

Abstract
The natural product curacin A, a potent anticancer agent, contains a rare cyclopropane group. The five enzymes for cyclopropane biosynthesis are highly similar to enzymes that generate a vinyl chloride moiety in the jamaicamide natural product. The structural biology of this remarkable catalytic adaptability is probed with high-resolution crystal structures of the curacin cyclopropanase (CurF ER), an in vitro enoyl reductase (JamJ ER), and a canonical curacin enoyl reductase (CurK ER). The JamJ and CurK ERs catalyze NADPH-dependent double bond reductions typical of enoyl reductases (ERs) of the medium chain dehydrogenase reductase (MDR) superfamily. Cyclopropane formation by CurF ER is specified by a short loop which, when transplanted to JamJ ER, confers cyclopropanase activity on the chimeric enzyme. Detection of an adduct of NADPH with the model substrate crotonyl-CoA provides indirect support for a recent proposal of a C2-ene intermediate on the reaction pathway of MDR enoyl-thioester reductases.
Introduction
The remarkable structural diversity provided by the bacterial polyketides makes them pharmacologically attractive targets for drug development (Clardy et al., 2006; Gerwick and Fenner, 2013; Gerwick and Moore, 2012; Newman and Cragg, 2012). The unique structures of many polyketides are assembled by multi-modular type I polyketide synthases (PKSs), or by hybrid pathways of PKS and non-ribosomal peptide synthetase (NRPS) modules (Fischbach and Walsh, 2006; Hertweck, 2009; Jones et al., 2009). Polyketide biosynthesis takes place by repeated decarboxylative condensation of acyl-CoA esters in a process similar to that used by fatty acid synthase (FAS). Type I PKS pathways operate sequentially as an ordered assembly line of synthase modules. A minimal type I PKS module consists of an acyltransferase (AT) to select a building block, an acyl carrier protein (ACP) to carry the polyketide intermediate, and a ketosynthase (KS) domain to catalyze chain extension to a β-keto intermediate. Further reductive modifications of the polyketide intermediate may result in β-hydroxyl, enoyl or fully saturated products through the action of ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) domains within the module. Onto this canonical organization of sequential type I PKS modules, nature has grafted domains with a variety of other catalytic activities to create a vast number of distinct pathways that enrich the chemical diversity of polyketide natural products.
The marine cyanobacterium Moorea producens (formerly Lyngbya majuscula; (Engene et al., 2012)) is a rich source of potent anticancer and neurotoxic agents, with strains that synthesize a number of interesting chemical groups in compounds such as curacin A, the barbamides and the jamaicamides (Chang et al., 2002; Chang et al., 2004; Edwards et al., 2004). For example, the potent antimitotic curacin A has a unique combination of cyclopropane, thiazoline, cis-double bond, and terminal alkene (Chang et al., 2004; Verdier-Pinard et al., 1999). The jamaicamides, which block sodium channels, contain unusual groups including a vinyl chloride and an alkynyl bromide (Edwards et al., 2004). Early segments of the Cur and Jam biosynthetic gene clusters exhibit high sequence identity and include a 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase-like (HCS) gene cassette encoding enzymes to create a β-branch in the polyketide intermediate (Figure 1A) (Calderone, 2008; Gu et al., 2006). The high-identity regions of the cur and jam gene clusters encode a Fe2+/α-ketoglutarate (α-KG)-dependent halogenase (Hal) and tridomain ACP embedded in CurA and JamE; and the HMG cassette gene products: an HMG-ACP synthase in CurD and JamH, a dehydratase (ECH1) in CurE and JamI, a decarboxylase (ECH2) domain embedded in CurF and JamJ, and an enoyl reductase (ER) domain embedded in CurF and JamJ (Chang et al., 2004; Edwards et al., 2004; Gu et al., 2006; Gu et al., 2009). Remarkably, the biosynthetic products of these highly similar enzymes are a cyclopropane in curacin and a vinyl chloride in jamaicamide. Our biochemical and structural studies have identified the enzymes and the reaction order and have illuminated key features of the catalytic mechanisms (Figure 1B) (Akey et al., 2012; Gu et al., 2006; Gu et al., 2009). In the curacin pathway, CurA Hal produces γ-chloro-HMG-ACP (compound 1, Figure 1B) (Khare et al., 2010; Smith and Khare, 2015), which is dehydrated by ECH1 to form α,β-enoyl-γ-chloroglutaryl-ACP (2) and then decarboxylated by ECH2 (Geders et al., 2007). The analogous chemical steps in the jamaicamide and curacin pathways are identical through ECH1 dehydration, but the pathways diverge at the decarboxylation step where the CurF ECH2 generates an α,β-enoyl-γ-chloro-ACP product (3) and the 59% identical JamJ ECH2 forms a β,γ-enoyl-γ-chloro-ACP product (4) (Geders et al., 2007; Gu et al., 2009). The CurF ER catalyzes formation of cyclopropane (5) in an unprecedented mechanism involving hydride addition from NADPH and chloride elimination. In contrast, the 65% identical JamJ ER has no apparent function in jamaicamide synthesis as it is unreactive towards the β,γ-enoyl-γ-chloro-ACP JamJ ECH2 product. However, JamJ ER functions as a reducing ER to form β-methyl-γ-chlorobutyryl-ACP (6) from an unnatural substrate: the CurF ECH2 product (3) (Gu et al., 2009). The two pathways provide a prime example of how diversification of single enzymes can lead to different functional groups in natural product biosynthesis.
Figure 1.
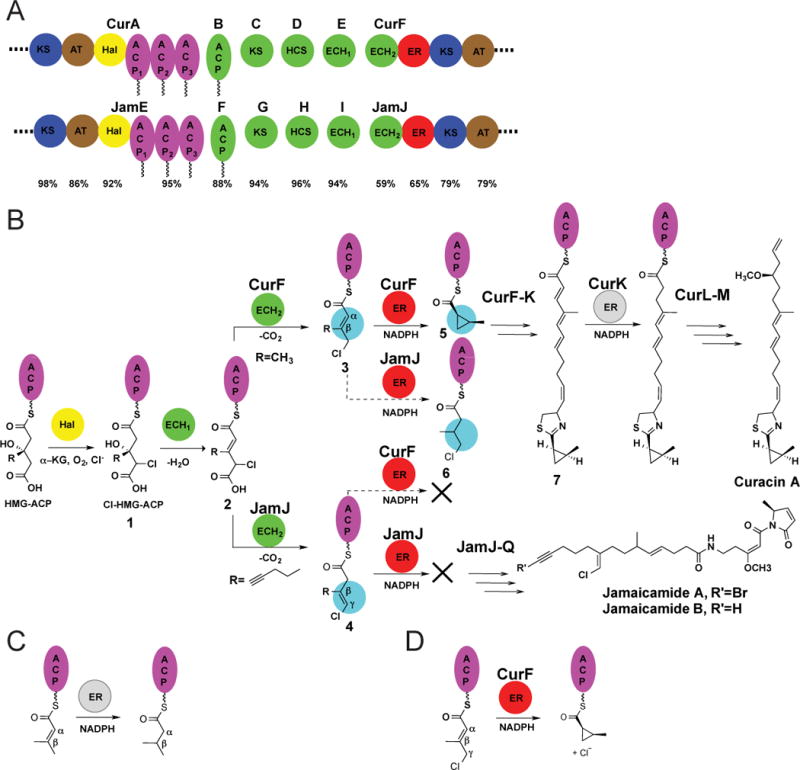
A. Highly similar regions of the curacin and jamaicamaide biosynthetic pathways. The HCS cassette proteins (green), triple-ACP (purple), halogenase (yellow), enoylreductase (red) and flanking regions (blue, brown) are shown for CurA-CurF and the homologous region in JamE-JamJ, with sequence identities below. KS, ketosynthase; AT, acyl transferase; Hal, halogenase; ACP, acyl carrier protein; HCS, HMG-ACP synthase; ECH1 and ECH2, enoyl-CoA hydratases; and ER, enoyl reductase.
