

Abstract
To provide new and expanded proteome documentation of the opportunistically pathogen Candida albicans, we have developed new protein extraction and analysis routines to provide a new, extended and enhanced version of the C. albicans PeptideAtlas. Two new datasets, resulting from experiments consisting of exhaustive subcellular fractionations and different growing conditions, plus two additional datasets from previous experiments on the surface and the secreted proteomes, have been incorporated to increase the coverage of the proteome. High resolution precursor mass spectrometry (MS) and ion trap tandem MS spectra were analyzed with three different search engines using a database containing allele-specific sequences. This approach, novel for a large-scale C. albicans proteomics project, was combined with the post-processing and filtering implemented in the Trans Proteomic Pipeline consistently used in the PeptideAtlas project and resulted in 49372 additional peptides (3-fold increase) and 1630 more proteins (1.6-fold increase) identified in the new C. albicans PeptideAtlas with respect to the previous build. A total of 71310 peptides and 4174 canonical (minimal non-redundant set) proteins (4115 if one protein per pair of alleles is considered) were identified representing 66% of the 6218 proteins in the predicted proteome. This makes the new PeptideAtlas build the most comprehensive C. albicans proteomics resource available and the only large-scale one with detections of individual alleles.
Graphical abstract

Introduction
Candida albicans, an inhabitant fungus of the gastrointestinal and genitourinary tracts in the human microbiota, may under certain circumstances (e.g., present in patients with a weakened immune system from AIDS or patients in an intensive care unit), become opportunistically pathogenic and hence become the etiological agent of a severe type of infection called candidiasis. In the search for diagnostic and prognostic biomarkers for candidiasis, a large assortment of proteomics studies have been performed [1], or with the objective to better understand clinically relevant biological processes such as the interaction with cells of the immune system [2-4], apoptosis [5,6] or the virulence-associated morphological yeast-to-hypha switch [7,8]. However, despite the extensive efforts on clinical aspects from the proteomics view [9-12], online public proteomic repositories were sparse. Our previously published C. albicans PeptideAtlas [13] described the first large-scale public proteomic resource for the study of this opportunistic pathogenic fungus. With over 2500 proteins and representing approximately 41% of the predicted proteome, the C. albicans PeptideAtlas attained unprecedented proteome coverage and is still the first human fungal pathogen present in the PeptideAtlas project [14,15]. However, this coverage lags far behind the coverage for other species, such as the yeast species Saccharomyces cerevisiae (61%) [16] and Schizosaccharomyces pombe (71%) [17]. To bridge this gap we have added additional high resolution precursor MS datasets based on purification strategies that collectively strive for maximizing the coverage of the detectable proteome. One of the approaches consists of an exhaustive subcellular fractionation based on differential sequential centrifugations followed by protein-level separation on SDS-PAGE; the other approach is based on a peptide-level separation by OFFGEL preparative isoelectric focusing of peptides [18,19] from a pool from five different culture conditions. In addition, two high-resolution C. albicans MS datasets corresponding to published works on the secreted proteome [20] and on the yeast and hyphal cell forms surface proteome [21] have also been included in the compilation of MS data files. These new datasets, along with the previously stored raw MS datafiles comprising the former version of the C. albicans PeptideAtlas [13], have all been reprocessed and analyzed through the Trans Proteomics Pipeline, TPP [22,23] using multi-search search engine approach utilizing a sequence database with allele-specific sequences to generate the most comprehensive existing C. albicans proteomics resource with unprecedented proteome coverage.
Material and Methods
Cell strains and culture media
The Candida albicans strain used throughout this development is the widely adopted wild-type SC5314, the strain used as the reference sequence in Candida Genome Database, CGD [24], which was also used as the reference sequence database for spectra to peptide assignment. The exhaustive subcellular fractionation is derived from protoplasts obtained from cells cultivated in YED culture medium (1% D-glucose, 1% Difco yeast extract and 2% agar, w/v).
For fractionation based on preparative isoelectric focusing of tryptic peptides, the following culture conditions were used: YED medium + 5% H2O2 at 30° C; YED medium at 42° C; YED medium at 30 °C until stationary phase; RPMI 1640 medium (supplemented with 5% Fetal Bovine Serum, v/v) at 37° C and Minimal Medium at 30° C.
Cell lysis and protein digestion
C. albicans cells from the different culture conditions were washed three times with ice-cold PBS and then scraped and collected by centrifugation at 5000g. Pellets were resuspended in lysis buffer (30mM Tris-HCl pH 8.5, 7M urea, 2M thiourea, 4% CHAPS, 1% protease inhibitor cocktail -Roche- and 0.5% PMSF). An equal volume of 0.4-0.6 mm diameter glass beads was added. Subsequently, cells were disrupted in a FastPrep cell breaker. Supernatants were separated by centrifugation at 3000g for 10 min and protein quantitation was measured using a Bradford assay (Biorad). Equal amounts of each condition (250 μg/sample) were pooled and denatured by adding 25 mM DTT for 30 min at 60°C. Then, samples were loaded into an Amicon (Nanosep 10K Omega; Pall Corporation) and centrifuged 45 min at 12000 g. Samples were washed twice with DB2 buffer (20mM TEAB, 0,5% sodium deoxycolate, w/v) and alkylated with 50 mM Iodoacetamide during 20 min in the dark. After twice washes with DB2, digestion was performed by adding sequence grade-modified trypsin (Roche) at an enzyme to substrate ratio of 1:50. After 12h in the dark at RT, peptides were collected into a clean collection tube and the Amicon was washed with DB2 and the flow-through was collected with samples acidified with 0.5% (v/v) TFA. Any protein precipitation was separated by centrifugation for 5 min at 16000 g.
