP, Upland CA, USA), revealing two bands of equal intensity, with the slower migrating band resulting from mRNA secondary structure. Heat denaturation resulted in loss of the slower migrating native band, resulting in a single denatured band for mRNA. A clear upward band shift was used to distinguish heat denatured tailed mRNA from non-tailed mRNA.
PEG-Peptide Synthesis and Formulation of mRNA PEG-Peptide Polyplexes
(Acr-Lys4)3-Acr-Lys-Cys (peptide 1) and Cys-Trp-Lys20 (peptide 2) were prepared by solid phase peptide synthesis using an Apex 396 Synthesizer (Advanced ChemTech, Louisville, KY, USA), using standard FMOC procedures as previously described34,35. PEGylation of the Cys residue was performed by reacting 1 µmol of peptide with 1.1 µmol of PEG5kDa-maleimide in 4 ml of 100 mM HEPES buffer pH 7 for 12 hours. PEGylated peptides were purified by semi-preparative RP-HPLC on a Vydac C18 (2 × 25 cm) column eluted at 10 ml/min with 0.1 v/v % TFA and an acetonitrile gradient of 20–60 v/v % while monitoring acridine by absorbance at 409 nm. The major peak was collected and pooled from multiple runs, concentrated by rotary evaporation, lyophilized, and stored at −20°C. The counter ion was exchanged by chromatography on a Sephadex G-25 column (2.5 × 50 cm) equilibrated with 0.1 v/v % acetic acid to obtain the peptide in an acetate salt form. The major peak corresponding to the PEG-peptide eluting in the void volume (100 ml) was pooled, concentrated by rotary evaporation, and freeze-dried. PEG-peptides were reconstituted in water and quantified by Abs409nm (each acridine ε409 nm = 9266 M−1 cm−1, whereas Trp ε280 nm = 5600 M−1 cm−1)34,35.
The binding of PEG-peptides to mRNA was monitored by agarose gel electrophoresis. Peptide 1 or 2 (0, 0.01, 0.05, 0.10, 0.3, 0.7, or 1 nmol) was combined with mRNA (1 µg) then loaded onto a 1% agarose gel and electrophoresed and imaged as described above. mRNA polyplexes were prepared for particle size and zeta potential analysis by combining 30 µg of mRNA with 24 nmol of PEG-peptide in a total volume of 2 mL of 5 mM HEPES pH 7.5. The particle size and zeta potential were determined by dynamic light scattering on a Brookhaven Zetaplus (Brookhaven Instruments, Holtsville, NY, USA).
The ability of mRNA polyplexes to withstand a direct digestion with RNase was determined by gel electrophoresis. PEG-peptide polyplexes were prepared by combining 2 µg of mRNA polyplexes with 1.6 nmol of peptide 1 or 2. Polyplexes were digested with 0, 3, 10, 30, 100, 300, 1000, or 3000 ng/ml of RNase A (Thermo Fisher Scientific, Pittsburgh, PA, USA) in total volume of 20 µl of 5 mM HEPES pH 7.4 for 10 min at 37°C. The RNase digestion was terminated by addition of proteinase K (500 µl of 0.5 mg/ml in 100 mM NaCl, 50 mM Tris, and 1% SDS, pH 8.0), followed by digestion at 37°C for 30 min. mRNA was extracted using phenol:chloroform:isoamyl alcohol followed by ethanol precipitation and electrophoresis as described above.
HD-dosing PEG-Peptide mRNA Polyplexes
All animal experiments were carried out with the approval of the University of Iowa Institutional Animal Care and Use Committee. Naked mRNA or mRNA polyplexes (1 µg of mRNA and 0.8 nmol of PEG-peptide) were prepared in 20 µl of 5 mM HEPES pH 7.0, then diluted with 1.8 ml of normal saline. Triplicate 5 – 8 week old 20 g male ICR mice (Harlan Laboratories, Indianapolis, IN, USA) were restrained and hydrodynamically dosed via the tail vein in 5–7 seconds. At 24 hours post mRNA delivery, mice were anesthetized with 2.5% isofluorane and intraperitoneally dosed with 80 µL of 30 µg/µL D-Luciferin (Gold Biotechnology, St. Louis, MO, USA) in phosphate buffered saline. At 5 min post luciferin injection, mice were imaged for bioluminescence (BLI) in an IVIS Imaging 200 Series (Xenogen, Hopkins, MA, USA), with 2.5% isofluorane, medium binning, 24.6 cm field of view, and 10 sec acquisition time.