B. Divergence in the biosynthetic pathways of curacin A and jamaicamaide A. In both pathways, CurA (or JamE) Hal chlorinates S-HMG-ACP, and CurE (or JamI) ECH1 dehydrates the chlorinated product. The Cur and Jam pathways diverge at the β-branching ECH2 step, where the CurF ECH2 forms an α-β double bond and the Jam ECH2 a β-γ double bond. The CurF ER then catalyzes cyclopropane ring formation. JamJ ER is unreactive towards the JamJ ECH2 product, however it reduces the CurF ECH2 product. CurK ER is a canonical PKS ER in the curacin A biosynthetic pathway, catalyzing reduction of an α-β double bond.
C. Canonical ER reductase reaction. Hydride transfer from the cofactor NADPH to the β carbon of the unsaturated substrate is followed by protonation at the α carbon atom.
D. CurF ER cyclopropanation. The unique CurF ER catalyzes the NADPH-dependent nucleophilic displacement of chloride to form the strained cyclopropane ring, presumably by a mechanism analogous to the canonical enoyl reduction (Gu et al., 2009).
In addition to CurF ER and JamJ ER, the Cur and Jam pathways also include ER domains embedded within PKS modules for enoyl reduction of polyketide intermediates: CurH ER, CurK ER, a second ER domain within JamJ, and JamL ER. These PKS ERs transfer a hydride ion from NADPH to the substrate β-carbon atom (C3) and a proton to the substrate α-carbon (C2) (Figure 1C). The CurF ER cyclopropanase also catalyzes hydride transfer from NADPH. However, in this unique ER domain, a highly strained cyclopropane ring forms upon displacement of chloride from the substrate γ-carbon atom (C4) (Figure 1D). Our structural studies were motivated by this unprecedented activity, which results from modest changes at the primary sequence level to an otherwise canonical PKS enoyl reductase domain. Moreover, we considered that the structural changes that enable generation of the rare cyclopropane ring system would provide new insights into subtle sequence variations that expand both natural product chemical diversity and biological activity.
The ER domains from PKS pathways belong to the medium chain dehydrogenase/reductase (MDR) superfamily of proteins including metazoan fatty acid synthase ERs, quinone oxidoreductases, the well-studied Zn2+-dependent alcohol dehydrogenases (Hedlund et al., 2010; Nordling et al., 2002; Persson et al., 2008), and a recently identified crotonyl-CoA carboxylase (Erb et al., 2007; Quade et al., 2012). MDR reduction of thioester-linked substrates was thought to proceed through an enolate intermediate that formed upon hydride transfer from the nicotinamide C4 atom to the substrate β-carbon atom (C3). Recently a transient “C2-ene” adduct between the nicotinamide C2 atom and the thioester α-carbon (C2) was identified in two MDRs, raising the possibility that the reaction proceeds through a C2-ene adduct and not through an enolate (Rosenthal et al., 2014).
Crystal structures of two PKS ERs have been reported, including the excised cis-acting KR-ER2 didomain from the bacterial spinosyn (Spn) pathway (Zheng et al., 2012) and the standalone trans-acting ER, LovC, from the fungal lovastatin pathway (Ames et al., 2012), however the structural basis of PKS enoyl reduction remains unclear. Site-directed mutagenesis of PKS ERs identified amino acids that may control ER stereochemistry in the α-branched polyketides (Kwan et al., 2008), but did not identify a proton donor for enoyl reduction (Ames et al., 2012; Kwan and Leadlay, 2010; Zheng et al., 2012). The PKS ER structures together with a structure-based sequence alignment reveal that a conserved proton donor does not exist in the PKS ERs. Identification of a proton donor by mutagenesis has been a general problem for MDR reductases and may be confounded by build-up of a dead-end NADP-substrate C4 adduct when substitutions are made at the proton donor position, as identified recently in a fatty acid synthase ER (Rosenthal et al., 2015).
To understand the structural biology of the unprecedented CurF ER cyclopropanase, we solved the crystal structure of the CurF ER with bound cofactor at 0.96 Å, as well as structures for the homologous (65% sequence identity) JamJ ER as the cofactor complex at 2.25 Å and in the apo form at 1.8 Å, and for the apo form of the canonical CurK ER reductase at 1.8 Å. These high-resolution structures demonstrate for the first time that the CurF cyclopropanase is strikingly different from the JamJ and CurK ERs in the substrate-binding region. This enabled the design of a functionally mixed chimeric JamJ variant that catalyzes both enoyl reduction and cyclopropanation reactions on the natural curacin A substrate. Furthermore, we assessed the possibility that one or more of the Cur/Jam ER reactions proceeds through a C2-ene adduct and not through an enolate. An NADP-substrate covalent adduct was directly detected with the CurK ER reductase.
Results
Overall architecture
The CurF ER, CurK ER and JamJ ER domains (Table 1, Figure 2 and 3, Figure S1 and S2) have the typical fold of the MDR enzyme superfamily (Hedlund et al., 2010). The ER is comprised of two domains (Figure 2A): a core cofactor (NADPH)-binding domain (CurF ER amino acids 135–274) and a discontinuous substrate-binding or catalytic domain (amino acids 1–134 and 275–337). The most similar structures (Holm et al., 2008) in the MDR superfamily include the spinosyn PKS ER (PDB 3SLK (Zheng et al., 2012); 2.1 Å rmsd, 34% identity), the human p53-inducible quinone oxidoreductase (2J8Z (Porte et al., 2009); 2.8 Å rmsd, 24% identity), a bacterial quinone oxidoreductase (1QOR (Thorn et al., 1995); 2.4 Å rmsd, 24% identity) and the mammalian fatty acid synthase ER domain (2VZ8 (Maier et al., 2008); 2.1 Å rmsd, 29% identity). The trans-acting ER from the lovastatin PKS, LovC, is a more distantly related MDR (3B70, (Ames et al., 2012); 3.2 Å rmsd, 25% identity). MDR superfamily members exist predominantly as dimers or tetramers in which a continuous β-sheet forms between the nucleotide-binding domains of two subunits through antiparallel pairing of adjacent β strands. However, in the PKS ERs, this association is blocked by a short helix (CurF ER α8, residues 258–265), and the ER is a monomer both in solution and in crystal structures (Figure 2) (Ames et al., 2012; Zheng et al., 2012). Apart from differences associated with cofactor binding, the new Cur and Jam ER domain structures are highly similar (CurF ER-NADP+ superimposes with an rmsd of 0.7 Å for 271 of 332 Cα atoms of JamJ ER-NADPH, and 1.3 Å for 290 of 332 Cα atoms in CurK apo-ER).
Table 1.