OFFGEL peptide fractionation
For peptide isoelectric focusing (IEF) separation, the resulting peptide mixture (1.2mg in total) was resuspended in a buffer containing 6% glycerol and 1.2% ampholytes in the 3-10 pH linear OFFGEL buffer (7M Urea, 2M thiourea, 1% DTT w/v) (GE Healthcare, Uppsala, Sweden). Sample volumes of 150 μl were loaded onto a commercially available 24-cm IPG strip with a linear 3-10 pH gradient (GE Healthcare) after rehydration of the gel for 20 min in a well of 40 μl rehydration solution. Cover fluid (mineral oil, Agilent Technologies) was applied to both ends of the gel strip. Electrofocusing of the peptides was performed at 20°C and 50 μA until 50 kVh were reached using an Agilent 3100 OFFGEL fractionator (Agilent Technologies) following the manufacturer instructions. Fractions were recovered, peptides extracted from each well with 2% TFA (v/v) and desalted by passing through a home-made column packed with Poros 50 R2 resin (Applied Biosystem, Foster City, CA, USA). Peptides were eluted with 50% ACN (v/v) in 0.1% TFA (v/v) and the fractions were dried and reconstituted in 0.1% formic acid (v/v) just before LC-MS analysis.
Subcellular fractionation
Sequential incremental centrifugations were used to selectively enrich for different types of membranes and organelles in the pellets, while also collecting soluble proteins from the supernatant (Figure 1). First, protoplasts were obtained as described in Pitarch et al [25] and then lysed using a combination of osmotic shock and Dounce homogenization. Protoplast lysates were then subjected to a low centrifugal force, 300 g, resulting in a pellet, P300, containing unlysed cells and large debris (Fraction A), and a supernatant, S300, that is subsequently, subjected to increasing centrifugation speeds of 13,000 and 100,000 g. These steps, respectively, generate a pellet, P13000, containing vacuolar and plasma membrane and other structures such as ER, mitochondria and nuclei (Fraction B), and a pellet, P100000, containing Golgi membranes and transport vesicles (Fraction C). The supernatant of the last centrifugation containing soluble proteins was also collected, S100000 (Fraction D). For each of these 4 fractions (P300, P13000, P100000 and S100000), 120μg of protein was separated by one-dimensional SDS-PAGE 4 20% Bis-Tris gels (mini-protean TGX Stain-free precast Gels, BioRad). The gel was stained with Coomassie blue and each lane was cut into 20 bands. Gel slices were cut into 1mm3 cubes, washed twice with water, dehydrated with 100% ACN (v/v), and incubated with 10 mM DTT in 50 mM NH4HCO3 for 30 min at 56°C for protein reduction. The resulting solution was subsequently alkylated by incubation with 55 mM iodoacetamide in 50 mM NH4HCO3 for 20 min at room temperature in the dark. The gel pieces were washed with 50% ACN (v/v), and then washed again with 10 mM NH4HCO3, dehydrated with 100% ACN (v/v), and then dried in a vacuum concentrator. The gel pieces were rehydrated by adding sequence grade-modified trypsin (Roche) 1:20 in 50 mM NH4HCO3 and incubated overnight at room temperature in the dark for protein digestion. Supernatants were transferred to clean tubes, and gel pieces were incubated in 50 mM NH4HCO3 at 50° C for 1h. The supernatants were collected and the remaining peptides were extracted by incubation with 5% formic acid for 15 min and with 100% ACN for 15 min more. The extracts were combined, and the organic solvent was removed in a vacuum concentrator.
Figure 1.

The strategies implemented ad hoc to maximize the coverage of the proteome consist of a subcellular fractionation based on increasing centrifugation speeds followed by exhaustive protein-level separation on SDS-PAGE; and OFFGEL peptide-level separation from a pool of peptides from different growing and culture conditions. The MS output results, combined with additional MS datasets from works on the secreted and surface proteomes, along with the datasets in the first atlas were all processed with the TPP.
Compilation of additional C. albicans MS datasets
In addition to the current datasets, two high-resolution precursor C. albicans MS datasets were compiled in order to contribute to the new PeptideAtlas build, extending the coverage in two more specific niches, the set of secreted proteins obtained following the method described in [20] and the set of surface-exposed proteins, also termed surfacome, of both hyphal and yeast form cells as described in [21].
LC-MS/MS
All the samples obtained in the exhaustive subcellular fractionation and the OFFGEL peptide separation were analyzed in an LTQ XL Orbitrap (ThermoFisher) equipped with a nanoESI ion source. Samples were loaded into a chromatographic system consisting in a C18 preconcentration cartridge (Agilent Technologies) connected to a 60 cm long, 100 μm i.d. C18 column (NanoSeparations) for the OFFGEL samples and a 15 cm long, 100 μm i.d. C18 column (Nikkyo Technos Co.) for the subcellular fractionation samples.
