

SUMMARY
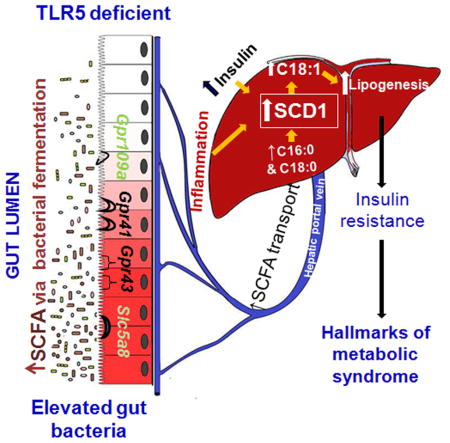
The gut microbiota plays a key role in host metabolism. Toll-Like Receptor 5 (TLR5), a flagellin receptor, is required for gut microbiota homeostasis. Accordingly, TLR5 deficient (T5KO) mice are prone to develop microbiota-dependent metabolic syndrome. Here we observed that T5KO mice display elevated neutral lipids with a compositional increase of oleate [C18:1 (n9)] relative to wild-type littermates. Increased oleate contribution to hepatic lipids and liver SCD1 expression were both microbiota-dependent. Analysis of short chain fatty acids (SCFA) and 13C-acetate label incorporation revealed elevated SCFA in ceca and hepatic portal blood and, increased liver de novo lipogenesis in T5KO mice. Dietary SCFA further aggravated metabolic syndrome in T5KO mice. Deletion of hepatic SCD1 not only prevented hepatic neutral lipid oleate enrichment but also ameliorated metabolic syndrome in T5KO mice. Collectively, these results underscore the key role of the gut microbiota-liver axis in the pathogenesis of metabolic diseases.
Keywords: Toll-like receptor 5, Gut bacteria, Short chain fatty acids, Hepatic neutral lipids, Monounsaturated fatty acids, Low-grade inflammation, Metabolic diseases
Graphical Abstract
Introduction
Metabolic syndrome is a constellation of metabolic abnormalities associated with insulin resistance that leads to metabolic diseases such as obesity, type II diabetes, and hepatic and cardiovascular diseases. The etiology of metabolic syndrome is influenced by genetics and a number of environmental factors including gut microbiota (Ley et al., 2006; Turnbaugh et al., 2006). Potential mechanisms by which the microbiota influences host metabolism include increased calorie extraction (Ley et al., 2006; Turnbaugh et al., 2006), metabolic endotoxemia (Cani et al., 2008), and generation of toxic metabolites (Wang et al., 2011). Previously, we have demonstrated that mice lacking TLR5 (T5KO) are susceptible to spontaneous gut inflammation, metabolic syndrome, and exhibit microbiotal dysbiosis (Carvalho et al., 2012; Vijay-Kumar et al., 2010; Vijay-Kumar et al., 2007). Further, T5KO mice maintained on a high-fat diet (HFD) develop hepatic steatosis and insulitis. Specifically, the gut microbiota in T5KO mice induces low-grade, chronic, systemic inflammation resulting in desensitization of insulin receptors culminating in hyperphagia and metabolic syndrome (Vijay-Kumar et al., 2010).
Microbiota exerts a substantial influence on host lipid metabolism (Rabot et al., 2010; Velagapudi et al., 2010). One such pathway is the generation of short chain fatty acids (SCFA) such as acetate, butyrate and propionate via anaerobic fermentation of dietary fiber. SCFA not only serve as a major fuel (i.e. butyrate) for the colonocytes but also are absorbed via enterohepatic circulation and contribute to hepatic lipogenesis (den Besten et al., 2013a; Samuel et al., 2008; Wolever et al., 1989). The major de novo synthesized lipotoxic saturated fatty acids (SFA) in the liver are palmitic acid (C16:0) and stearic acid (C18:0) which are converted into the less lipotoxic monounsaturated fatty acids (MUFA) palmitoleate (C16:1 n7) and oleate (C18:1 n9), a steatogenic agent. The addition of a double bond is catalyzed by hepatic microsomal, lipogenic rate limiting enzyme stearoyl CoA desaturase-1 (SCD1), a key regulatory enzyme in the homeostasis of SFA and MUFA (Hodson and Fielding, 2013). Despite oleate being a major MUFA from the diet, the expression of SCD1 is highly regulated by developmental, dietary, hormonal, and environmental factors. MUFA participates in the regulation of diverse processes, serve as better substrates for the synthesis of hepatic neutral lipids [triglycerides (TG) and cholesterol esters (CE)] and thus play a role in increasing tissue lipid load and may initiate insulin resistance (Miyazaki et al., 2000). Accordingly, SCD1 deficiency protects against high-fat–, high-carbohydrate–, and leptin deficiency–induced obesity and hepatic steatosis (Miyazaki et al., 2004; Miyazaki et al., 2009). Leptin-deficient mice exhibit increased SCD1 expression and associated with elevated levels of C16:1 and C18:1 in liver lipids.
SCD1 negatively regulates inflammation possibly by controlling the homeostasis of MUFA and SFA. Specifically, inhibition of SCD1 exacerbates responses to exogenous pro-inflammatory challenges such as lipopolysaccharide (LPS), C. rodentium, and a chemical colitogen DSS (Chen et al., 2008). Further, accumulation of SFA in Scd1KO mice promote inflammation via TLR4 signaling and promote atherosclerosis (Fessler et al., 2009), whereas loss of SCD1 attenuates adipose tissue inflammation (Liu et al., 2010), suggesting that its activity in diverse tissues has distinct outcomes.
In this study, we hypothesized that T5KO mice, which exhibit microbiota-dependent metabolic syndrome may have altered hepatic lipogenesis that might promote an obese phenotype. Accordingly, we demonstrate that T5KO mice displayed elevated hepatic C18:1 in neutral lipids when compared to WT littermates. This difference was not seen in microbiota-ablated and germ-free (GF) T5KO mice. 1H NMR metabolic profiling of cecal contents indicated decreased cecal oligosaccharides and elevated SCFA, suggesting profound changes in bacterial fermentation. Further, colonic SCFA receptors, hepatic lipogenic enzymes including SCD1, and pro-inflammatory genes were upregulated in T5KO mice. Using 13C-acetate labeling we showed that gut derived SCFA reached to the liver and incorporated into both liver and plasma palmitate (C16:0) relatively to a greater extent in T5KO mice. Dietary SCFA further aggravated metabolic syndrome while hepatic deletion of SCD1 in T5KO mice prevented most of the indices of metabolic syndrome. Hence, the ability of T5KO mice to synthesize higher MUFA, probably as a ‘metabolic adaptation’ to protect from lipotoxicity of SFA synthesized from SCFA but then potentiate metabolic syndrome. Even though numerous beneficial properties have been attributed to SCFA, their excess in conditions of innate immune deficiency coupled with bacterial overgrowth may increase susceptibility to metabolic diseases.
Results
Insulin resistance in T5KO mice is associated with oleate-enriched hepatic neutral lipids
T5KO mice exhibit hallmark features of metabolic syndrome including hyperlipidemia. To investigate the extent to which serum hyperlipidemia is reflected in the liver, we analyzed liver triglycerides (TG), cholesterol esters (CE), free cholesterol (FC), phospholipids (PL), insulin sensitivity, and signaling in 20-week-old T5KO mice and their WT littermates. As observed in other facilities (Emory and Georgia State University), T5KO mice housed at PSU displayed higher body weights, abdominal adiposity, hyperglycemia (Fig 1A–C) with associated splenomegaly (Fig. S1A), and elevated colonic expression of pro-inflammatory genes [Lipocalin 2 (Lcn2), iNos, Pro-Il-1β and Kc] (Fig. S1B–E), indicates a low-grade chronic inflammatory state. Further, T5KO mice were hyperinsulinemic and hyperlipidemic (Fig 1D–F). T5KO mice displayed elevated hepatic neutral lipids TG and CE compared to WT littermates (Fig. 1G&H). There was no significant difference in the liver FC, but a significant decrease in PL levels in T5KO mice was observed (Fig. 1I&J respectively). A similar trend of elevated lipid parameters was observed in the female T5KO mice (data not shown).