The metabolic stability of mRNA polyplexes was determined by incubation in mouse serum. mRNA (1 µg) and PEG-peptide (1 nmol) were combined to form mRNA polyplexes in a total volume of 5 µL of 5 mM Hepes pH 7.0. Mouse serum (15 µL) was added and allowed to incubate at room temperature for 30 min. mRNA polyplexes in serum were diluted with 1.8 ml of normal saline then dosed hydrodynamically and imaged at 24 hours by BLI as described above. The kinetics of luciferase expression in liver was determined by hydrodynamically dosing 1 µg of UTR mRNA polyplex in triplicate mice. Luciferase expression was measured by BLI at 4, 24, 36, 48, 60, 72, 84, and 96 hours.
Statistical Analysis
Bioluminescence imaging results were analyzed for statistical significance by calculating the base 10 logarithm of each luminescence measurement. The log transformed data was analyzed by ordinary one-way analysis of variance (ANOVA) using GraphPad Prism version 6.02 (GraphPad Software, La Jolla, CA, USA), with Dunnet’s Multiple Comparisons Test.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgement
The authors gratefully acknowledge support from NIH Grants GM097093 and T32 GM008365 (STC and JAP).
Footnotes
Conflict of Interest
The authors declare no financial conflict of interest with results presented in the study.
Supplemental
Supplementary information is available at Gene Therapy's website
References
- 1.Wu GY, Wu CH. Receptor-mediated Gene Delivery and Expression In Vivo. J Biol Chem. 1988;263:14621–14624. [PubMed] [Google Scholar]
- 2.Wooddell CI, Rozema DB, Hossbach M, John M, Hamilton HL, Chu Q, et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol Ther. 2013;21:973–985. doi: 10.1038/mt.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chuah MK, Evens H, VandenDriessche T. Gene therapy for hemophilia. J Thromb Haemost. 2013;11(Suppl 1):99–110. doi: 10.1111/jth.12215. [DOI] [PubMed] [Google Scholar]
- 4.Richard M, Arfi A, Seguin J, Gandolphe C, Scherman D. Widespread biochemical correction of murine mucopolysaccharidosis type VII pathology by liver hydrodynamic plasmid delivery. Gene Ther. 2009;16:746–756. doi: 10.1038/gt.2009.36. [DOI] [PubMed] [Google Scholar]
- 5.Pun SH, Davis ME. Development of a nonviral gene delivery vehicle for systemic application. Bioconjugate Chemistry. 2002;13:630–639. doi: 10.1021/bc0155768. [DOI] [PubMed] [Google Scholar]
- 6.Lenter MC, Garidel P, Pelisek J, Wagner E, Ogris M. Stabilized nonviral formulations for the delivery of MCP-1 gene into cells of the vasculoendothelial system. Pharmaceutical Research. 2004;21:683–691. doi: 10.1023/b:pham.0000022416.33048.81. [DOI] [PubMed] [Google Scholar]
- 7.Read ML, Logan A, Seymour LW. Barriers to Gene Delivery Using Synthetic Vectors. In: Hall JC, Dunlap JC, Friedmann T, van Heyningen V, editors. Advances in Genetics. Academic Press; 2005. pp. 19–46. [Google Scholar]
- 8.Hu Y, Haynes MT, Wang Y, Liu F, Huang L. A highly efficient synthetic vector: nonhydrodynamic delivery of DNA to hepatocyte nuclei in vivo. ACS Nano. 2013;7:5376–5384. doi: 10.1021/nn4012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 10.AlDosari MS, Knapp JE, Liu D, Leaf Huang M-CHaEW. Advances in Genetics. Academic Press; 2005. Hydrodynamic Delivery; pp. 65–82. [DOI] [PubMed] [Google Scholar]
- 11.Zhang G, Gao X, Song YK, Vollmer R, D.B S, Gaskowski JZ, et al. Hydroporation as the mechanism of hydrodynamic delivery. Gene Ther. 2004;11:675–682. doi: 10.1038/sj.gt.3302210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrianaivo F, Lecocq M, Wattiaux-De Coninck S, Wattiaux R, Jadot M. Hydrodynamics-based transfection of the liver: entrance into hepatocytes of DNA that causes expression takes place very early after injection. Journal of Gene Medicine. 2004;6:877–883. doi: 10.1002/jgm.574. [DOI] [PubMed] [Google Scholar]
- 13.Hodges BL, Scheule RK. Hydrodynamic delivery of DNA. Expert Opin Biol Ther. 2003;3:911–918. doi: 10.1517/14712598.3.6.911. [DOI] [PubMed] [Google Scholar]