For samples obtained using the OFFGEL approach, the injected volume was 8% of the volume from each fraction whereas in the subcellular fractionation, one-third of the volume of each digested gel band was injected.
High-resolution LC separation was performed at 0.25 μL/min using a 360-min acetonitrile gradient (OFFGEL samples) and at 0.4 μL/min in a 90-min acetonitrile gradient (subcellular fractionation samples). Both gradients ranged from 3 to 40% (solvent A: 0.1% formic acid, solvent B: acetonitrile 0.1% formic acid). The HPLC system was composed of an Agilent 1200 capillary nano pump, a binary pump, a thermostated micro injector and a micro switch valve. The LTQ XL Orbitrap was operated in the positive ion mode with a spray voltage of 1.8 kV. The spectrometric analysis was performed in a data dependent mode, acquiring a full scan followed by 10 MS/MS scans (CID, collision energy 35%) of the 10 most intense signals detected in the MS scan. The full MS (range 300-1800) was acquired in the Orbitrap with a resolution of 60.000. The MS/MS spectra were acquired in the linear ion trap. Precursor ion charge state screening was set up to select monoisotopic ions and reject singly charged ions. In all cases, dynamic exclusion was enabled with a repeat count of 1 and exclusion duration of 30s.
Post spectra acquisition processing
LC-MS/MS spectra files resulting from the subcellular fractionation and the OFFGEL approaches and the secreted and surface proteomes (Figure 1), in their native vendor-specific format, along with the meta data corresponding to each approach, were submitted to PeptideAtlas via the PeptideAtlas Submission System (PASS) on-line submission form with dataset identifications PASS00402, PASS00447, PASS00408 and PASS00446. LC-MS/MS spectra files were converted to XML-based HUPO-PSI-adopted standard format for mass spectrometry output, mzML [26]. The protein sequence fasta file was obtained from the Candida Genome Database (C_albicans_SC5314_version_A22-s05-m01-r01). Unlike the previous C. albicans PeptideAtlas, for this new build the sequences in the database are allele-specific, taking advantage of the recent assembly of phased diploid C. albicans [27]. Sequences were appended with a set of common contaminant proteins from the cRAP (common Repository of Adventitious Proteins) set from the GPM (http://www.thegpm.org/crap/) and decoy counterparts for every entry to add up a total of 25168 entries.
Then database searches was performed using three different search engines: Comet [28], an open-source, freely available version of SEQUEST [29], X!Tandem [30] with the k-score algorithm plugin [31], and OMSSA [32]. The search parameters were established depending on the type of experiment and instrument (see supplementary table ‘database_search_parameters’ for a list of parameters).
Following sequence database searching, we used the TPP tool suite to validate the results. First, PeptideProphet [33] creates a discriminant search engine-independent score, models distributions of correctly and incorrectly assigned peptide spectrum matches (PSMs) and computes PSM posterior probabilities. Next, iProphet [34], was used to further refine the PSM-level probabilities and calculated distinct peptide-level probabilities using corroborating information from other PSMs in the dataset. ProteinProphet [35] then was used to further refine peptide probabilities based on the Number of Sibling Peptides (NSP) that each peptide shares within a protein; it also groups and reports proteins with a protein-level probability estimated from peptide-level probabilities.
To assemble the C. albicans PeptideAtlas, all individual iProphet files from the 20 compiled datasets (16 corresponding to the previous build plus 4 new extensive datasets) were filtered at a variable probability threshold to reach a constant PSM-level FDR threshold of 0.001 across all datasets. The new C. albicans PeptideAtlas is made available at https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/buildDetails?atlas_build_id=443 The Mayu [36] software designed for large-scale protein FDR estimation was used to report FDR values at different levels (PSM, unique peptides and protein-level) for the whole build based on a strategy that estimates the number of false positive protein identifications from the number of proteins containing false positive PSMs, including a correction for high proteome coverage.
Functional analysis and estimation of protein abundances
To perform functional analyses, we used the resources available at CGD GOSlim, a tailor made subset of GO terms specific to Candida biology, and GOSlimMapper, a software that maps a provided list of genes to the high-level set of GOSlim terms in either of the three ontologies. Protein abundances were estimated using the emPAI method [37] and the online software emPAI Calc [38] for the set of proteins identified in the subcellular fractionation approach since it represents the largest contribution to this PeptideAtlas build. The emPAI values were log transformed in order to obtain a normalized, symmetrical around 0 abundance scale that was apportioned in the group of the most abundant proteins (from the largest value up to the one representing percentile 0.85 in the scale); the group of proteins with high abundance (between 0.85 and 0.75 in the scale); proteins with abundance around the median (the two central quartiles); low abundant proteins (those with emPAI values between percentiles 0.25 and 0.15); and the least abundant proteins (those corresponding to percentile 0.15 and lower in the emPAI scale).
Results and Discussion
Strategies for exhaustive proteome characterization
The experimental approaches were specifically designed to improve as much as possible of the C. albicans proteome, especially considering its great plasticity and variability.