Figure 1. Elevated oleate (C18:1)-enriched hepatic neutral lipids and insulin resistance in T5KO mice.

Age-matched T5KO male mice and their WT littermates (20-week-old, n=10–11) were monitored for A. Body weight, B. Fat pad, C. 15 h fasting blood glucose, D. Fasting serum insulin, E. Serum total cholesterol (TC) and F. Serum triglycerides (TG). Hepatic lipids were analyzed after 5 h of fasting. G. TG, H. Cholesterol esters (CE), I. Free cholesterol (FC) and J. Phospholipids (PL). Fatty acid composition of hepatic lipid fractions were analyzed by gas chromatography and represented as (%) C18:1 K. C18:1 in total lipid, L. C18:1 in CE, M. C18:1 in TG and N. C18:1 in PL. O. Immunoblot representing insulin-induced phosphorylation of Akt (Thr308) and total Akt in liver, soleus skeletal muscle and adipose tissue. P. Insulin sensitivity, Q. Liver total lipid oleate (%) levels were quantified in male T5KO and their WT littermates (n=6) at different age 3, 5, 8 and 12 weeks. Data are represented as mean ± SEM. *P<0.05.
Obesity in leptin-deficient mice is associated with elevated tissue MUFA levels and is SCD1 dependent (Sekiya et al., 2003). Similar to leptin-deficient mice, T5KO mice exhibited elevated plasma insulin, an anabolic hormone known to upregulate SCD1 (Jeffcoat et al., 1979). We therefore examined whether obese T5KO mice displayed similar MUFA-enriched lipids. Liver lipid analysis in T5KO mice showed an increase (25.0%) in C18:1 in the total liver lipids (Fig. 1K), mainly attributed to increased C18:1 in the CE (16.0%) and TG (9.0%) fractions (Fig. 1L&M). The C18:1 level in PL fraction was comparable between WT and T5KO mice (Fig. 1N). Interestingly, palmitoleate (C16:1), a minor product of SCD1, was comparable in the total lipid (although with large variation), TG, CE, and PL fractions of T5KO mice and WT littermates (Fig. S1 F–I). Notably, mRNA transcript of lipogenic genes in adipose and skeletal muscle remained unaltered in T5KO mice (Fig. S1 J–K). In general, female mice tend to have higher levels of MUFA in the liver when compared to males (Lee et al., 1996), but we did not observe such sex based differences (data not shown). In the total liver lipids, a decreasing trend in essential fatty acids (EFA) linoleic acid (C18:2 n6) and arachidonic acid (C20:4 n6) was observed but not significant (data not shown). Similarly, in the liver CE, but not the TG fraction, C18:2 levels were decreased in T5KO mice relative to WT littermates (data not shown). T5KO mice with elevated hepatic lipids displayed insulin resistance, as measured by phosphorylation of Akt, an insulin responsive protein, and whole body insulin sensitivity test (Fig. 1O&P). Similar to male, female T5KO mice also exhibited metabolic syndrome with hepatic lipid oleate enrichment (Fig. S2A–J).
Given that both diet and gut microbiota composition change drastically at the suckling-to-weaning transition, we next asked whether the elevated C18:1 in hepatic lipids of T5KO mice is present beginning from birth or occurs after weaning. At the age of 3 weeks mice were weaned on chow diet and liver total C18:1 measured at 3, 5, 8 and 12 weeks of age. At the time of weaning, both WT and T5KO mice displayed similar levels of C18:1 in liver, which increased within 2 weeks of weaning in both WT and T5KO mice (Fig. 1Q). However, the percent increase in C18:1 in T5KO mice was significantly higher than in WT littermates. In addition to C18:1, liver total TG levels increased with age in T5KO mice (data not shown). Thus, the elevated C18:1 in the liver is an acquired characteristic after weaning perhaps due to the dietary switch from suckling to lab chow. In contrast to leptin-deficient mice, the elevated C18:1 was not seen in either serum or adipose tissue, while fecal lipids of T5KO mice are comparable to WT littermates (Table S2).
Upregulation of hepatic lipogenic, pro-inflammatory, and insulin resistance genes in T5KO mice
Excessive hepatic lipogenesis may lead to liver inflammation and contribute to hepatic insulin resistance. Therefore, we next sought to determine whether increased lipid deposition associated with altered hepatic gene expression. Notably, the hepatic mRNA transcripts of lipogenic genes [sterol regulatory element binding protein 1c (Srebp1c), acetyl CoA carboxylase (Acc), fatty acid synthase (Fas), Scd1, 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase (Hmgcr), and HMG-CoA synthase (Hmgcs)] and enzymes responsible for the esterification of MUFA to cholesterol and TG via acyl CoA: cholesterol acyl transferase 2 (Acat2) and diacylglycerol acyl transferase 1 and 2 (Dgat1 & 2) were elevated in T5KO mice (Fig. 2A–D & F–J), including SCD1 at the protein level (Fig 2E). Interestingly, mRNA transcripts of key inflammatory cytokines Tnfα, Lcn2, and chemokine (C-X-C motif) ligand 1 (Kc) were augmented in T5KO mice (Fig. 2K–M). Further, the T5KO mice also showed increased expression of genes associated with insulin resistance such as Forkhead box protein O1 (Foxo1), peroxisome proliferator-activated receptor gamma coactivator 1 alpha (Pgc1α) transcription coactivator, and gluconeogenic enzyme phosphoenolpyruvate carboxykinase (Pepck) (Fig. 2N–P). Collectively, these results suggest that the elevated C18:1 in T5KO mice is a result of increased upregulation of a spectrum of lipogenic genes associated with markers of inflammation and insulin resistance. In accordance with unaltered fatty acid composition of adipose tissue in WT and T5KO mice, we did not observe any significant differences in the transcripts of above lipogenic genes in adipose tissue and skeletal muscles of these mice (Fig. S1 J&K).
Figure 2. Enhanced expression of hepatic lipogenic enzymes, pro-inflammatory, and insulin resistance genes in T5KO mice.

Hepatic expression of lipogenic enzymes, inflammatory, and insulin resistance marker genes were analyzed in male T5KO mice and their WT littermates (20-week-old, n=5). A. Sterol regulatory element binding protein 1c (Srebp1c), B. Acetyl CoA carboxylase (Acc), C. Fatty acid synthase (Fas), D. Stearoyl-CoA desaturase-1 (Scd1), E. Immunoblot for SCD1, F. Acyl CoA:cholesterol acyltransferase 2 (Acat2), G. Diacylglycerol acyltransferase 1 (Dgat1), H. Dgat2, I. 3-hydroxy-3-methyl-glutaryl-CoA synthase (Hmgcs) J. HMG- CoA reductase (Hmgcr). K. Tumor necrosis factor alpha (Tnfα), L. Lipocalin2 (Lcn2), M. Keratinocyte-derived chemokine (Kc), N. Forkhead box protein O1 (Foxo1), O. Peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (Pgc-1α), and P. Phosphoenolpyruvate carboxykinase (Pepck). Data are represented as mean ± SEM. *P<0.05.