- 14.Sahin U, Kariko K, Tureci O. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 15.Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct Gene Transfer into Mouse Muscle in Vivo. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 16.Deering RP, Kommareddy S, Ulmer JB, Brito LA, Geall AJ. Nucleic acid vaccines: prospects for non-viral delivery of mRNA vaccines. Expert Opin Drug Deliv. 2014;11:885–899. doi: 10.1517/17425247.2014.901308. [DOI] [PubMed] [Google Scholar]
- 17.Phua KK, Leong KW, Nair SK. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J Control Release. 2013;166:227–233. doi: 10.1016/j.jconrel.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ. Developing mRNA-vaccine technologies. RNA Biol. 2012;9:1319–1330. doi: 10.4161/rna.22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kariko K, Muramatsu H, Keller JM, Weissman D. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol Ther. 2012;20:948–953. doi: 10.1038/mt.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci U S A. 1989;86:6077–6081. doi: 10.1073/pnas.86.16.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 22.Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM, et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol. 2013;31:898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phua KK, Staats HF, Leong KW, Nair SK. Intranasal mRNA nanoparticle vaccination induces prophylactic and therapeutic anti-tumor immunity. Sci Rep. 2014;4:5128. doi: 10.1038/srep05128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perche F, Benvegnu T, Berchel M, Lebegue L, Pichon C, Jaffres PA, et al. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine. 2011;7:445–453. doi: 10.1016/j.nano.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Avci-Adali M, Behring A, Keller T, Krajewski S, Schlensak C, Wendel HP. Optimized conditions for successful transfection of human endothelial cells with in vitro synthesized and modified mRNA for induction of protein expression. J Biol Eng. 2014;8:8. doi: 10.1186/1754-1611-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng C, Convertine AJ, Stayton PS, Bryers JD. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials. 2012;33:6868–6876. doi: 10.1016/j.biomaterials.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Debus H, Baumhof P, Probst J, Kissel T. Delivery of messenger RNA using poly(ethylene imine)-poly(ethylene glycol)-copolymer blends for polyplex formation: biophysical characterization and in vitro transfection properties. J Control Release. 2010;148:334–343. doi: 10.1016/j.jconrel.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Su HH, Yang Y, Hu Y, Zhang L, Blancafort P, et al. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther. 2013;21:358–367. doi: 10.1038/mt.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida S, Itaka K, Uchida H, Hayakawa K, Ogata T, Ishii T, et al. In vivo messenger RNA introduction into the central nervous system using polyplex nanomicelle. PLoS One. 2013;8:e56220. doi: 10.1371/journal.pone.0056220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCaffrey AP, Ohashi K, Meuse L, Shen S, Lancaster AM, Lukavsky PJ, et al. Determinants of hepatitis C translational initiation in vitro, in cultured cells and mice. Mol Ther. 2002;5:676–684. doi: 10.1006/mthe.2002.0600. [DOI] [PubMed] [Google Scholar]
- 31.Wilber A, Frandsen JL, Geurts JL, Largaespada DA, Hackett PB, McIvor RS. RNA as a source of transposase for Sleeping Beauty-mediated gene insertion and expression in somatic cells and tissues. Mol Ther. 2006;13:625–630. doi: 10.1016/j.ymthe.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Baumhover NJ, Anderson K, Fernandez CA, Rice KG. Synthesis and In Vitro Testing of New Potent Polyacridine-Melittin Gene Delivery Peptides. Bioconjugate Chemistry. 2010;21:74–86. doi: 10.1021/bc9003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez CA, Baumhover NJ, Duskey JT, Khargharia S, Kizzire K, Ericson MD, et al. Metabolically Stabilized Long-Circulating PEGylated Polyacridine Peptide Polyplexes Mediate Hydrodynamically Stimulated Gene Expression in Liver. Gene Therapy. 