In the subcellular fractionation approach, (Figure 1) each of the fractions is enriched in different organelles and therefore ideally contributes with different subsets of proteins [39]. In Fraction A, the low centrifugal force enriches for larger membranes and complexes. The subsequent increasing centrifugation speed enriches Fraction B in other types of membranes (vacuolar, nuclear, endosomal and plasma) and also in Golgi complex and endoplasmic reticulum. Fraction C, the pellet resulting from the highest speed, contains some smaller structures like some endosomal membranes, parts of Golgi complex and transport vesicles. Finally, Fraction D, the final supernatant, contains soluble cytoplasmic and other released proteins. A total of 100 MS output files, corresponding to 20 slices from each of the four fractions run in SDS-PAGE (plus one extra replicate for fraction C) (Table 1) were obtained. This approach makes by itself the largest contribution to the C. albicans PeptideAtlas build with over 650,000 spectra of which 350,499 could be identified, and allocated to 28,599 peptides (5,839 identified exclusively in this dataset) corresponding to roughly 3,000 proteins, 48% of the full proteome.
Table 1.
Overview of the new datasets that were reprocessed together with the datasets from the previous version of the PeptideAtlas to produce the new build.
| Sample (as named in the web interface) | PASS id | Experiment | MS type | Fractions / Replicates | Number of spectra files |
|---|---|---|---|---|---|
| Calb_subcel_fract | PASS00402 | Differential sequential centrifugations and SDS-PAGE | LTQ XL Orbitrap |
FractionA × 20 SDS-PAGE slices | 100 |
| FractionB × 20 | |||||
| FractionC : 2 replicates | |||||
| FractionD × 20 | |||||
| Calb_offGel | PASS00447 | OFFGEL Preparative Isoelectric focusing of peptides | LTQ XL Orbitrap |
24 OFFGEL fractions | 24 |
| Calb_ves_secretome | PASS00408 | C.albicans secreted proteins [20] | LTQ-Orbitrap Velos |
Wt and RMLU2 strains:
|
12 |
| Calb_surfome | PASS00446 | Surface-exposed proteins from hyhpal and yeast form cells [21] | LTQ-Orbitrap Velos |
Yeast form: 3 replicates; | 14 |
| Serum-induced hyphae: 4 replicates; | |||||
| Inactivated serum-induced hyphae: 2 replicates; | |||||
| Lee medium-induced hyphae: 4 replicates |
Total MS files: 150
In the OFFGEL approach, the variability of the proteome was stimulated by the different growing conditions. The thermal and oxidative types of stress ideally enforce the cell to produce certain populations of proteins to face these culture conditions; the minimal medium makes cells adapt to deprivation of certain nutrients and therefore activate alternative mechanisms or pathways; hyphae generated in RPMI medium supplemented with FBS, provide a set of proteins inherent to this growing form; and finally, cells in the stationary phase also ideally contribute with proteins that would not be present in other more favorable conditions. These multiple growing conditions subjected to peptide-level separation by the OFFGEL system, generated 24 fractions and equivalent MS output files that make, as a dataset, the second largest contribution to the C. albicans PeptideAtlas with 460,000 spectra searched, 223,395 of them identified and assigned to 27,360 peptides (5,846 unique to this dataset) which, in turn, were assigned to more than 3,000 proteins. An overview of the contributions of each dataset to the entirety of the atlas is depicted in Figure 2.
Figure 2.

Contribution of the different constituent datasets of the C. albicans PeptideAtlas. The two implemented ad hoc strategies (subcel_fract and OFFGEL, as named in the web interface) have both the largest numbers of distinct peptides (height of blue bars) and contribute most to the increasing of the cumulative number of distinct peptides identified (total height of bars), and also represent the largest contributions in terms of spectra identified (width of bars). The experiments that constituted the previous PeptideAtlas are annotated for comparison.
Assessment of increment in proteome coverage
A total 229 MS runs (124 corresponding to the datasets implemented ad hoc, plus 26 corresponding to the additionally compiled datasets on secreted and surface proteomes, plus 79 datasets from the previous C. albicans PeptideAtlas build) generated 2,255,208 spectra of which more than one-third, 984,462, could be allocated to a peptide sequence. In the resulting outcome, for a PSM FDR threshold set at 0.10%, 71,310 peptides are detected which can be explained by the minimal non-redundant set of 4174 canonical C. albicans protein sequences (4115 if only one protein sequence per pair of alleles is considered), representing 66% of the 6218 (as of March 2015) predicted different protein sequences.
With respect to the 22,000 peptides and 2545 proteins reported in analogous manner in the first version of the C. albicans PeptideAtlas, the multi-search engine reprocessing with the new LC-MS/MS datasets represents an increment of more than 3-fold in terms of peptides and 1.6-fold in the number of identified proteins.
One remarkable additional value in the C. albicans PeptideAtlas is the report of highly confident (ProteinProphet probability > 0.9) identification of proteins corresponding to uncharacterized genes (following the terminology in CGD), i.e. those genes without previously known empirical evidence for a translated product. These amounted to 1564 in the previous PeptideAtlas build and has notably increased to 2860 (note that uncharacterized is unrelated to which of the alleles originates the protein product), representing an increment from one-third to almost two-thirds (63%) of the total uncharacterized genes in CGD (Figure 3). As for the verified set of genes, those that do have experimental evidence for a gene product, 76% are covered in the list of canonical proteins in this build. (See Supplementary Tables CGD_uncharacterized_vs_PA_canonical.xls and CGD_verified_vs_PA_canonical.xls)
Figure 3.

Share of the uncharacterized genes (genes for which there is no empirical evidence of a protein product) and Verified genes (those having a protein product with a given GO annotation) in CGD that are covered by canonical (with high confidence identification) proteins in the C. albicans PeptideAtlas.