Summary of Crystallography
| Crystal form | CurF ER (NADP+) | JamJ ER (free enzyme) | JamJ ER (NADPH) | Se-Met JamJ ER (NADPH) | CurK ER (free enzyme) |
|---|---|---|---|---|---|
| Diffraction data | |||||
| X ray source | APS 23ID-D | APS 23ID-D | APS 23ID-B | APS 23ID-D | APS 23ID-B |
| Wavelength (Å) | 0.72932 | 1.0332 | 1.0332 | 0.979 | 1.0332 |
| Space group | C2 | P1 | P31 | P31 | F222 |
| Cell dimensions | |||||
| a, b, c (Å) | 108.9, 47.3, 76.9 | 50.9, 57.2, 66.1 | 110.7, 92.8 | 111.3, 91.6 | 93.6, 127.1, 127.8 |
| α, β, γ (°) | β=123.4 | 95.3, 106.7, 105.1 | |||
| dmin (Å) | 0.96 (1.01–0.96)* | 1.80 (1.86–1.80) | 2.25 (2.33–2.25) | 2.6 (2.69–2.60) | 1.85 (1.92–1.85) |
| Rmerge (%) | 8.0 (66.0) | 7.4 (70.2) | 7.3 (60.9) | 12.1 (60.5) | 5.9 (62.2) |
| Avg I/σI | 9.5 (1.5) | 14.2 (2.0) | 12.3 (1.4) | 25.8 (3.3) | 21.0 (1.9) |
| Observations (#) | 197,326 | 63,164 | 59,866 | 19,823 | 32,609 |
| Completeness (%) | 99.9 (94.2) | 95.2 (95.8) | 99.6 (99.8) | 100.0 (100.0) | 99.0 (100.0) |
| Avg multiplicity | 3.7 (2.7) | 2.2 (2.2) | 2.1 (2.0) | 5.7 (5.8) | 3.8 (3.7) |
| Refinement | |||||
| Data range (Å) | 23.0–0.96 | 46.5–1.8 | 45.6–2.25 | 24.3–1.85 | |
| Reflections (#) | 187,338 | 58,496 | 56,585 | 30,569 | |
| Rwork/Rfree (%) | 11.7/13.1 | 18.6/22.7 | 17.9/21.5 | 18.5/22.0 | |
| Number of atoms | |||||
| Protein | 2843 | 5320 | 8064 | 2619 | |
| Water | 558 | 414 | 362 | 230 | |
| Ligands | 48 | – | 141 | – | |
| RMS deviations | |||||
| Bond lengths (Å) | 0.01 | 0.010 | 0.005 | 0.008 | |
| Bond angles (°) | 1.529 | 1.35 | 1.048 | 1.15 | |
| Avg B-factors (Å2) Protein NADP Water |
10.3 8.3 30.9 |
31 – 41 |
39.5 29.3 32.6 |
35.4 – 46.5 |
|
| MolProbity score | 1.1 | 1.23 | 1.32 | 1.18 | |
| MolProbity clashscore | 3.06 | 4.55 | 3.66 | 3.94 | |
| Ramachandran plot | |||||
| Favored (%) | 98.92 | 98.07 | 95.96 | 98.8 | |
| Allowed (%) | 1.08 | 1.1 | 3.74 | 0.9 | |
| Outliers (%) | 0.0 | 0 | 0.3 | 0.3 | |
| Molecular state | |||||
| Asymmetric unit | 1 monomer | 2 monomers | 3 monomers | 1 monomer | |
| Active site ligands | NADP+ | – | NADPH | – | |
| PDB code | 5DP2 | 5DOV | 5DOZ | 5DP1 |
Values in parentheses are for the highest-resolution shell.
Figure 2.
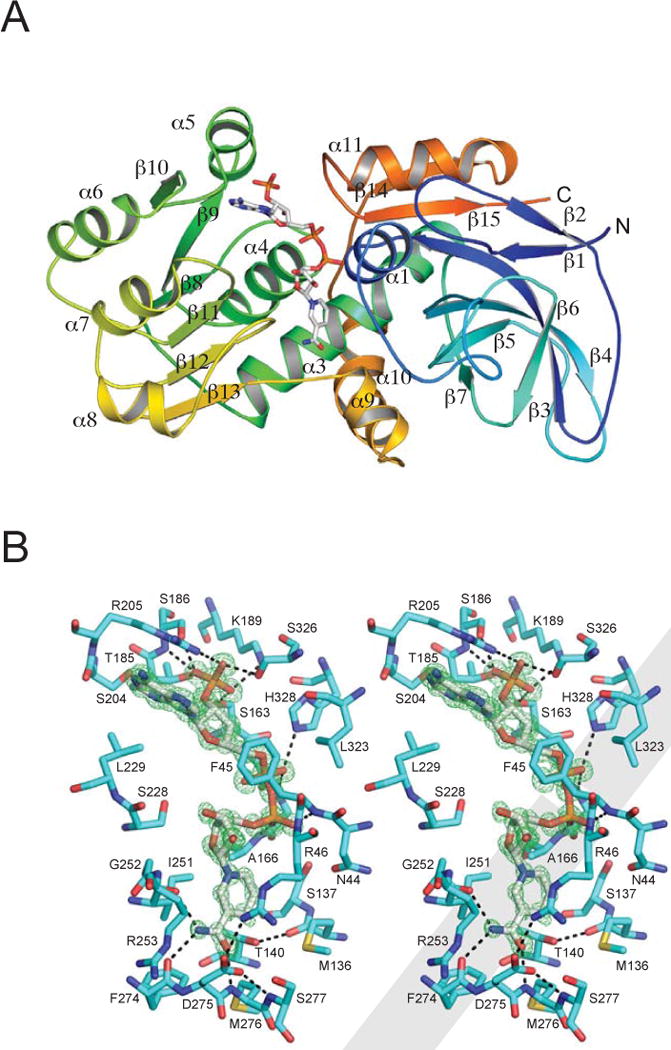
A. Overall structure of CurF ER. The polypeptide in the ribbon diagram is colored as a rainbow from blue (N-terminus) to red (C-terminus) and secondary structures are labeled. Cofactor NADP+ is shown as sticks with atomic colors (white C, red O, blue N, orange P).
B. Cofactor binding in CurF ER. The stereo view shows the environment around the cofactor NADP+ (Fo−Fc omit map at 0.96-Å resolution contoured at 4σ in green). NADP+ and the neighboring residues are shown as sticks (white C in NADPH; cyan C in the protein). Hydrogen bonds are shown as black dashed lines.
Figure 3. Comparison of ER structures.
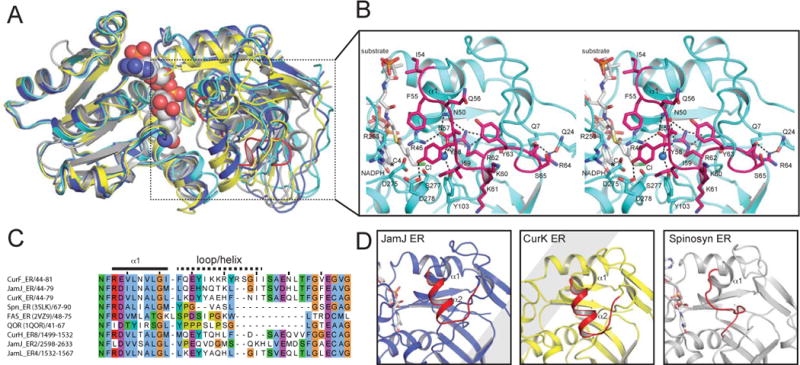
A. Superposition of nucleotide-binding domains. The similarity of nucleotide-binding domains and divergence of substrate-binding domains is apparent for NADPH-bound CurF ER (cyan), JamJ ER (blue), CurK ER (yellow) and spinosyn ER2 (grey).
B. CurF ER active site. The docked substrate (4-Cl-3-methylcrotonyl) shown in the active site in the stereo drawing (white carbon atoms). Selected residues tested by mutagenesis are shown in stick form in the cyan ER with the cyclopropanase loop highlighted in magenta. The NADPH C4 atom is marked with a star, and the active site water is rendered as a blue sphere.
C. Sequence alignment of the substrate loop region. The longer substrate loop of the cyanobacterial ERs is apparent.
D. Comparison of ER substrate loops. The longer substrate loops of the JamJ and CurK ERs include a helix, which does not exist in the shorter loop of the Spn ER2. The CurF ER cyclopropanase loop in (b) differs from the substrate loops of all canonical ERs.
Catalytic activity
We used the CurF ER substrate to assay cyclopropane ring formation by CurF ER and enoyl thioester α-β double-bond reduction by JamJ ER. The natural substrate was generated by loading a CurA ACP domain with 2-hydroxymethylglutarate, followed by chlorination with CurA Hal, dehydration with CurE ECH1 and decarboxylation with CurF ECH2 to generate γ-chloro-β-methylcrotonyl-ACP (3, Figure 1B) (Geders et al., 2007; Gu et al., 2009). Products were detected by Fourier transform ion cyclotron resonance (FT-ICR) tandem mass spectrometry (Figure 4). In the absence of the natural CurK ER substrate (7, Figure 1B), we employed an HPLC assay to evaluate JamJ ER and CurK ER reduction of crotonyl-CoA to butyryl-CoA (Figure S3). All three ER domains displayed catalytic activity (Table 2, Figures 4 and S3). JamJ ER had greater activity with the ACP-borne CurF ER substrate (3) than with crotonyl-CoA, but with the CoA substrate JamJ ER was more active than CurK ER.
Figure 4. Detection of ER products by FT-ICR mass spectrometry.