HFD-fed and calorie-restricted T5KO mice display elevated MUFA-enriched hepatic neutral lipids
Diets rich in saturated fat are a risk factor for insulin resistance, obesity, and hepatic steatosis. Accordingly, we demonstrated exacerbated metabolic syndrome and hepatic steatosis in T5KO mice fed HFD (Vijay-Kumar et al., 2010). Because of the substantial reduction in plant-derived fiber in HFD, we envisioned that such diets might influence hepatic lipids. After 8 weeks on a HFD, T5KO mice displayed hyperglycemia and insulin resistance (Fig. 3A–C) with concomitant increases in serum TC and TG (Fig. 3D&E). Moreover, elevated liver TG, total C18:1 and C18:1 in the TG fraction (Fig. 3F–H) were observed in comparison to HFD-fed WT mice. Interestingly, the HFD did not increase C18:1 in the CE fraction of liver lipids in HFD-fed T5KO mice (Fig. 3I) as was observed in T5KO mice fed lab chow. Another major difference observed with the HFD was a reduction in total lipid C18:2 in T5KO mice (Fig. 3J). A decreasing trend in C20:4 in the liver total lipid fraction of T5KO mice was observed in comparison to WT littermates (Fig. 3K), suggesting altered delta-6 desaturase activity. Additionally, mRNA transcripts of hepatic Fas, Scd1, Hmgcr, Foxo1 and Lcn2 were elevated in HFD-fed T5KO mice (Fig. 3L). There were no significant differences in the transcript levels of lipogenic genes in adipose and skeletal muscles of HFD-fed T5KO mice (Fig. S3A&B). Colonic expression of SCFA receptors and pro-inflammatory genes except iNos were also comparable to WT mice (Fig. S3C). Similar results were obtained with HFD-fed male T5KO mice (data not shown).
Figure 3. HFD-fed and calorie restricted T5KO mice exhibit elevated oleic acid in hepatic neutral lipids.

Age matched female T5KO mice and their WT littermates (12-week-old, n=5) maintained on a HFD for 8 weeks and 5 h fasting serum and liver were analyzed for following parameters. A. Blood glucose, B. Insulin sensitivity test, C. Fasting serum insulin, D. Serum TC and E. Serum TG. F. Liver TG, G. C18:1 in total lipid, H. C18:1in TG, I. C18:1 in CE, J. Total C18:2, and K. Total C20:4. L. mRNA expression of hepatic lipogenic, inflammatory, and insulin resistance genes in HFD fed WT and T5KO mice. Age-matched female T5KO mice and their WT littermates (n=5) were subjected to calorie restriction for 12 weeks and hepatic TG and C18:1 were analyzed after 5 h of fasting. M. Liver TG, N. C18:1 in total lipid, O. C18:1 in CE and P. C18:1 in TG. Data are represented as mean ± SEM. *P<0.05.
Metabolic syndrome in T5KO mice is largely driven by hyperphagia; calorie restriction for 12 weeks normalized most of the indices of metabolic syndrome in T5KO mice except insulin resistance (Vijay-Kumar et al., 2010). Analysis of liver lipids and fatty acid profiles of calorie-restricted WT and T5KO mice exhibited an increase in liver TG and liver total C18:1 mainly in TG and CE fractions (Fig. 3M–P) of T5KO mice in comparison to WT, suggesting that an increase in hepatic lipids might be contributing to insulin resistance. Further, the PUFA C18:2 and C20:4 levels showed a decreasing trend in the liver total lipids, TG and CE fractions as observed in ad libitum fed mice (data not shown).
Oleate enrichment in hepatic lipids of T5KO mice is microbiota-dependent
TLR5 is predominantly expressed on gut epithelial cells and plays a key role in microbiota homeostasis (Vijay-Kumar et al., 2007). Accordingly, T5KO mice exhibit three-fold higher fecal bacterial load (data not shown). To demonstrate the role of the microbiota in liver lipogenesis, we eliminated ~90% of bacteria via antibiotics. Microbiota ablation ameliorated the majority of symptoms of metabolic syndrome in T5KO mice, and moreover, normalized hepatic TG and C18:1 in total lipid, TG and CE fractions (Fig. 4A–D).
Figure 4. Hepatic neutral lipid enrichment of C18:1 in T5KO mice is microbiota dependent.

Age matched male T5KO mice and their WT littermates (4-week-old, n=5) were treated with broad-spectrum antibiotics (ampicillin and neomycin) in drinking water for 8 weeks and analyzed for liver lipids. A. Liver TG, B. C18:1 in total lipid, C. C18:1 in TG, D. C18:1 in CE. Hepatic lipids were analyzed in 12-week-old males (age and sex matched) GF-T5KO mice and their WT littermates (n=7). E. Liver TG, F. C18:1 in total lipid, G. C18:1 in TG, H. C18:1 in CE and, I. Immunoblot for hepatic SCD1 in conventional (conv), antibiotics treated and GF-T5KO mice and their WT littermates. Four-week-old male GF (Swiss Webster) WT mice (n=5) were orally administered cecal microbiota from either WT or T5KO mice and housed in specific pathogen–free conditions. Hepatic C18:1 in lipid fractions was analyzed 6 weeks after transplantation. J. C18:1 in total lipid, K. C18:1 in TG, and L. C18:1 in CE. Data are represented as mean ± SEM. *P<0.05.
These observations were further confirmed in GF-T5KO mice and their WT littermates as GF-T5KO mice did not exhibit either symptoms of colitis or indices of metabolic syndrome (Fig. S4A–E). Notably, hepatic TG, total C18:1 and C18:1 in TG and CE fractions in GF-T5KO mice were comparable to WT littermates (Fig. 4E–H). Interestingly, the levels of C18:1 (in both strains) was substantially reduced (50%) with concomitant reduction of hepatic SCD1 expression in microbiota-ablated and GF mice when compared to conventional mice (Fig. 4I). These results suggest that the microbiota or its metabolites likely play a key role in de novo synthesis of hepatic MUFA.
In a similar line, we asked whether the cecal microbiota of T5KO mice is sufficient to modulate hepatic lipogenesis upon transplant to GF-WT recipients. Cecal microbiota transplantation influenced hepatic lipid MUFA levels in GF-WT mice that received either WT or T5KO cecal microbiota. As shown in Fig. 4J–L, total lipid, TG, and CE C18:1 levels in GF-WT mice that received T5KO cecal microbiota were significantly higher than the WT cecal microbiota recipients. As observed in conventional mice, transplantation of cecal microbiota resulted in a greater than 50% increase in C18:1 level in GF-WT recipients. However, this effect was more profound in T5KO mice cecal recipients, suggesting that the T5KO microbiota is sufficient to induce hepatic neutral lipid C18:1enrichment (Fig. 4J–L).
