

Abstract
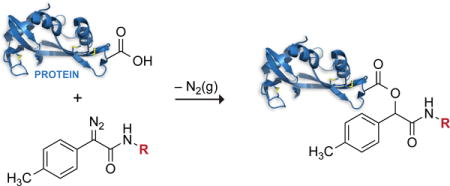
The O-alkylation of carboxylic acids with diazo compounds provides a means to esterify carboxylic acids in aqueous solution. A Hammett analysis of the reactivity of diazo compounds derived from phenylglycinamide revealed that the p-methylphenylglycinamide scaffold has an especially high reaction rate and ester:alcohol product ratio, and esterifies protein carboxyl groups more efficiently than does any known reagent.
Broad reactivity has made diazo compounds one of the most versatile functional groups in synthetic organic chemistry.1 Recently, this broad utility has been expanded into the field of chemical biology. For example, the diazo group has been shown to undergo 1,3-dipolar cycloadditions with strained alkynes in a tunable manner. The rates can greatly exceed those of the analogous azide,2 and the reactions are chemoselective in the presence of mammalian cells.3 In addition, diazo compounds have been used to label proteins via C–H, N–H, and S–H insertion reactions.4
Diazo compounds have another well-known mode of reactivity—esterification of carboxylic acids. We realized that this reactivity could provide unique opportunities in chemical biology. For example, unlike the alkylation of other functional groups, O-alkylation of a carboxyl group is bioreversible because mammalian cells contain non-specific esterases.5 The esterification of carboxyl groups in proteins and other biomolecules is, however, difficult to effect, as solvent water competes effectively with alcohols for eletrophilic acyl groups. In contrast, esterification reactions mediated by diazo groups rely on the carboxyl group serving as a nucleophile (Scheme 1).6
Scheme 1.
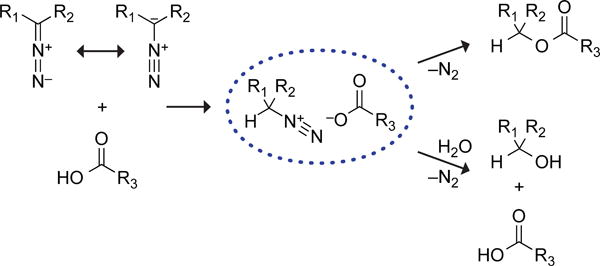
The use of diazo compounds to label proteins was attempted 60 years ago.7 These initial results were not compelling. A large molar excess (up to 103-fold) of diazo compound was required to overcome hydrolytic decomposition. Moreover, the reaction was not chemoselective, as amino, sulfhydryl, and phenolic side chains suffered alkylation. Such modifications are potentially deleterious to protein function and not bioreversible.8
Previous work in our laboratory suggested that the obstacles in reactivity can be overcome by tuning the reactivity of a diazo group. In particular, we found that the basicity of 9-diazofluorene endows this diazo compound with the ability to label a protein in an aqueous environment.9 The fluorenyl scaffold is, however, unduly large and not readily amenable to synthetic modification, and its reaction rate and chemoselectivity are not necessarily maximal.
Accordingly, we sought a scaffold that is optimal for the esterification of carboxyl groups in an aqueous environment. Towards that end, we have examined derivatives of phenylglycinamide (Figure 1A). This scaffold delocalizes the electron density on Cα into an amidic carbonyl group as well as a phenyl group that enables a Hammett analysis12 of the esterification reaction. Moreover, the amide linkage enables facile installation of useful moieties.
Figure 1.