2011;18:23–37. doi: 10.1038/gt.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kizzire K, Khargharia S, Rice KG. High-affinity PEGylated polyacridine peptide polyplexes mediate potent in vivo gene expression. Gene Ther. 2013;20:407–416. doi: 10.1038/gt.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khargharia S, Kizzire K, Ericson MD, Baumhover NJ, Rice KG. PEG length and chemical linkage controls polyacridine peptide DNA polyplex pharmacokinetics, biodistribution, metabolic stability and in vivo gene expression. J Control Release. 2013;170:325–333. doi: 10.1016/j.jconrel.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khargharia S, Baumhover NJ, Crowley ST, Duskey J, Rice KG. The uptake mechanism of PEGylated DNA polyplexes by the liver influences gene expression. Gene Ther. 2014 doi: 10.1038/gt.2014.81. [DOI] [PubMed] [Google Scholar]
- 37.Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL. The Vienna RNA websuite. Nucleic Acids Res. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pedroso de Lima MC, Simoes S, Pires P, Faneca H, Duzgunes N. Cationic lipid-DNA complexes in gene-delivery: from biophysics to biological applications. Adv Drug Deliv Rev. 2001;47:277–294. doi: 10.1016/s0169-409x(01)00110-7. [DOI] [PubMed] [Google Scholar]
- 40.Haynes MT, Huang L. Lipid-coated calcium phosphate nanoparticles for nonviral gene therapy. Adv Genet. 2014;88:205–229. doi: 10.1016/B978-0-12-800148-6.00007-9. [DOI] [PubMed] [Google Scholar]
- 41.Wagner E. Polymers for nucleic acid transfer-an overview. Adv Genet. 2014;88:231–261. doi: 10.1016/B978-0-12-800148-6.00008-0. [DOI] [PubMed] [Google Scholar]
- 42.Wolff JA, Budker V. The mechanism of naked DNA uptake and expression. Adv Genet. 2005;54:3–20. doi: 10.1016/S0065-2660(05)54001-X. [DOI] [PubMed] [Google Scholar]
- 43.Pante N, Kann M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell. 2002;13:425–434. doi: 10.1091/mbc.01-06-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pollard H, Remy JS, Loussouarn G, Demolombe S, Behr JP, Escande D. Polyethylenimine but not cationic lipids promotes transgene delivery to the nucleus in mammalian cells. Journal of Biological Chemistry. 1998;273:7507–7511. doi: 10.1074/jbc.273.13.7507. [DOI] [PubMed] [Google Scholar]
- 45.Rozema DB, Lewis DL, Wakefield DH, Wong SC, Klein JJ, Roesch PL, et al. Dynamic PolyConjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci U S A. 2007;104:12982–12987. doi: 10.1073/pnas.0703778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rettig GR, Behlke MA. Progress toward in vivo use of siRNAs-II. Mol Ther. 2012;20:483–512. doi: 10.1038/mt.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rettig G, McAnuff M, Kim J, Liu D, Rice KG. Quantitative Bioluminscence Imaging of Transgene Expression In Vivo. Analytical Biochemistry. 2006;335:90–94. doi: 10.1016/j.ab.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 48.Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood. 2006;108:4009–4017. doi: 10.1182/blood-2006-04-015024. [DOI] [PubMed] [Google Scholar]
- 49.Ward CM, Read ML, Seymour LW. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 2001;97:2221–2229. doi: 10.1182/blood.v97.8.2221. [DOI] [PubMed] [Google Scholar]
- 50.Oupicky D, Konak C, Dash PR, Seymour LW, Ulbrich K. Effect of albumin and polyanion on the structure of DNA complexes with polycation containing hydrophilic nonionic block. Bioconjug Chem. 1999;10:764–772. doi: 10.1021/bc990007+. [DOI] [PubMed] [Google Scholar]
- 51.Anderson BR, Muramatsu H, Jha BK, Silverman RH, Weissman D, Kariko K. Nucleoside modifications in RNA limit activation of 2'–5'-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011;39:9329–9338. doi: 10.1093/nar/gkr586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kariko K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim H, Kim J-S. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.