The increased hepatic MUFA in T5KO mice is associated with elevated cecal SCFA
Gut microbiota plays a key role in host energy homeostasis by generating SCFA via fermentation of dietary fiber which serves as substrate for lipid biosynthesis (den Besten et al., 2013a). The SCFA are utilized by colonocytes or absorbed efficiently in the colon (den Besten et al., 2013b). Therefore, we performed metabolic profiling of intestinal contents by 1H NMR. As shown in Fig. 5A–E, cecal contents of T5KO mice displayed increased SCFA, specifically butyrate and propionate. Similar results were observed in calorie restricted T5KO mice (data not shown). Surprisingly, there was no significant difference observed in the major luminal SCFA acetate in the cecal contents of both strains. As expected, fecal metabolites by 1H NMR and fatty acid analysis by GC did not reveal any significant differences in WT and T5KO mice (Fig. S5A and Table S2), indicating SCFA produced in ceca were absorbed in the colon. In support of this notion we observed elevated colonic mRNA transcripts of SCFA receptors [G protein coupled receptor (Gpr)41 and 43; Fig. 5F] and increased SCFA (propionate and butyrate) in the hepatic portal vein serum of T5KO mice (Fig. 5G). However, no change in Gpr109A and Na+-coupled transporter of SCFA Slc5a8 was observed between WT and T5KO mice (Fig. S5B&C). These results suggest that T5KO mice, which have an increased microbial burden, utilize more oligosaccharides and generate higher levels of SCFA. To extend our demonstration how increased availability of SCFA to liver affects de novo lipogenesis (DNL), we fed 13C-acetate to WT and T5KO mice to measure label incorporation into liver and plasma lipids. The feeding achieved an overall enrichment of the C16:0-McLafferty ion+1 (m/Z=75) of 1.17% ± 0.07 above background. Under these conditions, the proportion of label appearing in C16:0 was increased in T5KO mice (Fig. 5H&I) indicating SCFA contributes a greater fraction of TG in both the liver and plasma pools. This is apparent in serum TG of T5KO mice (Fig. 5I), where increased serum TG-C16:0 results from an increased utilization of SCFA above that observed in WT. This indicates that the increased delivery of SCFA to hepatocytes combines with the observed increase in synthetic capacity (Fig. 2) to result in increased DNL in T5KO mice.
Figure 5. Metabolic profiles of cecal contents are altered in T5KO mice.

Typical 600 MHz 1H NMR spectra of cecal contents from male A. T5KO, and B. WT mice (20-week-old, n=10–11). C. OPLS-DA score plot (left) and correlation coefficient loading plot (right) showing the discrimination between 1H NMR spectra of cecal contents from T5KO and WT mice, respectively (|r| cutoff value is 0.602, n=10 for WT group and n=11 T5KO mice group; C. R2X=0.56, Q2=0.58; CV-ANOVA: P = 1.89 × 10−3). Keys: 1, n-butyrate; 2, propionate; 3, isoleucine; 4, leucine; 5, valine; 6, ethanol; 7, lactate; 8, alanine; 9, lysine; 10, acetate; 11, proline; 12, bile acids; 13, succinate; 14, pyruvate; 15, trimethylamine (TMA); 16, creatine; 17, α-ketoglutarate; 18, methanol; 19, oligosaccharides & amino acids; 20, α-glucose; 21, raffinose; 22, stachyose; 23, uracil; 24, taurine; 25, glycine; 26, uridine; 27, fumarate; 28, tyrosine; 29, choline; 30, methionine; 31, phenylalanine; 32, histidine; 33, urocanate; 34, adenine; 35, adenosine; 36, nicotinurate. Relative abundance of D. SCFA (propionate & butyrate) & E. oligosaccharides, glucose, raffinose and stachyose in cecal content extracts from WT and T5KO mice. F. Colonic expression of SCFA receptor in T5KO and their WT littermates (n=10). G. SCFA concentration in hepatic portal vein serum (n=6–8). H–I) 13C-acetate label in animals fed 13C-sodium acetate (0.3 M) in drinking water for 4 days (n=5), H). Fractional enrichment of C16:0 using C16:0 as the primary end product of DNL and the fractional enrichment as an estimate of fractional synthesis; I. Absolute concentration of C16:0 in liver and plasma TG. Data are represented as mean ± SEM. *P<0.05.
Dietary SCFA upregulate hepatic SCD1 and aggravate metabolic syndrome in T5KO mice
Dietary SCFA can regulate gut inflammation (Maslowski et al., 2009; Smith et al., 2013). SCFA produced in the cecum partake in hepatic lipogenesis in both mice and human (den Besten et al., 2013a; Wolever et al., 1989). Therefore, we next asked whether dietary SCFA would protect from low-grade inflammation or promote metabolic syndrome in T5KO mice. We administered a combination of acetate, butyrate, and propionate in drinking water for 21 days, as previously described (Smith et al., 2013) and observed that T5KO mice on SCFA treatment exhibited further increase in body weight, fat pad, blood glucose and insulin resistance (Fig. 6A–D) compared to vehicle treated T5KO mice. In agreement with a recent report (Frost et al., 2014), we observed a significant reduction in food intake in both WT and T5KO mice receiving dietary SCFA (Fig. 6F). Dietary SCFA further elevated serum TC and TG in T5KO mice (Fig. 6G–H). Interestingly, no significant differences were observed in the above parameters in WT mice with or without dietary SCFA (Fig. 6A–H) except reduced food intake. Taken together, these results suggest that SCFA participate in hepatic lipogenesis and contributing to insulin resistance in T5KO mice.
Figure 6. Dietary SCFA aggravates metabolic syndrome in T5KO mice.

Age matched male T5KO mice and their WT littermates (16-week-old, n=6) received either a mixture of SCFA (67.5mM sodium acetate, 40mM sodium butyrate and 25.9mM sodium propionate) or equimolar sodium chloride in drinking water for 21 days. A. Percent increase in body weight, B. Fat pad, C. 15 h fasting blood glucose, D. Insulin sensitivity test, E. 5 h fasting serum insulin, F. Food intake, G. Serum TC, H. Serum TG. Hepatic lipids were analyzed in 5 h fasted mice. I. TG, J. CE, K. FC, L. Immunoblot represents hepatic SCD1, M. C18:1 in total lipid, N. C18:1 in CE, O. C18:1 in TG and P. mRNA expression of hepatic lipogenic, inflammatory and insulin resistance genes. Data are represented as mean ± SEM. *P<0.05.
We next analyzed how SCFA supplementation impacted the liver lipids. Similar to data from the serum, T5KO mice fed SCFA showed an increase in liver TG and CE except liver FC (Fig. 6I–K) compared to vehicle treated T5KO mice. Interestingly, dietary SCFA further increased liver SCD1 in T5KO mice (Fig. 6L). Serum insulin remains unaltered in SCFA-fed T5KO mice (Fig. 6E), suggesting that the increased hepatic SCD1 in these mice could be due to more SCFA availability as substrate. Further, dietary SCFA increased hepatic total C18:1, TG C18:1 and CE C18:1 in T5KO mice when compared vehicle given mice (Fig 6M–O). Hepatic palmitoleate (C16:1) was also elevated in T5KO mice that received SCFA (2.36 ± 0.19, T5KO vs 2.91 ± 0.1, T5KO+SCFA). However, no change in adipose tissue fatty acid composition was observed in T5KO mice given SCFA (data not shown). Expression of hepatic inflammatory genes and lipogenic genes in liver (Fig. 6P), adipose and skeletal muscles (Fig. S6A&B) were unaltered in SCFA-fed T5KO mice. Moreover, colonic expression of SCFA receptors/transporters and pro-inflammatory genes also remained unaltered, with the exception of Lcn2, which is reduced, in T5KO mice fed SCFA (Fig. S6C). Additionally, colonic MPO activity (Fig. S6D) and gut bacterial load (Fig. S6E) remained unchanged in T5KO mice fed with SCFA.