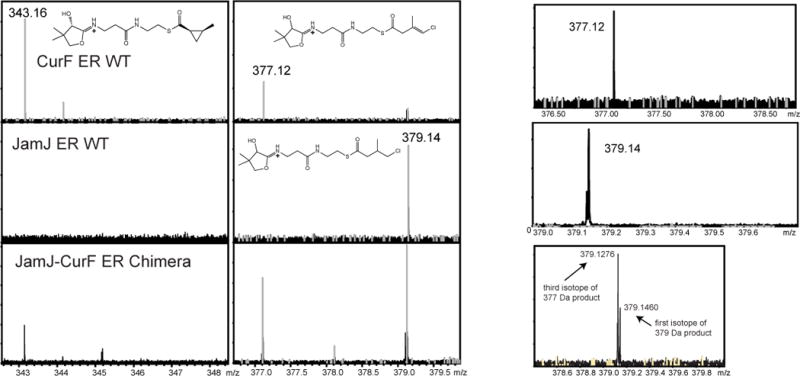
Mass spectra for phosphopantetheine ejection products of CurF ER, JamJ ER and the JamJ-CurF chimera. The substrate intermediates were ejected from the ACP by collision-induced dissociation (CID) and detected and quantified by FT-ICR MS. Calculated and observed m/z values are 343 for the cyclopropane product, 377 for the vinyl chloride, and 379 for the reduced product. Panels at the right show the isotopic distribution for the vinyl chloride and reduced products. Reactions were performed in duplicate.
Table 2.
Characterization of products by FT-ICR mass spectrometry.
| Cyclopropane product (%) | Reduced product (%) | ||
|---|---|---|---|
| Observed mass of monoisotopic peak of fragments (calculated mass) [Da] | 343.16 (343.16) | 379.14 (379.14) | |
| CurF ER | WT | 67 ± 2 | |
| R46A | 30 ± 7 | ||
| Y58A | 27.4 ± 0.1 | ||
| R62Q | 18.5 ± 0.5 | ||
| Y103A | 18 ± 3 | ||
| Y103F | 35.3 ± 0.2 | ||
| R253K | 15 ± 2 | ||
| D275A | 28 ± 23 | ||
| D275N | 18 | ||
| S277A | 76 ± 9 | ||
| S277L | 23 ± 9 | ||
| JamJ ER | WT | 96.9 ± 0.2 | |
| K251A | 98 ± 2 | ||
| K251R | 100 | ||
| D273A | 55 ± 4 | ||
| D273N | 70 | ||
| JamJ-CurF ER chimera | 13.3 ± 0.4 | 47 ± 4 | |
Cofactor binding
The NADPH cofactor binds in a narrow cleft between the catalytic and nucleotide-binding domains and contacts both domains through a number of hydrogen bonds and van der Waals interactions (Figure 2B and S4) in a similar manner to NADPH binding to other PKS ERs (Ames et al., 2012; Zheng et al., 2012). The nicotinamide group is at an open end of the active site cleft that defines the substrate entrance. Although NADPH was used in crystallization, the 0.96-Å electron density for the nicotinamide ring was flat with no pucker at the hydride donor (C4), indicating that oxidized NADP+ was the bound species in the CurF ER (Figure 2B). The nicotinamide C4 is in van der Waals contact (3.4 Å) with the Thr140 (Thr138 in JamJ ER and CurK ER) side chain hydroxyl, whose position is stabilized by a hydrogen bond to the Met136 backbone carbonyl. The fixed Thr140 position and close contact to C4 indicate that Thr140 restricts the nicotinamide pucker of NADPH, so that C4 must pucker away from Thr140, placing the 4-pro-R-hydride in an axial orientation optimal for transfer to the substrate and the 4-pro-S-hydride in an NADP+-like equatorial orientation. Thus, the Thr140 steric constraint on nicotinamide pucker provides a subtle assist to catalysis and explains the invariance of Thr140 among PKS ERs. Elimination of the steric constraint by mutagenesis of Thr140 to Ala (Figures S3B and S3C, T138A) or to Val (Kwan and Leadlay, 2010) reduced activity three- to four-fold. Consistent with this hypothesis, a Val substitution for Thr in LovC resulted in opposing effects on Km and kcat (25-fold and 35-fold, respectively) with little effect on catalytic efficiency (Ames et al., 2012).
The cofactor nicotinamide is positioned by hydrogen bonds of the amide group to backbone atoms of two ER loops (Ile251 in the β12-α8 loop; Phe274 and Met276 in the β13-α9 loop; Figure 2B). These loops are at the substrate entrance and also carry conserved basic and acidic side chains that are within reach of one another but do not interact in any of the structures (CurF Arg253, JamJ Lys251, CurK Lys250; CurF Asp275, JamJ Asp273, CurK Asp272). We speculated that interaction of the oppositely charged side chains may be critical to an ER state that exists only when substrates are bound, however mutagenesis did not support a critical role for either amino acid in CurF, CurK and JamJ ERs (Table 2, Figures 4 and S3), nor in Spn ER2, LovC ER or ER13 of the rapamycin pathway (Ames et al., 2012; Kwan and Leadlay, 2010; Zheng et al., 2012).
Comparison of the cofactor-bound and -free ER structures reveals flexibility in loops that contact NADPH, including the β11-α7 and β12-α8 loops near the nicotinamide and the α11-β15 loop near the adenine nucleotide. Substrate binding may induce additional changes to loops near the nicotinamide. At the other end of the cofactor site, amino acid side chains occlude the NADPH site in the cofactor-free JamJ (Phe45) and CurK (His325) ER structures (Figure S2). Loop flexibility in absence of cofactor also explains the inability to crystallize an NADPH complex of CurK ER, where a crystal contact trapped the α11-β15 loop in a position that blocks the NADPH adenine nucleotide site.
Substrate binding and catalysis
The three ER structures present views of an enzyme for canonical enoyl reduction (CurK ER), for cyclopropane ring formation (CurF ER), and without natural function but possessing robust reductase activity for the CurF ER substrate (JamJ ER) (Gu et al., 2009). The active site of a PKS ER should bind the substrate with its C3 (β) carbon near the NADPH C4 hydride donor, and the reductases should provide a proton donor (amino acid or water molecule) near the substrate C2 (Figure 1B). Furthermore, the cyclopropanase must rigorously exclude proton donors from C2 to allow formation of the strained ring. We modeled the CurF substrate into the active site by positioning its C3 atom near the nicotinamide C4 in order to identify amino acids that may be critical to catalysis (Figure 3B). Amino acids in the active site surrounding the nicotinamide and modeled substrate were probed by site-directed mutagenesis of JamJ ER, CurF ER and CurK ER. Like others (Ames et al., 2012; Kwan and Leadlay, 2010; Zheng et al., 2012), we found no amino acid that was essential for reductase activity (Table 2 and Figure S3). Similarly, no single amino acid in the CurF ER was essential for cyclopropane ring formation.
In light of the recent identification of an unstable covalent adduct between NADPH and an enoylthioester MDR substrate (Rosenthal et al., 2014), we searched for an adduct of NADPH with crotonyl-CoA in assays of the JamJ and CurK ERs. In reaction mixes of the CurK ER D272N variant, we detected such an adduct by HPLC, isolated it, and confirmed its existence by mass spectrometry (Figure S5A,B). Rosenthal et al. characterized two adducts of NADPH with crotonyl-CoA, a C2-ene (nicotinamide C2 to the thioester α-carbon (C2)), which is proposed to be a catalytic intermediate, and a dead-end C4 adduct (nicotinamide C4 to the thioester α-carbon (C2)), which is proposed to form when a proton donor is not available to resolve the C2-ene. The C2-ene and C4 adducts have identical mass, but are distinguished by UV absorbance bands at 370 nm and 328 nm, respectively (Rosenthal et al., 2014; Rosenthal et al., 2015). On this basis, CurK ER D272N yielded the C4 adduct under the assay conditions (Figure S5C). No adduct accumulated at levels sufficient for analysis with any other variant of CurK ER or any variant of JamJ ER.