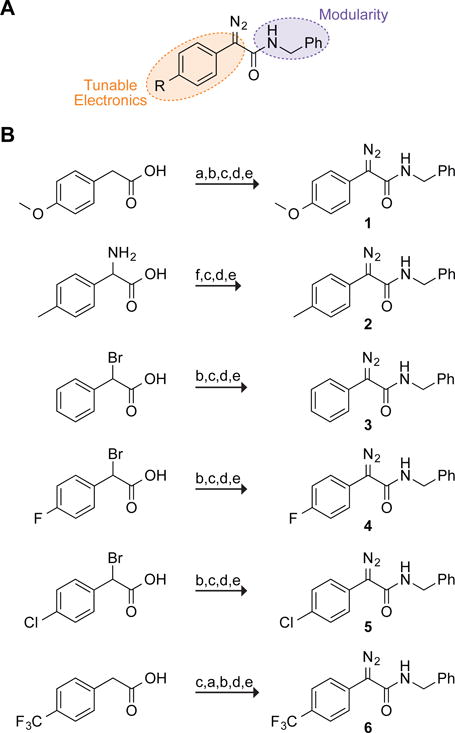
(A) Scaffold for testing the reactivity and selectivity of diazo compounds. (B) Synthetic route to diazo compounds 1–6. Steps: a) NBS, AIBN; b) NaN3, THF:H2O; c) NHS, DCC, THF; d) PhCH2NH2, DCM; e) N-succinimidyl 3-(diphenylphosphino)propionate, then NaHCO3 or DBU;10 f) imidazole-1-sulfonyl azide hydrochloride, DBU, CuSO4, MeOH.11
Diazo compounds 1–6 were accessed from derivatives of phenylacetic acid (Figure 1B). Briefly, an azide was installed at the benzylic position of the acid either through displacement of a bromide or by diazo transfer to an existing amine. The ensuing α-azido acids were then coupled to benzylamine and converted to the diazo compound by deimidogenation using a phosphinoester.8
In initial experiments, we probed the effect of electron distribution on the reactivity of diazo groups by measuring the rate of esterification in acetonitrile. To do so, we reacted diazo compounds 1–6 with BocGlyOH, and measured the second-order rate constants with 1H NMR spectroscopy (Figure S1). The effect of electron distribution on the reaction rate was dramatic: rate constants spanned over two orders of magnitude and increased with the electron-donating character of the phenyl substituents (Figure 2A). Hammett analysis of these rate constants gave a slope of ρ = −2.7 (Figure 2B). This value, which is comparable to those for typical SN1 reactions, indicates that the esterification reaction is highly sensitive to substituents and that substantial positive charge accumulates during its course,14 as expected from a mechanism involving an intermediate diazonium ion (Scheme 1).6
Figure 2.
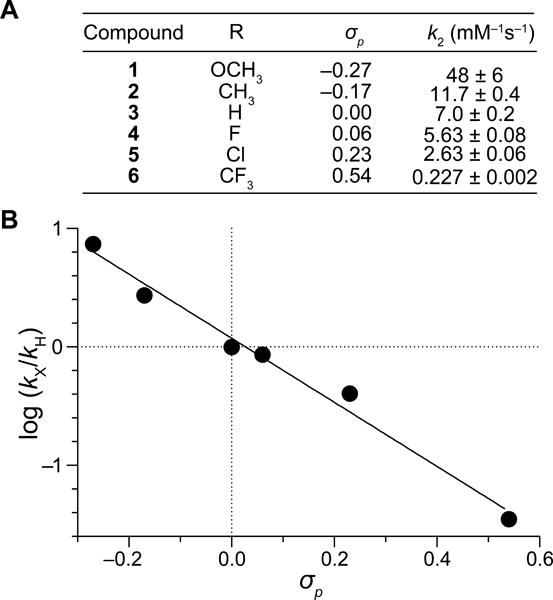
(A) Second-order rate constants for the esterification of BocGlyOH by diazo compounds 1–6 in CD3CN. (B) Hammett plot of the data in panel A. Values of σp are from ref. 13. ρ = −2.7.
Next, we sought to find the one compound that demonstrates the greatest selectivity for esterification over hydrolysis in an aqueous environment. Towards that end, we reacted diazo compounds 1–6 with equimolar BocGlyOH in a 1:1 mixture of acetonitrile and 2-(N-morpholino)ethanesulfonic acid (MES)–HCl buffer at pH 5.5, and we determined the ratio of ester-to-alcohol product with 1H NMR spectroscopy.
Surprisingly, the ester:alcohol ratio reached a maximum of 1.4:1 and remained unchanged despite increasing electron-withdrawal by the substituents (Figure 3). This result is consistent with a sharp cutoff for the formation of a carboxylate·diazonium intimate ion- pair intermediate that is maintained in a solvent cage by a Coulombic interaction (Scheme 1).6,15
Figure 3.
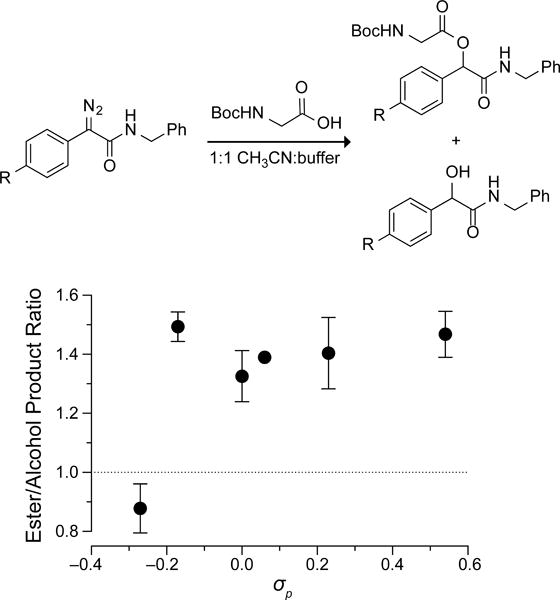
Effect of σp value on the chemoselectivity of diazo compounds 1–6 in aqueous solution.