Hepatic stearoyl-CoA desaturase 1 deficiency improves metabolic syndrome in T5KO mice
T5KO mice displayed substantial upregulation of hepatic SCD1 as well as increased presence of its product C18:1 in the hepatic lipid, suggesting its potential involvement in metabolic syndrome. Therefore, to investigate the role of hepatic SCD1, we generated liver specific Scd1 deficient T5KO mice (T5KO-Scd1ΔHep) and monitored for indices of metabolic syndrome in comparison to Scd1 floxed T5KO mice (T5KO-Scd1fl/fl). Most notably, T5KO-Scd1ΔHep mice exhibited reduced body weight and abdominal adiposity coupled with improved insulin resistance when compared to T5KO-Scd1fl/fl mice (Fig. 7A–D). Even though serum insulin, TC, and TG levels were unaltered, hepatic TGs and CEs were reduced in T5KO-Scd1ΔHep (Fig. 7E–I). However, there was no change in hepatic FC (Fig. 7J), indicating that non-hepatic SCD1 activity might be able to maintain serum lipids. As expected, liver total, TG and CE C18:1 level was substantially reduced (Fig. 7K–M) with a concomitant increase in total hepatic C18:0 and total C18:2 in T5KO-Scd1ΔHep (Fig. 7N&O). Moreover hepatic palmitoleate (C16:1), was reduced in T5KO-Scd1ΔHep mice (2.27 ± 0.07, T5KO-Scd1fl/fl vs 1.96 ± 0.09, T5KO-SCDΔHep). The hepatic transcripts of pro-inflammatory genes and the marker of insulin resistance Foxo1 were reduced in T5KO-Scd1ΔHep mice (Fig. 7P). Additionally, protein level of GLUT4, an insulin responsive gene, was augmented in skeletal muscle of T5KO-Scd1ΔHep mice (Fig. S7A), suggesting that liver Scd1 deletion in T5KO mice is able to rescue the skeletal muscle insulin resistance. However, GLUT4 expression was unaltered in adipose tissue of T5KO-SCDΔHep in comparison to T5KO-Scd1fl/fl mice (data not shown). Moreover, expression of lipogenic genes remained unaltered in liver, adipose and skeletal muscle of T5KO-Scd1ΔHep mice (Fig. 7P, S7B&C respectively). Similarly, the expression of colonic SCFA transporters and pro-inflammatory genes along with colonic MPO activity, food intake, and gut bacterial load remained unaltered in T5KO-Scd1ΔHep mice when compared to T5KO-Scd1fl/fl mice (Fig. S7D–G). As expected, C18:1 level was reduced in hepatic total and lipid fractions in WT-Scd1ΔHep mice when compared to WT-Scd1fl/fl. However, we did not observe any significant change in the metabolic syndrome-related parameters between these groups (Fig. 7A–P). Altogether, our results suggest that loss of TLR5 results in a microbiota-generated increase in SCFA. In turn, the increase in SCFA may drive hepatic SCD1-mediated lipogenesis, consequently promoting insulin resistance and inflammation in T5KO mice.
Figure 7. Deletion of hepatic Scd1 in T5KO mice protects against metabolic syndrome.

Liver Scd1 deficient WT (WT-Scd1ΔHep), T5KO (T5KO-Scd1ΔHep) and control (WT-Scd1fl/fl & T5KO-Scd1fl/fl) mice (male, n=4–6) were maintained on lab chow for 20 weeks A. Immunoblot (upper panel) and bar graph representing the hepatic expression of SCD1 at protein and mRNA level respectively, B. Body weight, C. Percent fat pad D. Insulin sensitivity test, E. Fasting serum insulin, F. Serum TC and G. Serum TG. Hepatic lipids were analyzed in 5 h fasted mice. H. Liver TG, I. CE, J. FC, K. Total C18:1, L. TG C18:1, M. CE C18:1, N. Total C18:0, O. Total C18:2 and P. mRNA expression of hepatic lipogenic, inflammatory and insulin resistance genes. Data are represented as mean ± SEM. *P<0.05.
Discussion
Gut microbiota is known to play a key role in the host energy homeostasis and is thus considered an environmental risk factor for metabolic diseases. Accordingly, mice deficient in TLR5 develop an altered microbiota that promotes metabolic syndrome. Herein, we report a potential molecular mechanism involving elevated C18:1 hepatic neutral lipids underlying the development of metabolic syndrome in T5KO mice. Such elevated hepatic C18:1 levels likely result, at least in part, from augmented SCD1 expression. We further observed that hepatic enrichment of oleate is microbiota-dependent.
The cecum is a transient reservoir for digested food and therefore contains the majority of complex dietary fiber that undergoes anaerobic bacterial fermentation which generates acetate, propionate, and butyrate in molar ratio of 60:20:20 (Cummings et al., 1979). Recent studies demonstrate a direct link of obesogenic dysbiosis i.e. altered Firmicutes/Bacteroidetes ratio with metabolic syndrome in both mice and humans (Ley et al., 2006; Turnbaugh et al., 2006). We observed altered species primarily in Firmicutes and Bacteroidetes phyla in T5KO mice when compared to their WT littermates, but relative abundance of these phyla was unaltered (Vijay-Kumar et al., 2010), suggesting that elevated gut microbiota along with altered bacterial species might contribute to increased cecal SCFA generation in T5KO mice. These SCFA not only serve as a fuel for intestinal epithelia (specifically butyrate) (Blottiere et al., 2003), but also contribute additional sources of energy to the host (den Besten et al., 2013b). A recent study using stable isotopes of SCFA demonstrated that acetate and butyrate are lipogenic whereas propionate is gluconeogenic (den Besten et al., 2013a) in mice. Specifically, this study showed that cecal acetate and butyrate contribute to synthesis of hepatic palmitate and cholesterol. In a similar vein, 1H NMR-based metabolic profiling of cecal content showed considerably elevated levels of butyrate and propionate accompanied with a decreasing trend of oligosaccharides in T5KO mice. Increased expression of colonic SCFA receptors, particularly Gpr41 and Gpr43, might affords efficient absorption of these SCFA and promoting the inflammatory response via MAP kinase signaling in T5KO mice (Kim et al., 2013). Acetate levels in cecal contents of T5KO mice did not differ significantly in comparison to WT, suggesting its rapid conversion into butyrate by the gut bacteria (den Besten et al., 2013a). Previously, we showed no difference in either energy content in feces (bomb calorimetry) or SCFA levels in the cecal contents of T5KO mice and their WT littermates via GC-MS (Vijay-Kumar et al., 2010). The discrepancy could be due to differences in collection of samples from previous studies, including (i) age of the mice, (ii) ad libitum fed mice, (iii) use of wet rather than dry stools, or (iv) differences in processing of samples for analysis with 1H NMR. In this study using a metabolomic approach, we found that T5KO mice with increased microbiotal load generated substantial levels of SCFA in the cecum. This is transferred to the liver and contributes to increased hepatic DNL. Notably, the relative contribution of acetate to lipogenesis was more in T5KO mice. It is likely that the gut-derived SCFA drive increased lipogenesis by provision of greater amounts of substrate for the elevated lipid synthetic machinery in T5KO mice.
Several studies have demonstrated that SFA C16:0 and C18:0, dietary or end products of DNL, are lipotoxic (Nolan and Larter, 2009; Shi et al., 2006). Increased SCFA serve as substrates for SFA synthesis which are converted into less lipotoxic MUFA via SCD1 (Miller et al., 2005) and plays a role in the development of obesity (Poudyal and Brown, 2011). Notably hepatic SCD1 level is substantially reduced in GF and microbiota ablated WT and T5KO mice, indicating that liver SCD1 is partly regulated via gut microbial metabolites. Accordingly, our results indicate that dietary SCFA aggravates the metabolic syndrome phenotype in T5KO mice probably via overproduction of SFA from SCFA.