One of the key issues relating to evolution of enzymes in secondary metabolism involves identification of specific amino acid changes that transform catalytic function. This issue is particularly relevant for the CurF ER ability to catalyze formation of a highly strained ring system rather than the expected and typical enoylreduction of an α,β unsaturated acylthioester. Interestingly, substitution of CurF ER Arg253 with the more typical Lys reduced cyclopropanation fourfold, but the converse substitution in JamJ ER (Lys251Arg) had no effect on reductase activity. We also tested CurF Arg46, another conserved basic residue near the substrate entrance that interacts with Asp275, which resulted in a modest (twofold) reduction in cyclopropane ring formation.
The three ERs have strikingly different pockets within the catalytic domain that appear tailored to the substrate length (Figure 5). The CurF ER and JamJ ER pockets are of similar size and end near the terminal chlorine atom of the modeled substrate. The CurK ER pocket is several Ångström units longer than the CurF ER pocket, consistent with CurK ER substrate (7, Figure 1B), which is 15 atoms longer than the CurF ER substrate (3).
Figure 5. Substrate binding cavity.
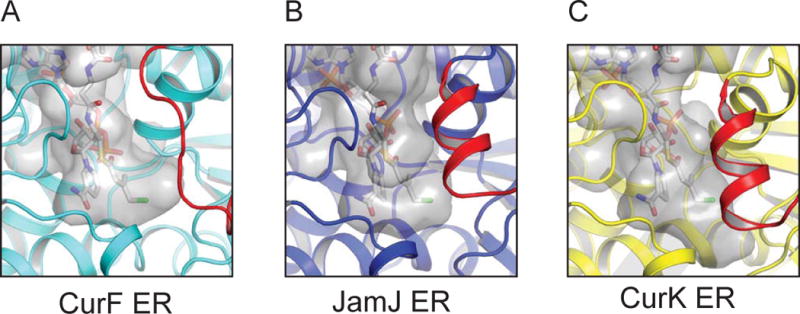
The transparent grey surface represents the interior surface of the presumed substrate tunnel in CurF ER, JamJ ER and CurK ER, calculated using the program HOLLOW (Ho and Gruswitz, 2008). NADPH and the modeled CurF ER substrate are shown in stick form in the active site.
Cyclopropanase loop and JamJ-CurF ER chimera
The substrate binding site is sandwiched between the NADPH nicotinamide and a region of the catalytic domain with the most divergent sequence among PKS ERs (CurF ER amino acids 46–75; Figure 3C). This region, which includes helix α1 and the following loop or helix, is about fifteen amino acids longer in the six ERs within the cyanobacterial curacin and jamaicamide pathways than in other bacterial cis-acting modular PKS ERs (CurF ER 46–75 vs. Spn ER2 232–247). Moreover, in this region, the CurF cyclopropanase structure differs substantially from the JamJ and CurK reductases despite their close primary sequence relationship to CurF ER (65% identity for JamJ ER, 51% for CurK ER) (Figure 3B–D). Accordingly, we designated CurF ER amino acids 54–68 as the “cyclopropanase loop”. Arg62 is a keystone of the cyclopropanase loop through four hydrogen bonds of its side chain to uncharged carbonyl oxygen atoms (Asn50 side chain and Thr74 main chain). Arg62 forms a fifth hydrogen bond with a water molecule, which is near the Cl atom of the modeled CurF substrate. Thus Arg62 may stabilize the chloride ion that forms during cyclopropanation. Consistent with its predicted role, substitutions for Arg62 destabilized the CurF ER protein. Only the Gln substitution yielded quasi-stable protein, in which cyclopropane ring formation was reduced three- to fourfold (Figure 4 and Table 2).
Next, we tested the function of the cyclopropanase loop by creating a chimeric JamJ-CurF ER protein. The CurF ER cyclopropanase loop (amino acids 55–68) was transplanted into JamJ ERK251R in place of amino acids 55–66 (Figure 3C and D). The chimeric enzyme was nearly as stable as JamJ ER and displayed gain-of-function activity as a cyclopropanase (Table 2 and Figure 4). Using 3-chloromethyl-crotonyl-ACP (3, Figure 1B), the natural CurF ER substrate, we detected the formation of both the reduced product (6) and also the cyclopropanated product (5) at levels of about one-quarter of the total product. No cyclopropanated product was detected from the singly substituted JamJ ERK251R (Table 2). To generate a JamJ ER chimera even more similar to CurF ER, additional single-residue substitutions were introduced at sites that contact the cyclopropanase loop, but these uniformly led to unstable protein. Cyclopropanation by the chimeric JamJ ER protein demonstrates that the cyclopropanase loop carries the determinants of this unique activity.
Discussion
The three high-resolution crystal structures of PKS ER domains from the cyanobacterial curacin and jamaicamide pathways illuminate important features of the CurF ER cyclopropanase and, more generally, the PKS ER reductases. The ER active site, defined by the position of bound nicotinamide, includes no invariant amino acids, with the notable exception of a threonine (CurF ER Thr140) whose hydroxyl group contacts the hydride-donating C4 atom of the cofactor on the opposite side of the nicotamide ring from the substrate. The Thr140 side chain is anchored in this position by a hydrogen bond of the hydroxyl to the ER backbone. We suggest that the function of Thr140 is to direct the nicotinamide pucker of NADPH towards the substrate, placing the pro-R-hydride in an axial orientation optimal for transfer to the β-carbon atom of the thioester substrate. An electrostatic assist to catalysis has also been suggested for the threonine hydroxyl group (Ames et al., 2012). Site-directed mutagenesis of this position resulted in moderate loss of activity (Figure S3) (Kwan and Leadlay, 2010), in accord with the proposed conformational assist to catalysis. The threonine assist appears to be a general feature of the MDR family as the same short contact of the hydroxyl to the nicotinamide C4 occurs in structures of several PKS ERs (Ames et al., 2012; Zheng et al., 2012) and other MDR enzymes (for example, (Gibbons and Hurley, 2004; Hori et al., 2006; Rosenthal et al., 2015).
Our experiments provide indirect support for the hypothesis of a C2-ene intermediate on the reaction pathway of enoyl-thioester reductases of the MDR superfamily (Rosenthal et al., 2014). With CurK ER D272N and a crotonyl-CoA substrate, we detected a C4 adduct (Figure S5), which had been identified previously in experiments using the C2-ene adduct as a substrate (Rosenthal et al., 2015). It is likely that CurK ER D272N first formed a C2-ene adduct, which spontaneously converted to the observed C4 adduct. With an unnatural acyl group (crotonyl-CoA vs. 7, Figure 1B) and an unnatural carrier (CoA vs. ACP), CurK ER is a highly inefficient enzyme. The extended reaction times required to accumulate sufficient adduct for analysis may have been too long to detect a C2-ene adduct. An aim of the mutagenesis experiments was to identify the proton donor in the reductase reaction. As with other PKS ERs, we found no amino acid to be essential to catalytic activity (Table 2) (Ames et al., 2012; Kwan and Leadlay, 2010). Formation of the unstable, dead-end C4 adduct, which spontaneously collapses to the reduced product and NADP+, is proposed to mask the identity of the proton donor in mutagenesis experiments (Rosenthal et al., 2015). Our detection of the C4 adduct in CurK ER D272N is consistent with Asp272 as the proton donor in CurK ER. However, other possibilities cannot be excluded. No adduct was detected in CurK ER D272A nor in analogous substitutions in JamJ ER. Given the high sequence similarity of PKS ERs, a common protein proton donor would be expected if such exists. It is likely that the direct proton donor is a water molecule, and that mutagenesis perturbs amino acids that position and orient the water appropriately for proton donation.
