

Abstract
Pancreatic ductal adenocarcinoma (PDA) is characterized by an extremely poor prognosis, since it is usually diagnosed at advanced stages. In order to employ tools for early detection, a better understanding of the early stages of PDA development from its main precursors, pancreatic intraepithelial neoplasia (PanIN), and intraductal papillary mucinous neoplasm (IPMN) is needed. Recent studies on murine PDA models have identified a different exocrine origin for PanINs and IPMNs. In both processes, developmental pathways direct the initiation of PDA precursors from their cellular ancestors. In this review, the current understanding of early PDA development is summarized.
1. Introduction
The idea that cancer cells share properties of their embryonic predecessors is, scientifically speaking, ancient [1]. Within the last decade, this conception has mostly fueled the work in the field of cancer stem cells. As controversial as this theory might be, it is one way to explain tumor heterogeneity, and solid tumors, especially pancreatic ductal adenocarcinomas (PDA), are heterogeneous [2, 3]. Similar to embryonic development, tumor cells, or at least a subset of them, have the ability to maintain indefinite growth as well as cellular plasticity. Terminal differentiation most likely has to be disadvantageous for tumor cells in order to adapt to the hostile environment within the primary tumor, the circulation, or at the metastatic site.
In the pancreas, different cell types harbor distinct susceptibilities towards oncogenic insults. Recently, new light has been shed on the cell-of-origin question of PDA. Historically PDA was thought to arise from pancreatic ductal epithelium. Instead, murine models revealed that both ductal and acinar cells are capable of transforming into distinct precursor lesions that develop into biologically different PDA subsets [4]. The parent cell transformation in both processes is characterized by dedifferentiation with recapitulation of elements of pancreatic development. Recent data investigating the role of Sox9 in PDA initiation suggest that ductal but also centroacinar cells (CACs) are more refractory to transformation mediated by a mutated Kras allele compared to acinar cells [5]. In contrast, Pten loss results in rapid formation of invasive carcinoma, which is preceded by significant expansion of CACs [6]. This suggests that CACs, ductal cells, and acinar cells may have the potential to initiate invasive carcinoma but that each cellular context may require a different repertoire of genetic alterations for tumour initiation. Cell-specific induction of different oncogenic mutations in mice may define morphologically and molecularly distinct tumours, which may help to identify human PDA subtypes that respond differently to therapeutic intervention.
In the following, we will give an overview of pancreas organogenesis and discuss how pancreatic cancer cells exploit developmental programs during cancer initiation with respect to their cellular origin.
2. Pancreatic Morphogenesis and Lineage Segregation
The development of the murine pancreas is initiated around embryonic day 8.5 (e8.5) after gastrulation, when a pancreatic and duodenal homeobox 1- (Pdx1-) expressing (Pdx1+) population within endodermal gut tube gives rise to both the ventral and dorsal pancreas anlage [7]. A subset of Pdx1+ cells that arises from the ventral foregut eventually loses its Pdx1-expression, eventually, to form the extrahepatic bile duct [8]. Stringent genetic studies in the mouse have demonstrated that Pdx1 ablation leads to pancreatic agenesis [9, 10]. The Pdx1+ multipotent progenitor cells (MPCs), directed by cues from the surrounding mesenchyme, establish distinct cellular lineages in order to produce and drain digestive enzymes as well as maintaining glucose homeostasis [11].
In order to execute these processes, three main lineages are required: the acinar cells, producing a plethora of digestive enzymes; the ductal cells, forming a hierarchical conduit system; and the endocrine cells, organized in the islet of Langerhans, producing hormones like insulin, glucagon, pancreatic polypeptide, somatostatin, and ghrelin. Careful designed genetic lineage-tracing studies using Cre/LoxP technology have provided insight into the spatiotemporal organization of these compartments.
For example, Gu et al. utilized Pdx1-CreER TM mice to demonstrate that the Pdx1+ population truly harbors multipotent progenitors since tamoxifen administration at E9.5 labels exocrine, endocrine, and duct cells [7]. Interestingly, it is the number of Pdx1+ progenitor cells that determines the size of the pancreas in the adult mouse, suggesting that it is rather an intrinsic program of the progenitor population than growth compensation limiting organ size, like in other organs, for example, the liver [12]. Besides Pdx1, numerous transcription factors have been employed to investigate pancreatic lineage commitment in the developing embryo. Slightly later than Pdx1 (around e9.5–10.5), Ptf1a (pancreatic transcription factor 1) is expressed in MPCs, further seizing segregation from a duodenal fate while the pancreatic bud evaginates [13]. These two transcription factors are certainly the most prominent members during this early phase of organogenesis, termed primary transition. It is at the end of primary transition, approximately around e12.5, when a primitive trunk epithelium with a continuous lumen as well as tip-structures emerges [14]. This spatial tip-trunk organization was thought to be accompanied by a loss of multipotency of the primitive duct at the beginning of secondary transition [14]. However, more recent studies, employing Sox9Cre ER as well as Hnf1β CreERT2, with both transcription factors localizing to the trunk, demonstrated that either population still harbors the ability to give rise to the endocrine, acinar, and ductal lineage during secondary transition to a varying degree [15, 16].