The conversion of SFA into MUFA is known to prevent negative feedback inhibition of fatty acids (palmitate) on ACC (Ogiwara et al., 1978). The blockade of SFA removal as MUFA in Scd1KO mice underlies its lean phenotype. Although T5KO mice exhibited elevated C18:1-enriched hepatic neutral lipids, those lipid species were not enriched with palmitoleate (C16:1 n7), a minor product of SCD1. It is interesting to note that C16:1 has been shown to act as a lipokine, strongly induces muscle insulin action, and suppresses hepatic lipogenesis in mice (Cao et al., 2008). In addition, adipocyte-derived oleate (18:1 n9), but not palmitoleate (16:1 n7), mediates inflammation in macrophages and adhesion responses in endothelial cells (Liu et al., 2010).
Deletion of SCD1 or supplementing leptin to Ob/Ob mice ameliorates obesity (Cohen et al., 2002). Interestingly, T5KO mice exhibited upregulation of a spectrum of hepatic lipogenic enzymes, including SCD1 even though they are hyperleptinemic (Vijay-Kumar et al., 2010), suggesting they may be in a state of leptin resistance. Elevated hepatic SCD1 in T5KO mice may be driven by increase in: (i) stability of this protein; (ii) insulin level; (iii) precursor (e.g., SCFA) availability (Alberts et al., 1974; Jeffcoat et al., 1979; Mziaut et al., 2000); (iv) low-grade hepatic inflammation; and (v) Srebp1c, a transcription factor known to stimulate lipogenic genes including Scd1 in hepatocytes. Accordingly, we demonstrated that deletion of hepatic Scd1 prevented accumulation of C18:1 and improved metabolic syndrome in T5KO mice. Taken together these results reinforce the concept that the microbiota-generated SCFA contribute, at least in part, to hepatic lipogenesis in T5KO mice. MUFA generated via SCD1 activity, but not dietary MUFA, function as endogenous fatty acid amide hydrolase (FAAH) inhibitor and thereby increases hepatic arachidonoylethanolamide (AEA) which induces insulin resistance by activating liver endocannabinoid/CB1 receptor (Liu et al., 2013). Endogenous MUFA, synthesized by SCD1, but not dietary MUFA is required for hepatic TG synthesis as oleate enriched diet failed to restore hepatic TG level in global Scd1 deficient mice (Miyazaki et al., 2001). These observations support the notion that there is a difference in the metabolic fate of endogenously-produced vs. ingested metabolites. HFD aggravated metabolic syndrome and caused insulitis and hepatic steatosis in T5KO mice. Surprisingly HFD-fed T5KO mice displayed modest increase in C18:1, this could reflect a reduced supply of SCFA as the HFD lacks plant-derived complex carbohydrates. However, the HFD used in this study is rich in palmitate (26%) and oleate (44%), which together may be enough to maintain C18:1 level on a higher side in T5KO mice. A recent study by Wichmann et al. demonstrated that total SCFA in cecal contents is greatly reduced (>50%) in HFD–fed mice compared to chow-fed mice (Wichmann et al., 2013). Although there was a substantial reduction in EFA C18:2 and C20:4 in PL fractions of HFD-fed T5KO mice, they were far from EFA deficiency, as indicated by no detectable levels of eicosatrienoic acid (C20:3 n9), a surrogate marker of EFA deficiency.
Altogether, metabolic syndrome in T5KO mice seems to be orchestrated by increased fermentation and low-grade chronic inflammation with elevated MUFA in hepatic neutral lipids. Abrogation of spontaneous inflammation and metabolic syndrome in GF T5KO mice indicates that the microbiota is involved in both of these processes. Previous studies elegantly demonstrate that dietary SCFA prevent obesity and associated metabolic abnormalities by suppressing hepatic lipogenesis, fat accumulation and reducing appetite in mice (den Besten et al., 2015; Frost et al., 2014; Kimura et al., 2013) and improve colonic inflammation (Arpaia et al., 2013; Furusawa et al., 2013; Singh et al., 2014; Smith et al., 2013). Conversely, elevated fecal SCFA correlate with obesity metabolic syndrome factors in humans (Fernandes et al., 2014; Teixeira et al., 2013). Similar to our finding that SCFA aggravate metabolic syndrome in T5KO mice, long term adaptation of serum lipid responses occur in type 2 diabetic subjects receiving fiber rich diet; serum acetate correlates with plasma TG and cholesterol in these subjects (Wolever et al., 2002). In the present study, we observed that continuous over-production of SCFA by gut bacterial fermentation contribute in hepatic lipid accumulation and thus metabolic syndrome in mice with elevated gut bacterial load, low grade chronic inflammation and, augmented expression of hepatic lipogenic genes. According to the available literature, SCFA are beneficial in metabolic syndrome, but in certain context where uncontrolled, prolonged generation of SCFA due to increased microbiotal load as in T5KO mice may have detrimental effects. Herein, we envisage that this prolonged overproduction of SCFA in T5KO mice may lead to “metabolic adaptation” as observed with elevated hepatic SCD1, to convert them into lipid species.
Although our favored interpretation of our data is that elevated gut microbial metabolites (i.e. SCFA) and low-grade chronic inflammation promote augmented hepatic lipogenesis and thus metabolic syndrome of T5KO mice, alternative mechanisms are possible. Notably, we cannot exclude the important role of selective hepatic insulin resistance and elevated serum insulin which are shown to promote lipogenesis in liver (Biddinger et al., 2008) in driving the metabolic syndrome in T5KO mice. In any case, the microbiotal-hepatic axis may not account for all the reasons for which T5KO mice develop metabolic syndrome and there is a possibility that other yet unknown processes/pathways (e.g. leptin resistance), which are not examined by our study design, may also contribute to the observed metabolic syndrome in T5KO mice, the relative extent of contribution by the microbiotal-hepatic axis to the observed metabolic syndrome will remain unknown unless and until these other processes/pathways either within the liver or elsewhere are deciphered. Present study highlights the role of the gut microbiota–liver axis in driving metabolic diseases, specifically in conditions with innate immune deficiency coupled with microbiotal overload and susceptibility to spontaneous inflammation. Collectively our results suggest that balanced pharmacologic manipulation of hepatic SCD1 (Brown and Rudel, 2010) may be useful in the treatment of metabolic diseases with a caution to avoid secondary accumulation of lipotoxic SFA.
Experimental Procedures
All reagents and detailed methods are described in supplementary file.
Mice, diet, treatments and metabolic parameter analysis
T5KO mice and their WT littermates were maintained in the specific pathogen–free condition in Emory, Georgia State and Pennsylvania State Universities. Mice were maintained on lab chow (Lab Diets 5001) or HFD (D12492, Research Diets Inc.) or on calorie restriction.
Serum lipids were monitored in 5 h fasted serum using commercial kits (Randox Laboratories). All lipid and fatty acid quantification were performed at mouse metabolic phenotyping centers (MMPC) lipid laboratory, Vanderbilt University. Serum insulin was quantified by an ELISA kit from EMD Millipore. To measure insulin sensitivity, insulin was injected (0.25 units/kg body weight, i.p.) in 5 h fasted mice and then blood glucose was monitored at 0, 30, 60, 90, and 120 min after the insulin injection (Vijay-Kumar et al., 2010).
Quantitative RT-PCR
Total RNA was isolated from tissues using Tri-reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized by cDNA synthesis kit (Superscript VILO, Invitrogen). Real time PCR was performed using Step One Plus Real-Time PCR System (Applied Biosystems) in a reaction mixture of cDNA, SYBR green master mix and mouse specific oligonucleotides (Table. S1).