Several features of the structures bear on substrate selectivity. For example, the hydrophobic acyl-binding pocket may be a selectivity filter, as its size is correlated with the size of the substrate acyl group in the CurF ER cyclopropanase and CurK ER reductase. The pocket extends from the catalytic cleft adjacent to the nicotinamide ring into the hydrophobic core of the substrate-binding domain. A key element of substrate recognition and selectivity is a “substrate loop” at the entrance to the hydrophobic pocket. In contrast to the other loops surrounding the active site, which are well conserved, the substrate loop stands out as the most variable region of PKS ER sequences. The substrate loop is longer and differently structured in the cyanobacterial ERs (amino acids 55–74 in CurF, CurK and JamJ ERs) compared to cis-acting ERs from actinomycetes PKSs (amino acids 241–247 in Spn ER2, Fig. 3C). The cyanobacterial reductases, CurK ER and JamJ ER, have very similar helix-loop structures in this region in contrast to the analogous region of the actinomycete Spn ER2, which consists of a much shorter, well ordered loop (Fig. 3D). In all ER structures, the substrate loop is anchored at both ends by hydrogen bonds from the ER backbone to a conserved acidic side chain (Glu47 in CurF ER, Asp47 in CurK and JamJ ERs, Asp233 in Spn ER2). This acidic side chain was a candidate catalytic residue, however mutagenesis results were consistent with disruption of the structure of the substrate loop (Kwan and Leadlay, 2010).
A major motivation for our study was to gain new insights into the structural basis for the remarkable cyclopropanase activity of CurF ER, and here the substrate loop is a major player. The CurF and JamJ ERs have a recent common ancestor, presumably a canonical PKS enoyl reductase (Gu et al., 2009). The JamJ ER is an ideal comparative domain as it is 65% identical to CurF ER, possesses robust reductase activity, and lacks cyclopropanase activity toward the natural CurF ER substrate (3, Figure 1B). The substrate loop of the CurF ER cyclopropanase is strikingly different from the substrate loops of the JamJ and CurK ER reductases. On this basis, we reasoned that this unique CurF ER loop contains the determinants of cyclopropanase activity.
The 20-residue cyclopropanase loop has a well ordered structure supported by at least 15 hydrogen bonds. Arg62 anchors the loop structure through six hydrogen bonds to protein atoms. The positively charged side chain points into the substrate-binding domain where four of the five guanidinium protons are in hydrogen bonds with uncharged protein oxygen atoms and the fifth is in a hydrogen bond with a bound water, which is near the Cl atom of the modeled CurF ER substrate. Thus, Arg62 is ideally situated to stabilize a chloride leaving group positioned near the bound water (3 Å from Arg62 NH1, 9 Å from nicotinamide C4). The lack of interaction with a formal negative charge sets the stage for chloride ion stabilization. We also considered the nearby Arg46 as a candidate to stabilize the chloride, as it is highly conserved among cis-acting PKS ERs. However, Arg62 is far more accessible than Arg46, which is further from the modeled Cl atom and interacts with the carboxylates of both Glu57 and Asp275. Other features of the CurF ER that promote cyclopropane formation were not apparent from our study, in particular whether an adduct of NADPH with the substrate might be on the reaction pathway.
The importance of the cyclopropanase loop was demonstrated in the chimeric JamJ-CurF ER where the CurF ER loop (amino acids 54–68) was engineered into the wild type JamJ protein (replacing amino acids 54–66). The chimera displayed cyclopropanase activity with the authentic CurF ER substrate as well as reductase activity. This result shows that, in the enoyl reductases, the “substrate loop” is not the sole determinant of reductase activity as the chimera also generated the reduced product from the authentic CurF ER substrate, an activity lacking in the wild type CurF ER domain (Gu et al., 2006; Gu et al., 2009).
In conclusion, this study provides new insights into the structural evolution of a bacterial modular PKS enoylreductase to catalyze a unique NADPH-dependent cyclopropanation reaction through discrete changes to a highly variable part of the protein. It also provides evidence for an adduct of NADPH with the enoyl-thioester substrate on the reaction pathway of PKS enoyl reductases.
Materials and Methods
Cloning, expression and protein purification
The CurK ER domain (CurK amino acids 1482–1815, here referred to as 1–334) was amplified by PCR from cosmid pLM9 (Chang et al., 2004). CurF ER (amino acids 275–611, here 1–337) and the first ER domain within JamJ (amino acids 260–594, here 1–335) were amplified by PCR from plasmids encoding CurF ER and JamJ ER (Gu et al., 2009). The amplified products were inserted into the pMCSG7 (CurK ER and JamJ ER) and pMocr (CurK ER, JamJ ER and CurF ER) vectors and expressed in E. coli BL21 (DE3) cells (DelProposto et al., 2009; Stols et al., 2002). Cells were grown to an OD600 of 1.0 at 37°C in TB (Tartoff and Hobbs, 1987) supplemented with 4% glycerol, incubated at 20°C for 1 hr, induced with 0.4 mM IPTG, and grown for an additional 20 hr. The cell pellet from a 0.5-L culture was resuspended in 40 mL Buffer A (100 mM Tris pH 7.8, 500 mM NaCl, 5 mM imidazole and 10% glycerol) and lysed by ultrasonication. All purification steps were carried out at 4°C. The soluble fraction was loaded to a HisTrap Ni2+ NTA column (GE Healthcare), washed with Buffer A and eluted with a linear gradient of 5–500 mM imidazole. The His6-Mocr tag was cleaved by TEV protease and the tag-free ER purified on a Ni2+ NTA column. Further purification was done on a Superdex 200 size exclusion column equilibrated with Buffer B (10 mM Tris pH 7.8, 50 mM NaCl and 10% glycerol). Selenomethionyl labeled JamJ ER was purified similarly to the wild type protein, except that the pMocr::JamJ ER plasmid was transformed into the E. coli met- auxotroph strain B834 (DE3), cells were grown in minimal media (Molecular Dimensions), and 1 mM DTT was added in all subsequent purification steps. All excised PKS ER domains eluted with an apparent molecular weight of 40 kDa, corresponding to a monomer. A JamJ-CurF ER chimera was generated by using overlap PCR to replace amino acids 55–66 of JamJ ER with the amino acids 55–68 of CurF ER. Mutants of CurK, JamJ and CurF ER in the pMocr vector were generated by site directed mutagenesis and purified as described above.
Crystallization and data collection
All crystallization experiments were carried out at 20°C using the sitting drop vapor diffusion technique. Prior to crystallization, the protein was pre-incubated with NADPH (5:1 NADPH:protein) 1 hr at 4°C. A single crystal of His6-CurK ER was obtained in 4 weeks, from a 1:1 mixture of 16 mg/mL protein mixed with reservoir solution (1.2 M NaH2PO4, 0.8 M K2HPO4, 0.2 M Li2SO4 and 0.1 M CAPS pH 10.5). Microseeding was necessary to reproduce the His6-CurK ER crystals. JamJ ER at 6.5 mg/mL crystallized overnight from a number of conditions in a sparse matrix screen. Further optimization using 2:1 protein:reservoir mix (20% PEG 3350, 0.25 M sodium acetate, 100 mM Bis Tris propane pH 8.5) gave the best diffracting crystals. A second crystal form of His6-JamJ ER grew in 3 weeks from a 1:1 mixture of protein:reservoir (30% PEG 4000, 0.2 M MgCl2 and 0.1 M Tris pH 8.5). Prior to crystallization, CurF ER was dialyzed 3 hr against 50 mM Tris pH 7.8, 100 mM NaCl to remove glycerol, and crystallized in about 5 days from a 1:1 mixture of 12 mg/mL protein with 0.1 M HEPES pH 7.5, 28% PEG 3350. Crystals were cryoprotected in the corresponding reservoir solution with 20% glycerol (v/v) and flash cooled in liquid nitrogen. Diffraction data were collected at 100K on the GM/CA beamlines at the Advanced Photon Source (Table 1). Data from CurK and JamJ ER crystals were integrated and scaled with HKL2000 (Otwinowski, 1997). CurF ER data consist of a high-resolution and a low-resolution pass, which were integrated separately in XDS (Kabsch, 2010) and subsequently scaled together and merged (Table 1).