Gene Ontology enrichment analysis of the covered and undetected proteome subsets
The set of 4174 identified canonical proteins was subjected to a GO term enrichment analysis using GOSlimMapper in CGD. This analysis revealed no specific bias towards any particular biological process in the covered part of the proteome showing a very similar (no statistically significant difference) histogram of frequencies of GO Slim biological process terms as that for the entire genome at CGD.
The undetected set of 2103 proteins, obtained by subtracting the 4115 canonical proteins in PeptideAtlas from the 6218 predicted proteins in CGD, was similarly enriched in GO Slim biological processes revealing, as expected, very heterogeneous annotations with a majority of the genes that cannot be grouped under a more precise category (in the Slim pruned GO tree) than “biological process” which means these are likely uncharacterized genes. This undetected set is where the focus should be laid on to further extend the proteome coverage in future builds of the C. albicans PeptideAtlas by designing specific strategies to detect, at least, those proteins that do have some specific biological process or cellular component annotations.
Both GO analyses for the covered and undetected subsets are available in supplementary material (GO_SLIM_PA_201503.xls and GO_SLIM_undetectedCGD_201503.xls)
Assessment of protein abundance and functional analysis
Once the abundance clusters were established based on the emPAI method for the set of 3,000 proteins identified in the subcellular fractionation approach (supplementary file ‘emPAI_results.xls’), a functional analysis was carried out on them. First, they were mapped onto the ergosterol biosynthesis pathway (Figure 4), which is of great interest since it is specific to fungi (the functional equivalent in mammalian cell membranes is cholesterol) and is therefore the target of many antifungal drugs that exploit selective toxicity. In addition, farnesol, a by-product in this pathway, has been shown to have a role in quorum sensing [40] and apoptosis induction [41]. As depicted in figure 4, most of the proteins, representing 18 out of 22 steps in the pathway, were detected, with a majority of them belonging in the high abundance groups.
Figure 4.

The C. albicans PeptideAtlas contains information on the detection and estimated abundances (emPAI) of proteins representing 18 out of 22 steps in the biosynthesis of ergosterol, an essential pathway comprising targets of many antifungal drugs.
Then, a GO enrichment analysis was also applied to the abundance sets, in this case combining the two high abundance protein groups on one hand, and the two low abundance groups on the other. The top 3 enriched and under-represented GO-Slim annotations of each the Biological Process (BP), Cellular Component (CC) and Molecular Function (MF) ontologies were selected and shown in figure 5. Interestingly, low abundance proteins appear to be enriched in the BP terms ‘pseudohyphal growth’ and ‘cell budding’ and consistently in ‘hyphal tip’ and ‘site of polarized growth’ CC annotations. Signal transduction and kinase activities are also enriched in this set of low abundance proteins, in agreement with the described low quantities of the proteins that carry out these functions. The set of high abundance proteins, as expected, are enriched in some of the terms for which the low abundance proteins are under-represented, such as ‘cell wall’ and ‘extracellular region’, and conversely are under-represented in some other processes and functions in which low abundance proteins seem to be involved, like ‘pseudohyphal growth’ and ‘kinase activity’.
Figure 5.
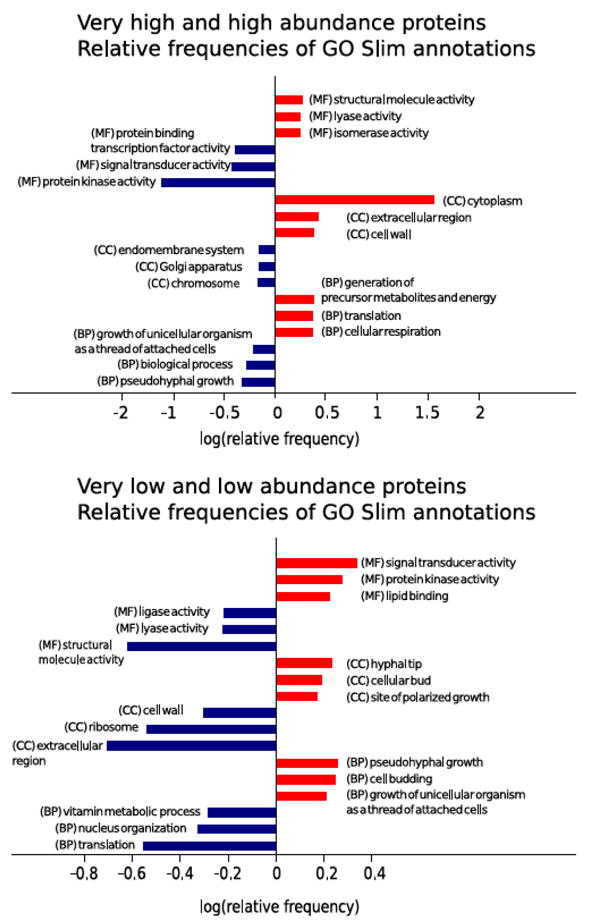
Top three enriched and under-represented annotations for the Molecular Function, Cellular Component and Biological Process ontologies for the sets of very low and low abundance proteins (a) and high and very high abundance proteins (b) in the subcellular fractionation approach.