Immunobloting
Tissues (50 mg) were homogenized in 0.5ml RIPA buffer (Cell Signaling) with protease inhibitors (Roche) and fractionated on SDS-PAGE and then transferred to PVDF membrane. After blocking with non-fat milk (5%) blots were incubated overnight at 4°C with anti-mouse SCD-1, Akt, pAktThr308 or GLUT4. The blots were developed by chemiluminiscent reagent. β-actin and GAPDH was used as a loading control.
Transplantation of cecal microbiota into germ-free (GF) WT mice
Microbiota transfer was performed as described previously (Vijay-Kumar et al., 2010). Briefly, cecal contents from age and sex matched T5KO mice and their WT littermates were suspended in 2.5ml PBS (at 37°C) and administered (0.1ml/mouse) to male GF-WT mice.
SCFA estimation
Hepatic portal serum was collected from age and sex matched T5KO mice and their WT littermates and its SCFA content was quantified as in ref. (Zheng et al., 2013). In brief, serum was mixed with NaOH (0.005M) containing internal standard (caproic acid-d3, 5 ug/mL). A mixture of 1-propanol: pyridine (3:2, v/v) and propyl chlorformate were subsequently added to the glass tube. After derivatization, at 60°C for 75 minutes, the derivatives were extracted with hexane. The upper hexane layer was transferred to a autosampler vial and SCFA was measured using an Agilent 7890A GC-MS spectrometer (Agilent Instruments, USA).
NMR Spectroscopy
Mice cecal and fecal contents were homogenized and extracted in PBS containing 50% D2O, 0.002% (w/v) of sodium 3-(trimethylsilyl) propionate-2,2,3,3-d4 (TSP) as described previously (Wu et al., 2010). All 1H NMR spectra of sample extracts were recorded at 298 K using a Bruker Avance III 600 MHz NMR spectrometer. Multivariate data analysis was carried out with the SIMCAP+ software (version 13.0, Umetrics, Sweden).
Analysis of 13C label acetate in fatty acids
Male T5KO and their WT littermates were fed with 13C sodium acetate (0.3 M, in drinking water) upto 4 days. Plasma and liver fatty acids were extracted and methylated using a modification of Morrison et al (Morrison and Smith, 1964) and quantified by through GC-MS (Shimadzu, Japan). Tri-heneicosanoin was used as a surrogate in plasma samples to adjust for extraction efficiency. To detect 13C enrichment in palmitic acid (C16:0) area counts were obtained for the McLafferty ion (MCI) and the MCI+1 (m/z =74 and 75 respectively) (Sharkey et al., 1959) using GC MS solution software (Shimadzu Corporation, Japan).
Supplementary Material
Acknowledgments
M.V-K. is supported by NIH grants K01, (DK083275), R03 (DK094864), R01 (DK097865-01A1), A.T.G by DK061417 and DK083890, and BC by Crohn’s and Colitis Foundation of America (CCFA) Research Fellowship award. B.S.Y. is supported by NIH grant T32AI07445. A.D.P. is supported by NIH grant ES022186 and grant with the Pennsylvania Department of Health using Tobacco CURE Funds. B.J. is supported by NIH grants R01HL11264 and R01HL020176. We thank the Mouse Metabolic Phenotyping Center (MMPC)/DTRC Lipid laboratory (DK59637), Vanderbilt University, Nashville, TN. We thank Philip Smith, Director, Penn State Metabolomics Core Facility. We thank Jesse Aitken, Gayathri Srinivasan, Alyssa Grube, Jackie Yun Ying and Rodrigo Aguilera for technical assistance.
Abbreviations
- CE
Cholesterol ester
- FC
Free Cholesterol
- PL
Phospholipid
- TLR5
toll-like receptor 5
- SCD1
Stearoyl CoA Desaturase 1
- SCFA
Short chain fatty acids
- FAS
Fatty acid synthase
- EFA
Essential fatty acid
- DNL
De novo lipogenesis
Footnotes
The authors have no conflict of interest.
Author contributions:
V.S., B.C., M.V-K. designed the study and wrote the manuscript. L.Z. performed NMR metabolomics and analyzed the data. V.S., B.S.Y., X.X., M.K., M.T.B. performed in vivo experimental work and dietary studies. J.C. performed SCFA analysis by GC-MS and analyzed the data. R.W., K.B., K.H., N.S., G.C.S. performed acetate labeling and lipid analysis. J.M.N., B.J., A.D.P., A.T.G. supported the work with key suggestions, provided valuable feedback on the manuscript and participated in discussion and editing. M.V-K. directed the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberts AW, Ferguson K, Hennessy S, Vagelos PR. Regulation of lipid synthesis in cultured animal cells. J Biol Chem. 1974;249:5241–5249. [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biddinger SB, Hernandez-Ono A, Rask-Madsen C, Haas JT, Aleman JO, Suzuki R, Scapa EF, Agarwal C, Carey MC, Stephanopoulos G, et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008;7:125–134. doi: 10.1016/j.cmet.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blottiere HM, Buecher B, Galmiche JP, Cherbut C. Molecular analysis of the effect of short-chain fatty acids on intestinal cell proliferation. Proc Nutr Soc. 2003;62:101–106. doi: 10.1079/PNS2002215. [DOI] [PubMed] [Google Scholar]
- Brown JM, Rudel LL. Stearoyl-coenzyme A desaturase 1 inhibition and the metabolic syndrome: considerations for future drug discovery. Curr Opin Lipidol. 2010;21:192–197. doi: 10.1097/MOL.0b013e32833854ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho FA, Koren O, Goodrich JK, Johansson ME, Nalbantoglu I, Aitken JD, Su Y, Chassaing B, Walters WA, Gonzalez A, et al. Transient Inability to Manage Proteobacteria Promotes Chronic Gut Inflammation in TLR5-Deficient Mice. Cell Host Microbe. 2012;12:139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Shah YM, Morimura K, Krausz KW, Miyazaki M, Richardson TA, Morgan ET, Ntambi JM, Idle JR, Gonzalez FJ. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008;7:135–147. doi: 10.1016/j.cmet.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Miyazaki M, Socci ND, Hagge-Greenberg A, Liedtke W, Soukas AA, Sharma R, Hudgins LC, Ntambi JM, Friedman JM. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- Cummings JH, Hill MJ, Bone ES, Branch WJ, Jenkins DJ. The effect of meat protein and dietary fiber on colonic function and metabolism. II. Bacterial metabolites in feces and urine. Am J Clin Nutr. 1979;32:2094–2101. doi: 10.1093/ajcn/32.10.2094. [DOI] [PubMed] [Google Scholar]
- den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, Oosterveer MH, Jonker JW, Groen AK, Reijngoud DJ, et al. Short-Chain Fatty Acids protect against High-Fat Diet-Induced Obesity via a PPARgamma-dependent switch from lipogenesis to fat oxidation. Diabetes. 2015 doi: 10.2337/db14-1213. [DOI] [PubMed] [Google Scholar]
- den Besten G, Lange K, Havinga R, van Dijk TH, Gerding A, van Eunen K, Muller M, Groen AK, Hooiveld GJ, Bakker BM, et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am J Physiol Gastrointest Liver Physiol. 2013a doi: 10.1152/ajpgi.00265.2013. [DOI] [PubMed] [Google Scholar]