Structure determination and refinement
CurK ER crystals belong to the orthorhombic space group F222 and the structure was solved by molecular replacement using the program BALBES (Long et al., 2008), which used the ER domain from the mammalian fatty acid synthase (PDB code 2vz9) as a starting template. The initial BALBES model (Rwork/Rfree 37.4/42.6%; quality factor 0.69) was subjected to iterative rounds of manual model building in Coot (Emsley and Cowtan, 2004) followed by refinement in Refmac5 (Murshudov et al., 1997) in the CCP4 suite (Winn et al., 2011). The asymmetric unit contains one molecule (VM = 2.6 Å3/Dalton, 52% solvent). Structures of the His6-JamJ ER and the tagless JamJ ER in the apo form were solved by molecular replacement using the CurK ER structure. The His6-JamJ ER crystals are in triclinic space group P1 with two molecules in the asymmetric unit (VM = 2.3 Å3/Dalton, 47% solvent).
Crystals of the NADPH complex of JamJ ER appeared to be hexagonal with Laue symmetry 6/m. However, the apparent hexagonal symmetry arises from nearly perfect merohedral twinning of trigonal crystals following the twin law -h, -k, l, as revealed by an H-test in phenix.xtriage (Adams et al., 2010). Molecular replacement using Phaser (McCoy et al., 2007) revealed the space group to be P31 and placed two molecules per asymmetric unit (A and B), corresponding to a high VM of 4.4 Å3/Dalton and 72% solvent. Subsequent crystallographic refinement stalled at Rwork of 23% and Rfree of 26%, and unoccupied weak density suggested an additional molecule in the asymmetric unit. However, extensive molecular replacement searches yielded no additional molecule. Ultimately, data were collected from selenomethionine-substituted JamJ ER crystals, and the phased anomalous difference map clearly showed Se sites in the unassigned density region, corresponding to a molecule C. The twinning is explained by the packing of molecules A and B, which are related by nearly perfect 61 symmetry. However molecule C is incompatible with hexagonal symmetry as it would clash with a 61 symmetry mate. The structure was refined with three molecules in the asymmetric unit using the twin law (-h, -k, l) with a final Rwork of 18.5% and Rfree of 20.6%. Molecule C has substantially higher atomic B factors (average 53 Å2) than do molecules A and B (average 33 Å2).
The CurF ER structure was solved by molecular replacement with Phaser using the apo form of the 65% identical JamJ ER. Refinement at 0.96 Å was performed using Refmac5 with anisotropic atomic temperature factors, and multiple side-chain conformations were built for 33 amino acids based on electron density. |Fo|-|Fc| difference density was visible for ~17% of the hydrogen atoms. Several water molecules were included with partial occupancy because they are mutually exclusive with partially occupied sites of amino acids or other water molecules. A summary of data and refinement statistics are in Table 1. All X-ray structures were evaluated with MolProbity (Davis et al., 2007) and figures were prepared using PyMOL (Schrodinger, 2010) and LIGPLOT for protein-ligand interactions (Laskowski and Swindells, 2011).
HPLC-based assay to detect conversion of crotonyl-CoA to butyryl-CoA
The 20 μL reaction mixture containing 1 μM JamJ ER (100 μM CurK ER), 1 mM NADPH, 500 μM crotonyl-CoA, 10 mM Tris (pH 7.8), 50 mM NaCl and 10% glycerol was incubated 8 min at 25°C (3 hr with CurK ER), and quenched by addition of 10 μL of 1.0 M acetic acid. Crotonyl-CoA and butyryl-CoA were separated using reversed phase HPLC (Agilent 1200 series) on an Eclipse XDB-C18 column (4.6×150 mm). The column was run at a flow rate of 1 mL min−1 and 26 μL of sample was injected onto the column equilibrated with 5% methanol and 95% 10 mM ammonium acetate, followed by a linear gradient to 95% methanol over 22 min. Crotonyl-CoA eluted at 8 min and butyryl CoA at 8.6 min.
MS analysis of NADPH adduct with crotonyl-CoA
HPLC purification of reaction intermediate was carried out as described in previous section. MS analysis on the metabolite peak eluting at 6.4 min was performed on a Shimadzu 2010 EV APCI Spectrometer using a Luna 5μm C8(2) 100 Å, 50 × 1.0 mm column (Phenomenex). The pure intermediate was subjected to a gradient of 0–60 % methanol in 10 mM ammonium acetate over 11 min. The isolated metabolite eluted at 9.2 min with the [M+2H]2+ ion peak m/z 791.95 and the [M+2Na]2+ ion peak m/z 813.95, consistent with an adduct of NADPH with crotonyl-CoA (calculated mass 1578.21 Da, m/z 789.11 for the +2 ion) from the reaction mix.
ACP linked substrate and ER activity assay
The ACP-linked HMG substrate was generated and ER was assayed as described previously (Gu et al., 2009). Briefly, 100 μM of ACP-linked substrate was incubated sequentially with 10 μM enzymes (CurA halogenase + CurE ECH1 + CurF ECH2) and the corresponding cofactors at 25°C for 25 min each. The halogenase reaction was initiated by exposing the reaction mixture to O2. The γ-chloro-β-methylcrotonyl-ACP sample was further treated with CurF ER (100 nM) or JamJ ER (100 nM) for 5 min at 25°C. The reactions were quenched by addition of 10% formic acid (CH2O2) and the ACP-loaded sample was separated from the reaction mix by reverse phase HPLC using a Source 15RPC ST4.6/100 column (GE Healthcare). The proteins were eluted using a linear gradient from 30% to 70% of CH3CN (0.05% CF3COOH and 0.05% CH2O2)/H2O (0.05% CF3COOH and 0.05% CH2O2).
ACP sample analysis by FT-ICR mass spectrometry
HPLC fractions were diluted at a 40:100 ratio in 50/50 HPLC-grade H2O (ThermoFisher Scientific) and CH3CN (ThermoFisher Scientific) with 0.1% CH2O2 (ThermoFisher Scientific). Samples were directly infused into the electrospray source of an actively shielded 7T SolariX quadrupole FT-ICR mass spectrometer (Bruker Daltonics). Data were gathered from m/z 200–2000 in positive ion mode. Electrospray was conducted at 4,500V with 128 scans per spectrum and the transient set to 1M data points. External ion accumulation in a hexapole was 0.5 s with one ICR fill prior to excitation and detection. External calibration utilized HP-mix (Agilent) mixed with ubiquitin (Sigma Aldrich). Collision-induced dissociation (CID) was performed in the collision cell hexapole with ultra-high purity argon (Cryogenic Gases) at 10 V and all charge states present were fragmented. Quantification was performed as previously described (Khare et al., 2010), using the natural abundances of all isotopes.
Supplementary Material
Highlights.
An NADPH-dependent cyclopropanase (CurF ER) crystal structure at 0.96 Å is reported.
Two thioester enoyl reductases (JamJ and CurK ERs) are highly similar to CurF ER.
Insertion of a CurF ER “cyclopropanase loop” confers the unusual activity on JamJ ER.
A covalent adduct of NADPH and the enoyl thioester substrate is detected in CurK ER.
Acknowledgments
This work was supported by NIH grants DK-042303 to J.L.S. and CA-0108874 (to D.H.S., J.L.S. and W.H.G.), by the Martha L. Ludwig Professorship to J.L.S, by the Hans W. Vahlteich Professorship to D.H.S., and by NIH grant GM107148 to K.H. The GM/CA @ APS beamlines are supported by the NIH National Institute of General Medical Sciences (GM) and the National Cancer Institute (CA) at the Advanced Photon Source (APS), which is supported by the US Department of Energy Office of Science.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
Atomic coordinates and structure factors were deposited in the Protein Data Bank under the accession codes 5DP2 for CurF ER- NADP+ complex, 5DOZ for the JamJ ER-NADPH complex, 5DOV for the JamJ ER free enzyme, and 5DP1 for CurK ER.