Allele specific proteins
Taking advantage of the database containing allele specific sequences, we have examined the lists of proteins that can be allocated to their specific originating allele.
Of the 4174 identified canonical C. albicans protein sequences, there are 59 pairs of alleles with different sequences for which both protein products have been identified through their differentiating peptides.
There are 354 proteins from allele B that have been labeled canonical without a similar detection of their corresponding allele A counterparts. This means there is solid evidence for the presence of the protein originating from allele B, but does not necessarily imply that only the form from allele B was present. Proteins from allele A might have also been present in the samples but are not included in the minimal non-redundant list either because all their identified peptides are shared and can be explained by the canonical B which do have additional exclusive peptide evidence (the ‘A’ protein forms are subsumed, in PeptideAtlas terminology); or because they have a too weak peptide evidence that could distinguish them from the canonical identification (termed possibly distinguished and conservatively excluded from the canonical list).
Three more proteins (C5_01745W_B, C5_01765C_B and C5_01775C_B) from allele B were identified but are related to the mating type locus (MTL), and therefore exist only in haplotype B. And 11 more proteins, encoded by mitochondrial DNA, cannot be allocated to either allele. Subtracting the canonical proteins from allele B and the described particular cases from the 4056 protein count that excludes identifications from both alleles, there remain 3688 (4056 – 354 – 3 – 11) identifications that could be originated from allele A. However, within these, 2070 have identical allele A and B sequences. In those cases, either allele is equally likely the origin of the identified protein and any one allele can be chosen as representative. Notably, this does not imply that the remaining 1618 (3688 – 2070) proteins necessarily correspond exclusively to allele A. Out of these, 1205 allele B forms are indistinguishable to that from A, which means their identified peptides are the same and mapped to common parts of the protein sequence. And lastly, 413 do have independent peptide evidence of being originated from allele A, but yet again this is not exclusive, the form from allele B might be subsumed or possibly distinguished. An overview of how the canonical proteins in the C. albicans PeptideAtlas are distributed by genomic origin and the presence level is summarized in Table 2.
Table 2.
Distribution of the canonical proteins in PeptideAtlas by genomic origin. In the PeptideAtlas terminology, subsumed refers to proteins whose peptides are also present in another canonical protein which has additional independent peptide evidence; possibly distinguished means a weak peptide evidence that could possibly distinguish the protein from the canonical identification (these are conservatively excluded from the canonical list); and indistinguishable refers to proteins having different sequence but with peptide evidence only in the common parts.
| Allele A | Allele B | Sequences A and B | mtDNA |
|---|---|---|---|
|
| |||
| 59 canonical | 59 canonical | different | |
|
| |||
| subsumed or possibly distinguished | 354 canonical | different | |
|
| |||
| 3 canonical from MTL exist only in B | |||
|
| |||
| 11 canonical | |||
|
| |||
| 2070 canonical (chosen as representative) | identical | identical | |
|
| |||
| 1205 canonical | indistinguishable | different | |
|
| |||
| 413 canonical | subsumed or possibly distinguished | different | |
Total : 4174 canonical proteins / 4115 (only one canonical protein per pair of alleles)
The significance of the characterization and study of allelic variant proteins in C. albicans has previously been highlighted for the case of the ALS gene family [42]. This gene family encodes eight cell-wall glycoproteins (Als1p to Als7p and Als9p) involved in adhesion to host surfaces, a key virulence factor [43]. In particular, Als3p allelic protein isoforms have been shown to have functional differences [44]. In this PeptideAtlas build, 3 proteins from the ALS family have been identified with independent peptide evidence from either allele. Als2p and Als4p have peptide evidence with single genome mapping to allele A, whereas Als9p has exclusive peptide evidence from allele B. This information could be used as a foundation to enable targeted proteomics assays to independently monitor each of the allelic variants and provide some insights on whether these proteins, like Als3p, contribute differently to adhesion.
Conclusion
We have described the new C. albicans PeptideAtlas 2015-02 which represents a great increase in the number of characterized peptides and proteins with respect to the previous build. A total of 71,310 peptides and 4174 protein sequences make it the most comprehensive proteomics resource available up to date with a coverage of 66% of the total predicted proteome. In addition, highly confident protein identifications have been reported for 63% of the genes termed uncharacterized (without a known protein product) in CGD. Furthermore, for the first time in a large-scale C. albicans proteomics project, an allele-specific protein sequence database has been searched and integrated into the resource enabling the ability to trace the identified proteins back to their originating allele. This, for example, enables the development of targeted assays to distinguish protein isoforms via the PeptideAtlas web interface [45] to select candidate proteotypic peptides for the basis of the best peptides to use.
While this effort provides an unbiased representative picture of the whole C. albicans proteome, there is still room for further improvement. Future PeptideAtlas builds may include other C. albicans datasets generated by the community reusing for instance spectra deposited in ProteomeXchange, or datasets specifically generated to detect the elusive proteins that may be expressed only under very particular circumstances, difficult to extract proteins, or may be translated in very low quantities.
Finally, improvements in the software pipeline used for post-acquisition analysis or in the protein sequence database will also motivate the construction of new C. albicans PeptideAtlas builds in the future.
Supplementary Material
Highlights.
Different culture conditions and fractions for exhaustive proteome characterization
C. albicans proteome coverage reaches 66% of predicted proteome
First large-scale proteomic dataset including detection of individual alleles
Web interface aids in selection of proteotypic peptides for targeted experiments
Significance.