- den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013b;54:2325–2340. doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes J, Su W, Rahat-Rozenbloom S, Wolever TM, Comelli EM. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr Diabetes. 2014;4:e121. doi: 10.1038/nutd.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol. 2009;20:379–385. doi: 10.1097/MOL.0b013e32832fa5c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost G, Sleeth ML, Sahuri-Arisoylu M, Lizarbe B, Cerdan S, Brody L, Anastasovska J, Ghourab S, Hankir M, Zhang S, et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat Commun. 2014;5:3611. doi: 10.1038/ncomms4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- Hodson L, Fielding BA. Stearoyl-CoA desaturase: rogue or innocent bystander? Prog Lipid Res. 2013;52:15–42. doi: 10.1016/j.plipres.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Jeffcoat R, Roberts PA, Ormesher J, James AT. Stearolyl-CoA desaturase: a control enzyme in hepatic lipogenesis. Eur J Biochem. 1979;101:439–445. doi: 10.1111/j.1432-1033.1979.tb19737.x. [DOI] [PubMed] [Google Scholar]
- Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145:396–406. e391–310. doi: 10.1053/j.gastro.2013.04.056. [DOI] [PubMed] [Google Scholar]
- Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. 2013;4:1829. doi: 10.1038/ncomms2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KN, Pariza MW, Ntambi JM. Differential expression of hepatic stearoyl-CoA desaturase gene 1 in male and female mice. Biochim Biophys Acta. 1996;1304:85–88. doi: 10.1016/s0005-2760(96)00145-2. [DOI] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Liu J, Cinar R, Xiong K, Godlewski G, Jourdan T, Lin Y, Ntambi JM, Kunos G. Monounsaturated fatty acids generated via stearoyl CoA desaturase-1 are endogenous inhibitors of fatty acid amide hydrolase. Proc Natl Acad Sci U S A. 2013;110:18832–18837. doi: 10.1073/pnas.1309469110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Miyazaki M, Flowers MT, Sampath H, Zhao M, Chu K, Paton CM, Joo DS, Ntambi JM. Loss of Stearoyl-CoA desaturase-1 attenuates adipocyte inflammation: effects of adipocyte-derived oleate. Arterioscler Thromb Vasc Biol. 2010;30:31–38. doi: 10.1161/ATVBAHA.109.195636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TA, LeBrasseur NK, Cote GM, Trucillo MP, Pimentel DR, Ido Y, Ruderman NB, Sawyer DB. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem Biophys Res Commun. 2005;336:309–315. doi: 10.1016/j.bbrc.2005.08.088. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Dobrzyn A, Man WC, Chu K, Sampath H, Kim HJ, Ntambi JM. Stearoyl-CoA desaturase 1 gene expression is necessary for fructose-mediated induction of lipogenic gene expression by sterol regulatory element-binding protein-1c-dependent and -independent mechanisms. J Biol Chem. 2004;279:25164–25171. doi: 10.1074/jbc.M402781200. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Kim YC, Gray-Keller MP, Attie AD, Ntambi JM. The biosynthesis of hepatic cholesterol esters and triglycerides is impaired in mice with a disruption of the gene for stearoyl-CoA desaturase 1. J Biol Chem. 2000;275:30132–30138. doi: 10.1074/jbc.M005488200. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Kim YC, Ntambi JM. A lipogenic diet in mice with a disruption of the stearoyl-CoA desaturase 1 gene reveals a stringent requirement of endogenous monounsaturated fatty acids for triglyceride synthesis. J Lipid Res. 2001;42:1018–1024. [PubMed] [Google Scholar]
- Miyazaki M, Sampath H, Liu X, Flowers MT, Chu K, Dobrzyn A, Ntambi JM. Stearoyl-CoA desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochem Biophys Res Commun. 2009;380:818–822. doi: 10.1016/j.bbrc.2009.01.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison WR, Smith LM. Preparation of Fatty Acid Methyl Esters and Dimethylacetals from Lipids with Boron Fluoride--Methanol. J Lipid Res. 1964;5:600–608. [PubMed] [Google Scholar]
- Mziaut H, Korza G, Ozols J. The N terminus of microsomal delta 9 stearoyl-CoA desaturase contains the sequence determinant for its rapid degradation. Proc Natl Acad Sci U S A. 2000;97:8883–8888. doi: 10.1073/pnas.97.16.8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan CJ, Larter CZ. Lipotoxicity: why do saturated fatty acids cause and monounsaturates protect against it? J Gastroenterol Hepatol. 2009;24:703–706. doi: 10.1111/j.1440-1746.2009.05823.x. [DOI] [PubMed] [Google Scholar]
- Ogiwara H, Tanabe T, Nikawa J, Numa S. Inhibition of rat-liver acetyl-coenzyme-A carboxylase by palmitoyl-coenzyme A. Formation of equimolar enzyme-inhibitor complex. Eur J Biochem. 1978;89:33–41. doi: 10.1111/j.1432-1033.1978.tb20893.x. [DOI] [PubMed] [Google Scholar]
- Poudyal H, Brown L. Stearoyl-CoA desaturase: a vital checkpoint in the development and progression of obesity. Endocr Metab Immune Disord Drug Targets. 2011;11:217–231. doi: 10.2174/187153011796429826. [DOI] [PubMed] [Google Scholar]
- Rabot S, Membrez M, Bruneau A, Gerard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010;24:4948–4959. doi: 10.1096/fj.10-164921. [DOI] [PubMed] [Google Scholar]
- Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. 2008;105:16767–16772. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya M, Yahagi N, Matsuzaka T, Najima Y, Nakakuki M, Nagai R, Ishibashi S, Osuga J, Yamada N, Shimano H. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology. 2003;38:1529–1539. doi: 10.1016/j.hep.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Sharkey AG, Shultz JL, Friedel RA. Mass Spectra of Esters. Formation of Rearrangement Ions. Anal Chem. 1959;31:87–94. [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira TF, Grzeskowiak L, Franceschini SC, Bressan J, Ferreira CL, Peluzio MC. Higher level of faecal SCFA in women correlates with metabolic syndrome risk factors. Br J Nutr. 2013;109:914–919. doi: 10.1017/S0007114512002723. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Boren J, Oresic M, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–1112. doi: 10.1194/jlr.M002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, Gewirtz AT. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328:228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann A, Allahyar A, Greiner TU, Plovier H, Lunden GO, Larsson T, Drucker DJ, Delzenne NM, Cani PD, Backhed F. Microbial modulation of energy availability in the colon regulates intestinal transit. Cell Host Microbe. 2013;14:582–590. doi: 10.1016/j.chom.2013.09.012. [DOI] [PubMed] [Google Scholar]
- Wolever TM, Brighenti F, Royall D, Jenkins AL, Jenkins DJ. Effect of rectal infusion of short chain fatty acids in human subjects. Am J Gastroenterol. 1989;84:1027–1033. [PubMed] [Google Scholar]
- Wolever TM, Schrade KB, Vogt JA, Tsihlias EB, McBurney MI. Do colonic short-chain fatty acids contribute to the long-term adaptation of blood lipids in subjects with type 2 diabetes consuming a high-fiber diet? Am J Clin Nutr. 2002;75:1023–1030. doi: 10.1093/ajcn/75.6.1023. [DOI] [PubMed] [Google Scholar]
- Wu J, An Y, Yao J, Wang Y, Tang H. An optimised sample preparation method for NMR-based faecal metabonomic analysis. Analyst. 2010;135:1023–1030. doi: 10.1039/b927543f. [DOI] [PubMed] [Google Scholar]
- Zheng X, Qiu Y, Zhong W, Baxter S, Su M, Li Q, Xie G, Ore BM, Qiao S, Spencer MD, et al. A targeted metabolomic protocol for short-chain fatty acids and branched-chain amino acids. Metabolomics. 2013;9:818–827. doi: 10.1007/s11306-013-0500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.