Author contributions
D.K. and J.L.S. designed research; D.K., W.A.H. and A.T. performed research; D.K., W.A.H., K.H., and J.L.S. analyzed data; and D.K., W.A.H., D.H.S., W.H.G., K.H., and J.L.S. wrote the paper.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey DL, Gehret JJ, Khare D, Smith JL. Insights from the sea: Structural biology of marine polyketide synthases. Nat Prod Rep. 2012;29:1038–1049. doi: 10.1039/c2np20016c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames BD, Nguyen C, Bruegger J, Smith P, Xu W, Ma S, Wong E, Wong S, Xie X, Li JW, et al. Crystal structure and biochemical studies of the transacting polyketide enoyl reductase LovC from lovastatin biosynthesis. Proc Natl Acad Sci U S A. 2012;109:11144–11149. doi: 10.1073/pnas.1113029109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone CT. Isoprenoid-like alkylations in polyketide biosynthesis. Nat Prod Rep. 2008;25:845–853. doi: 10.1039/b807243d. [DOI] [PubMed] [Google Scholar]
- Chang Z, Flatt P, Gerwick WH, Nguyen VA, Willis CL, Sherman DH. The barbamide biosynthetic gene cluster: a novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- Clardy J, Fischbach MA, Walsh CT. New antibiotics from bacterial natural products. Nat Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DelProposto J, Majmudar CY, Smith JL, Brown WC. Mocr: a novel fusion tag for enhancing solubility that is compatible with structural biology applications. Protein Expr Purif. 2009;63:40–49. doi: 10.1016/j.pep.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Engene N, Rottacker EC, Kastovsky J, Byrum T, Choi H, Ellisman MH, Komarek J, Gerwick WH. Moorea producens gen. nov., sp. nov. and Moorea bouillonii comb. nov., tropical marine cyanobacteria rich in bioactive secondary metabolites. Int J Syst Evol Microbiol. 2012;62:1171–1178. doi: 10.1099/ijs.0.033761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb TJ, Berg IA, Brecht V, Muller M, Fuchs G, Alber BE. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc Natl Acad Sci U S A. 2007;104:10631–10636. doi: 10.1073/pnas.0702791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- Geders TW, Gu L, Mowers JC, Liu H, Gerwick WH, Hakansson K, Sherman DH, Smith JL. Crystal structure of the ECH2 catalytic domain of CurF from Lyngbya majuscula. Insights into a decarboxylase involved in polyketide chain beta-branching. J Biol Chem. 2007;282:35954–35963. doi: 10.1074/jbc.M703921200. [DOI] [PubMed] [Google Scholar]
- Gerwick WH, Fenner AM. Drug discovery from marine microbes. Microb Ecol. 2013;65:800–806. doi: 10.1007/s00248-012-0169-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwick WH, Moore BS. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol. 2012;19:85–98. doi: 10.1016/j.chembiol.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons BJ, Hurley TD. Structure of three class I human alcohol dehydrogenases complexed with isoenzyme specific formamide inhibitors. Biochemistry. 2004;43:12555–12562. doi: 10.1021/bi0489107. [DOI] [PubMed] [Google Scholar]
- Gu L, Jia J, Liu H, Hakansson K, Gerwick WH, Sherman DH. Metabolic coupling of dehydration and decarboxylation in the curacin A pathway: functional identification of a mechanistically diverse enzyme pair. J Am Chem Soc. 2006;128:9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]
- Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hakansson K, Wipf P, Smith JL, Gerwick WH, et al. Metamorphic enzyme assembly in polyketide diversification. Nature. 2009;459:731–735. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund J, Jornvall H, Persson B. Subdivision of the MDR superfamily of medium-chain dehydrogenases/reductases through iterative hidden Markov model refinement. BMC Bioinformatics. 2010;11:534. doi: 10.1186/1471-2105-11-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed Engl. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- Ho BK, Gruswitz F. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct Biol. 2008;8:49. doi: 10.1186/1472-6807-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Ishijima J, Yokomizo T, Ago H, Shimizu T, Miyano M. Crystal structure of anti-configuration of indomethacin and leukotriene B4 12-hydroxydehydrogenase/15-oxo-prostaglandin 13-reductase complex reveals the structural basis of broad spectrum indomethacin efficacy. J Biochem. 2006;140:457–466. doi: 10.1093/jb/mvj176. [DOI] [PubMed] [Google Scholar]
- Jones AC, Gu L, Sorrels CM, Sherman DH, Gerwick WH. New tricks from ancient algae: natural products biosynthesis in marine cyanobacteria. Curr Opin Chem Biol. 2009;13:216–223. doi: 10.1016/j.cbpa.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare D, Wang B, Gu L, Razelun J, Sherman DH, Gerwick WH, Hakansson K, Smith JL. Conformational switch triggered by alpha-ketoglutarate in a halogenase of curacin A biosynthesis. Proc Natl Acad Sci U S A. 2010;107:14099–14104. doi: 10.1073/pnas.1006738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan DH, Leadlay PF. Mutagenesis of a modular polyketide synthase enoylreductase domain reveals insights into catalysis and stereospecificity. ACS Chem Biol. 2010;5:829–838. doi: 10.1021/cb100175a. [DOI] [PubMed] [Google Scholar]
- Kwan DH, Sun Y, Schulz F, Hong H, Popovic B, Sim-Stark JC, Haydock SF, Leadlay PF. Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem Biol. 2008;15:1231–1240. doi: 10.1016/j.chembiol.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordling E, Jornvall H, Persson B. Medium-chain dehydrogenases/reductases (MDR). Family characterizations including genome comparisons and active site modeling. Eur J Biochem. 2002;269:4267–4276. doi: 10.1046/j.1432-1033.2002.03114.x. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology 276: Macromolecular Crystallography, part A. 1997:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Persson B, Hedlund J, Jornvall H. Medium- and short-chain dehydrogenase/reductase gene and protein families: the MDR superfamily. Cell Mol Life Sci. 2008;65:3879–3894. doi: 10.1007/s00018-008-8587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porte S, Valencia E, Yakovtseva EA, Borras E, Shafqat N, Debreczeny JE, Pike AC, Oppermann U, Farres J, Fita I, et al. Three-dimensional structure and enzymatic function of proapoptotic human p53-inducible quinone oxidoreductase PIG3. J Biol Chem. 2009;284:17194–17205. doi: 10.1074/jbc.M109.001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quade N, Huo L, Rachid S, Heinz DW, Muller R. Unusual carbon fixation gives rise to diverse polyketide extender units. Nat Chem Biol. 2012;8:117–124. doi: 10.1038/nchembio.734. [DOI] [PubMed] [Google Scholar]
- Rosenthal RG, Ebert MO, Kiefer P, Peter DM, Vorholt JA, Erb TJ. Direct evidence for a covalent ene adduct intermediate in NAD(P)H-dependent enzymes. Nat Chem Biol. 2014;10:50–55. doi: 10.1038/nchembio.1385. [DOI] [PubMed] [Google Scholar]
- Rosenthal RG, Vogeli B, Quade N, Capitani G, Kiefer P, Vorholt JA, Ebert MO, Erb TJ. The use of ene adducts to study and engineer enoyl-thioester reductases. Nat Chem Biol. 2015;11:398–400. doi: 10.1038/nchembio.1794. [DOI] [PubMed] [Google Scholar]
- Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1 2010 [Google Scholar]
- Smith JL, Khare D. CHAPTER 18 Recent advances in the structural and mechanistic biology of non-haem Fe(ii), 2-oxoglutarate and O2-dependent halogenases. 2-Oxoglutarate-Dependent Oxygenases (The Royal Society of Chemistry) 2015:401–413. [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- Tartoff KD, Hobbs CA. Improved media for growing plasmid and cosmid clones. Bethesda Research Labs Focus. 1987;9:12. [Google Scholar]
- Thorn JM, Barton JD, Dixon NE, Ollis DL, Edwards KJ. Crystal structure of Escherichia coli QOR quinone oxidoreductase complexed with NADPH. J Mol Biol. 1995;249:785–799. doi: 10.1006/jmbi.1995.0337. [DOI] [PubMed] [Google Scholar]
- Verdier-Pinard P, Sitachitta N, Rossi JV, Sackett DL, Gerwick WH, Hamel E. Biosynthesis of radiolabeled curacin A and its rapid and apparently irreversible binding to the colchicine site of tubulin. Arch Biochem Biophys. 1999;370:51–58. doi: 10.1006/abbi.1999.1363. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Gay DC, Demeler B, White MA, Keatinge-Clay AT. Divergence of multimodular polyketide synthases revealed by a didomain structure. Nat Chem Biol. 2012;8:615–621. doi: 10.1038/nchembio.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.