Based on these experiments diazo compound 2 was selected for further study, as it demonstrated the fastest rate of those compounds that retain chemoselectivity in an aqueous environment. Because certain diazo compounds undergo O–H and S–H insertion reactions,1c,4 we sought to ensure that diazo compound 2 would esterify acids selectively in the presence of the sulfhydryl, hydroxyl, or phenolic moieties found on protein side chains. We were gratified to find that diazo compound 2 esterified BocSerOH, p-hydroxybenzoic acid, and 3-mercaptopropionic acid in 1:1 acetonitrile/100 mM MES–HCl buffer at pH 5.5, and that no other coupling products were observable by 1H NMR spectroscopy. We also attempted to esterify AlaOH to probe for reaction with an amino group. Consistent with previous observations,11 diazo compound 2 did not react with either the amino group or the carboxyl group of AlaOH, which was largely zwitterionic in the reaction mixture.
Finally, we compared diazo compound 2 to 9-diazofluorene for the labeling of a protein. To do so, we treated a well-known model protein, ribonuclease A,16 with 10 equiv of each diazo compound. The reactions were allowed to proceed for 4 h at 37 °C in 1:1 acetonitrile/10 mM MES–HCl buffer at pH 5.5. We then determined the extent of esterification with MALDI–TOF mass spectrometry. We found that diazo compound 2 was approximately twofold more efficient than was 9‐diazofluorene in effecting esterification (Figure 5).
Figure 5.
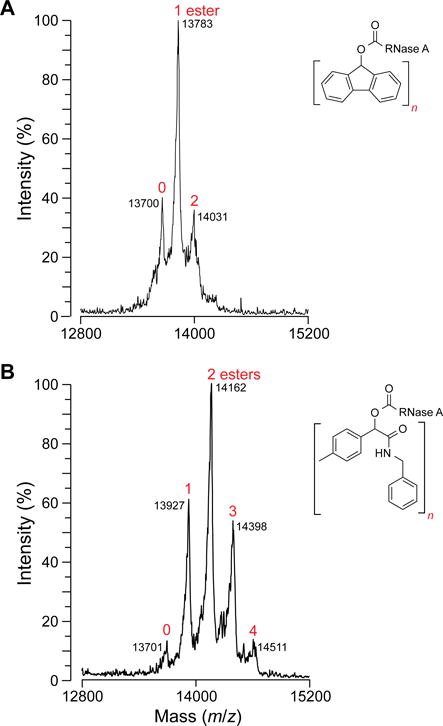
MALDI–TOF mass spectrometry data for esterification of RNase A with (A) 9-diazofluorene and (B) diazo compound 2.
We conclude that diazo compound 2 can be used to esterify proteins in an aqueous environment more efficiently than any other known reagent. Moreover, its modular design enables facile modification with useful moieties. We are now using this diazo compound to attach cell-type targeting, cell-penetration, and pharmacokinetic enhancing modules to proteins of interest.
Supplementary Material
Figure 4.
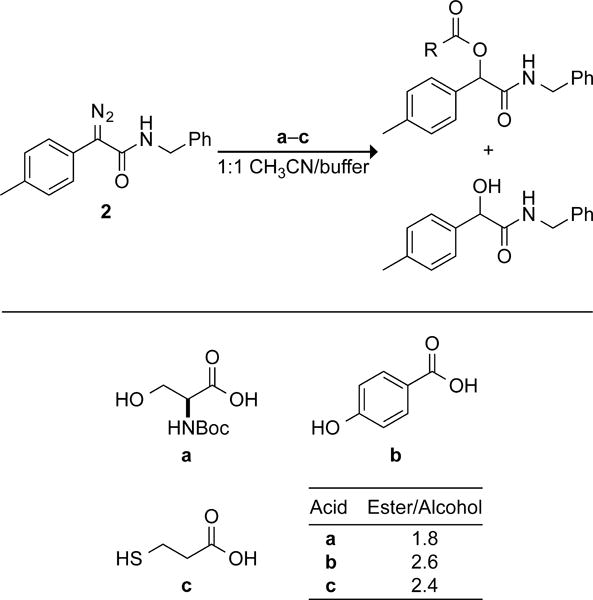
Chemoselectivity of esterification reactions in aqueous solution.
Acknowledgments
This work was supported by grant R01 GM044783 (NIH). K.A.M. was supported by Molecular Biosciences Training Grant T32 GM007215 (NIH). This work made use of the National Magnetic Resonance Facility at Madison, which is supported by grant P41 GM103399 (NIH), and the Biophysics Instrumentation Facility, which was established with grants BIR-9512577 (NSF) and S10 RR013790 (NIH). We thank Dr. N. A. McGrath (University of Wisconsin–La Crosse) for contributive discussions and critical reading of the manuscript, and Dr. B. VanVeller (Iowa State University) for suggesting the phenylglycine scaffold.