This C. albicans PeptideAtlas accounts for 66% of the known C. albicans proteome making it the most comprehensive online proteomics resource available for this clinically relevant fungus. It is, in addition, the only large-scale proteomics resource available that includes detection of individual alleles. Many useful features are available through the web interface allowing, for instance, selection of candidate proteotypic peptides for targeted experiments.
Acknowledgments
This work has been financially supported in part by project BIO2012-31767 Ministerio de Economía y Competitividad, Spain, PROPMT (S2010/BMD-2414) from the Comunidad Autónoma de Madrid, REIPI, Spanish Network for the Research in Infectious Diseases (RD12/0015/0004), and PRB2 projects PT13/0001/0004 and PT13/0001/0008 from the ISCIII. VV held a research contract associated to project BIO2012-31767.
This work was funded in part by the American Recovery and Reinvestment Act (ARRA) funds through National Institutes of Health from the NHGRI grant RC2HG005805, the NIGMS grants R01GM087221 and 2P50GM076547 to the Center for Systems Biology, the National Institute of Biomedical Imaging and Bioengineering grant U54EB020406, the National Science Foundation MRI grant 0923536, and the EU FP7 ‘ProteomeXchange’ grant 260558.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pitarch A, Nombela C, Gil C. Prediction of the clinical outcome in invasive candidiasis patients based on molecular fingerprints of five anti-Candida antibodies in serum. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.004010. M110.004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernández-Arenas E, Cabezón V, Bermejo C, Arroyo J, Nombela C, Diez-Orejas R, et al. Integrated proteomics and genomics strategies bring new insight into Candida albicans response upon macrophage interaction. Mol Cell Proteomics. 2007;6:460–78. doi: 10.1074/mcp.M600210-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Cheng S-C, Joosten L a B, Kullberg B-J, Netea MG. Interplay between Candida albicans and the mammalian innate host defense. Infect Immun. 2012;80:1304–13. doi: 10.1128/IAI.06146-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gow N, Veerdonk F van de. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat Rev. 2011;10:112–22. doi: 10.1038/nrmicro2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsdale M. Programmed cell death in pathogenic fungi. Biochim Biophys Acta (BBA)-Molecular Cell Res. 2008;1783:1369–80. doi: 10.1016/j.bbamcr.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 6.Hao B, Cheng S, Clancy CJ, Nguyen MH. Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis. Antimicrob Agents Chemother. 2013;57:326–32. doi: 10.1128/AAC.01366-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monteoliva L, Martinez-Lopez R, Pitarch A, Hernaez ML, Serna A, Nombela C, et al. Quantitative proteome and acidic subproteome profiling of Candida albicans yeast-to-hypha transition. J Proteome Res. 2011;10:502–17. doi: 10.1021/pr100710g. [DOI] [PubMed] [Google Scholar]
- 8.Vialás V, Perumal P, Gutierrez D, Ximénez-Embún P, Nombela C, Gil C, et al. Cell surface shaving of Candida albicans biofilms, hyphae and yeast form cells. Proteomics. 2012;12:2331–9. doi: 10.1002/pmic.201100588. [DOI] [PubMed] [Google Scholar]
- 9.Cabezón V, Llama-Palacios A, Nombela C, Monteoliva L, Gil C. Analysis of Candida albicans plasma membrane proteome. Proteomics. 2009;9:4770–86. doi: 10.1002/pmic.200800988. [DOI] [PubMed] [Google Scholar]
- 10.Pitarch A, Nombela C, Gil C. Proteomic profiling of serologic response to Candida albicans during host-commensal and host-pathogen interactions. Methods Mol Biol. 2009;470:369–411. doi: 10.1007/978-1-59745-204-5_26. [DOI] [PubMed] [Google Scholar]
- 11.Pitarch A, Nombela C, Gil C. Candida albicans biology and pathogenicity: insights from proteomics. Methods Biochem Anal. 2006;49:285–330. [PubMed] [Google Scholar]
- 12.Rupp S. Proteomics on its way to study host- pathogen interaction in Candida albicans. Curr Opin Microbiol. 2004;7:330–5. doi: 10.1016/j.mib.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Vialas V, Sun Z, Loureiro Y, Penha CV, Carrascal M, Abián J, Monteoliva L, et al. A Candida albicans PeptideAtlas. J Proteomics. 2013;97:62–8. doi: 10.1016/j.jprot.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, et al. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–8. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deutsch EW, Sun Z, Campbell D, Kusebauch U, Chu CS, Mendoza L, et al. The State of the Human Proteome in 2014/2015 as viewed through PeptideAtlas: enhancing accuracy and coverage through the AtlasProphet. J Proteome Res. 2015 doi: 10.1021/acs.jproteome.5b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King NL, Deutsch EW, Ranish JA, Nesvizhskii AI, Eddes JS, Mallick P, et al. Analysis of the Saccharomyces cerevisiae proteome with PeptideAtlas. Genome Biol. 2006;7:R106. doi: 10.1186/gb-2006-7-11-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunaratne J, Schmidt A, Quandt A, Neo SP, Sarac OS, Gracia T, et al. Extensive Mass Spectrometry-Based Analysis of the Fission Yeast Proteome: The S. pombe PeptideAtlas. Mol Cell Proteomics. 2013:1741–51. doi: 10.1074/mcp.M112.023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ros A, Faupel M, Mees H, Oostrum J van, Ferrigno R, Reymond F, et al. Protein purification by Off-Gel electrophoresis. Proteomics. 2002;2:151–6. doi: 10.1002/1615-9861(200202)2:2<151::aid-prot151>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 19.Hörth P, Miller CA, Preckel T, Wenz C. Efficient fractionation and improved protein identification by peptide OFFGEL electrophoresis. Mol Cell Proteomics. 2006;5:1968–74. doi: 10.1074/mcp.T600037-MCP200. [DOI] [PubMed] [Google Scholar]