Footnotes
Supporting Information Available. Experimental procedures for syntheses and kinetic analyses, additional kinetic data, and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Regitz M, Maas G. Diazo Compounds: Properties and Synthesis. Academic Press; London: 1986. [Google Scholar]; (b) Padwa A, Weingarten MD. Chem Rev. 1996;96:223–269. doi: 10.1021/cr950022h. [DOI] [PubMed] [Google Scholar]; (c) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. Wiley; New York, NY: 1998. [Google Scholar]; (d) Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861–2904. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (e) Candelas NR, Alfonso CA. Curr Org Chem. 2009;13:763–787. [Google Scholar]
- 2.McGrath NA, Raines RT. Chem Sci. 2012;3:3237–3240. doi: 10.1039/C2SC20806G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersen KA, Aronoff MR, McGrath NA, Raines RT. J Am Chem Soc. 2015;137:2412–2415. doi: 10.1021/ja5095815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Antos JM, Francis MB. J Am Chem Soc. 2004;126:10256–10257. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]; (b) Antos JM, McFarland JM, Lavarone AT, Francis MB. J Am Chem Soc. 2009;131:6301–6308. doi: 10.1021/ja900094h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen Z, Popp BV, Ball ZT. ACS Chem Biol. 2011;6:920–925. doi: 10.1021/cb2001523. [DOI] [PubMed] [Google Scholar]; (d) Kundu R, Ball ZT. Chem Commun. 2013;49:4166–4168. doi: 10.1039/c2cc37323h. [DOI] [PubMed] [Google Scholar]; (e) Vohidov F, Coughlin JM, Ball ZT. Angew Chem, Int Ed. 2015;54:4587–4591. doi: 10.1002/anie.201411745. [DOI] [PubMed] [Google Scholar]
- 5.(a) Testa B, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism. Verlag Helvetica Chimica Acta; Zurich, Switzerland: 2003. [Google Scholar]; (b) Liederer BM, Borchardt RT. J Pharm Sci. 2006;95:1177–1195. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]; (c) Lavis LD. ACS Chem Biol. 2008;3:203–206. doi: 10.1021/cb800065s. [DOI] [PubMed] [Google Scholar]; (d) Tian L, Yang Y, Wysocki LM, Arnold AC, Hu A, Ravichandran B, Stenerson SM, Looger LL, Lavis LD. Proc Natl Acad Sci USA. 2012;109:4756–4761. doi: 10.1073/pnas.1111943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Roberts JD, Watanabe W, McMahon RE. J Am Chem Soc. 1951;73:760–765. [Google Scholar]; (b) Roberts JD, Watanabe W, McMahon RE. J Am Chem Soc. 1951;73:2521–2523. [Google Scholar]
- 7.(a) Grossberg AL, Pressman D. J Am Chem Soc. 1960;82:5478–5482. [Google Scholar]; (b) Doscher MS, Wilcox PE. J Biol Chem. 1961;236:1328–1337. [PubMed] [Google Scholar]; (c) Riehm JP, Sheraga HA. Biochemistry. 1965;4:772–782. doi: 10.1021/bi00880a023. [DOI] [PubMed] [Google Scholar]; (d) Delpierre GR, Fruton JS. Proc Natl Acad Sci USA. 1965;54:1161–1167. doi: 10.1073/pnas.54.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris JM, Chess RB. Nat Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 9.McGrath NA, Andersen KA, Davis AKF, Lomax JE, Raines RT. Chem Sci. 2014;6:752–755. doi: 10.1039/c4sc01768d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Myers EL, Raines RT. Angew Chem Int Ed. 2009;48:2359–2363. doi: 10.1002/anie.200804689. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chou H-H, Raines RT. J Am Chem Soc. 2013;135:14936–14939. doi: 10.1021/ja407822b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goddard-Borger ED, Stick RV. Org Lett. 2007;9:3797–3800. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hammett LP. Chem Rev. 1935;17:125–136. [Google Scholar]; (b) Hammett LP. J Am Chem Soc. 1937;59:96–103. [Google Scholar]; (c) Hammett LP. Physical Organic Chemistry. McGraw-Hill; New York, NY: 1940. pp. 184–228. [Google Scholar]; (d) Shorter J. Chem Listy. 2000;94:210–214. [Google Scholar]
- 13.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–175. [Google Scholar]
- 14.Anslyn EV, Doughtery DA. Modern Physical Organic Chemistry. University Science Books; Sausalito, CA: 2006. [Google Scholar]
- 15.Szele I, Tencer M, Zollinger H. Helv Chim Acta. 1983;66:1691–1703. [Google Scholar]
- 16.Raines RT. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.