- 20.Gil-Bona A, Llama-Palacios A, Parra CM, Vivanco F, Nombela C, Monteoliva L, et al. Proteomics unravels extracellular vesicles as carriers of classical cytoplasmic proteins in Candida albicans. J Proteome Res. 2014;14:142–53. doi: 10.1021/pr5007944. [DOI] [PubMed] [Google Scholar]
- 21.Gil-Bona A, Parra-Giraldo CM, Hernáez ML, Reales-Calderon JA, Solis NV, Filler SG, et al. Candida albicans cell shaving uncovers new proteins involved in cell wall integrity, yeast to hypha transition, stress response and host-pathogen interaction. J Proteomics. 2015 doi: 10.1016/j.jprot.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller A, Eng J, Zhang N, Li X, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1 doi: 10.1038/msb4100024. 2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clin Appl. 2015 doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costanzo MC, Arnaud MB, Skrzypek MS, Binkley G, Lane C, Miyasato SR, et al. The Candida Genome Database: facilitating research on Candida albicans molecular biology. FEMS Yeast Res. 2006;6:671–84. doi: 10.1111/j.1567-1364.2006.00074.x. [DOI] [PubMed] [Google Scholar]
- 25.Pitarch A, Jiménez A, Nombela C, Gil C. Decoding serological response to Candida cell wall immunome into novel diagnostic, prognostic, and therapeutic candidates for systemic candidiasis by proteomic and bioinformatic analyses. Mol Cell Proteomics. 2006;5:79–96. doi: 10.1074/mcp.M500243-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Martens L, Chambers M, Sturm M, Kessner D, Levander F, Shofstahl J, et al. mzML--a community standard for mass spectrometry data. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.R110.000133. R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muzzey D, Schwartz K, Weissman JS, Sherlock G. Assembly of a phased diploid Candida albicans genome facilitates allele-specific measurements and provides a simple model for repeat and indel structure. Genome Biol. 2013;14:R97. doi: 10.1186/gb-2013-14-9-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–4. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 29.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 30.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 31.MacLean B, Eng J, Beavis R, McIntosh M. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics. 2006;22:2830–2. doi: 10.1093/bioinformatics/btl379. [DOI] [PubMed] [Google Scholar]
- 32.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, et al. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–64. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 33.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 34.Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, et al. iProphet: Multi-level Integrative Analysis of Shotgun Proteomic Data Improves Peptide and Protein Identification Rates and Error Estimates. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.007690. M111.007690–M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 36.Reiter L, Claassen M, Schrimpf SSP, Jovanovic M, Schmidt A, Buhmann JM, et al. Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol Cell Proteomics. 2009;8:2405–17. doi: 10.1074/mcp.M900317-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–72. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Shinoda K, Tomita M, Ishihama Y. emPAI Calc--for the estimation of protein abundance from large-scale identification data by liquid chromatography-tandem mass spectrometry. Bioinformatics. 2010;26:576–7. doi: 10.1093/bioinformatics/btp700. [DOI] [PubMed] [Google Scholar]
- 39.Harford JB, Bonifacino JS. Curr Protoc Cell Biol. John Wiley & Sons, Inc; 2001. Subcellular Fractionation and Isolation of Organelles. [DOI] [Google Scholar]
- 40.Albuquerque P, Casadevall A. Quorum sensing in fungi--a review. Med Mycol. 2012;50:337–45. doi: 10.3109/13693786.2011.652201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Léger T, Garcia C, Ounissi M, Lelandais G, Camadro J-M. The metacaspase (Mca1p) has a dual role in farnesol-induced apoptosis in Candida albicans. Mol Cell Proteomics. 2015;14:93–108. doi: 10.1074/mcp.M114.041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoyer LL, Green CB, Oh SH, Zhao X. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family – a sticky pursuit. Med Mycol. 2008;46:1–15. doi: 10.1080/13693780701435317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Groot PW, Bader O, de Boer AD, Weig M, Chauhan N. Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot Cell. 2013;12:470–81. doi: 10.1128/EC.00364-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh SH, Cheng G, Nuessen JA, Jajko R, Yeater KM, Zhao X, Pujol C, Soll DR, Hoyer LL. Functional specificity of Candida albicans Als3p proteins and clade specificity of ALS3 alleles discriminated by the number of copies of the tandem repeat sequence in the central domain. Microbiology. 2005;151:673–81. doi: 10.1099/mic.0.27680-0. [DOI] [PubMed] [Google Scholar]
- 45.Farrah T, Deutsch EW, Aebersold R. Serum/Plasma Proteomics vol. Vol. 728. Totowa, NJ: Humana Press; 2011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.