In the adult pancreas, under tissue homeostatic conditions, Pdx1 becomes restricted to insulin-producing β-cells maintaining a β-cell-phenotype by repressing an α-cell program [17, 18], while Ptf1a remains expressed exclusively in acinar cells [19]. On the other hand, Sox9 and Hnf1β remain expressed in the ductal tree including intercalated (terminal), intralobular, and interlobular ducts as well as the main duct [20]. Thus, a set of transcription factors defines pancreatic plasticity or differentiation capacity of pancreatic progenitors in a spatiotemporally regulated manner.
The fact that mature pancreatic lineages maintain a certain degree of plasticity becomes evident in nontissue homeostatic conditions, particularly in regeneration and carcinogenesis, and will be discussed in the following section.
3. Pancreatic Cancer and Its Precursors
PDA is characterized by an extremely poor prognosis with a mortality rate almost equaling the incidence rate [21]. The underlying reason for this dismal situation is the limited possibility for early detection of this disease and, therefore, diagnosis is often only made in advanced stages, where only few, insufficient treatment options exist [22]. Thus, a better understanding of the initial steps of PDA development is important in order to develop new tools for early detection. In addition, deciphering the factors important for PDA progression will help to identify novel treatment options.
It is believed that PDA can develop from three established precursor lesions [23]: (i) pancreatic intraepithelial neoplasia (PanIN) and the cystic lesions, (ii) intraductal papillary mucinous neoplasm (IPMN), and (iii) mucinous cystic neoplasm (MCN). These precursor lesions differ in their prevalence; the majority of PDA is thought to arise from PanINs and less frequently from IPMN, whereas MCNs are rare [24]. There is indirect evidence for PanINs as precursors for PDA, which is largely based on the fact that PDA is often associated with advanced PanIN, and both share common tumor promoting genetic alterations. In contrast, cystic lesions can directly be identified as the origin for PDA on histological examination and imaging techniques such as MRI scan or endoscopic ultrasound. The possibility of imaging cystic precursor lesions also offers the chance to detect the precursor before PDA has developed. In fact, probably due to more frequent and better diagnostic imaging as well as physicians' awareness, IPMN lesions are increasingly identified in the pancreas, and ideal management of these patients is still an ongoing debate [25].
Interestingly, PDA that is associated with IPMNs has a much more favorable prognosis than PDA that is thought to arise from PanINs [26–28]. The underlying reasons for this different biological behavior are largely unknown. One possibility could be different genetic mutations during evolution of PDA from its precursors. In fact, whereas a KRAS mutation occurs nearly universally during PanIN initiation [29], KRAS is less frequently mutated in IPMNs [30, 31]. Instead, IPMNs but not PanINs frequently harbor mutations in GNAS and RNF43 [32–34]. Apart from this difference, common genetic alterations in both precursors are found in TP53 and CDKN2A (reviewed by Xiao [35] and Gnoni et al. [24]).
An additional possibility for the different biology of PanIN and IPMN-associated PDA could be a different cellular origin of the precursors. In line with this hypothesis, recent evidence from genetically engineered mouse models (GEMM) revealed that the cellular origin of PanINs and IPMNs might be different [4, 5]. This work will be discussed below.
4. Mechanisms of PanIN Development from Its Cellular Origin
Whereas there is considerable knowledge about the molecular and genetic events during progression of PanIN lesions, the mechanisms of precursor initiation are still poorly understood. Historically, PDA and its precursors were thought to develop from pancreatic ductal cells because both have a ductal morphology and express ductal markers such as cytokeratin 19 (CK19). However, this assumption was challenged by studies in mice that revealed an acinar source for PanIN lesions [36–38].
4.1. ADM/ADR: The Precursor of the Precursor?
That pancreatic acinar cells have a marked plasticity was already noted 30 years ago in pancreatitis studies on rats [39, 40]. In these experiments an acute pancreatitis was induced by repetitive injections of cerulein, a cholecystokinin analogue, which causes autodigestion of the pancreas and a pronounced inflammatory reaction. It was found that in response to this damage acinar cells form a transient duct-like metaplasia before a complete regeneration of the organ occurs. The direct in vivo evidence for an acinar source of this acinar to ductal metaplasia (ADM) was brought in 2008 using lineage-tracing techniques on a murine pancreatitis model by Fendrich et al. [41]. Further characterization of ADM has shown that it is not only accompanied by downregulation of acinar and expression of ductal markers (e.g., CK19) but also resembles pancreatic embryonic progenitor cells evidenced by reexpression of pancreatic developmental factors, such as PDX1, Sox9, and Hes1 [41–43]. Following this transient phase in response to an acute damage, the duct-like cells of ADM resume an acinar morphology and expression profile [43, 44]. In contrast to an acute damage, chronic pancreatitis leads to a persistence of ADM [45] and a failure to regenerate the pancreas. However, although acute and chronic pancreatitis can induce duct-like structures originating from pancreatic acinar cells, both are not sufficient to induce PanIN lesions per se.
Importantly, an oncogenic mutation in Kras can induce ADM from acinar cells that resembles ADM formed in response to pancreatitis [37, 46]. However, the Kras-induced ADM is persistent and not transient and is also termed acinar to ductal reprogramming (ADR) in this context. In addition to ADR, acinar expression of mutant Kras is sufficient to induce PanIN lesions [36–38, 46]. Although direct lineage-tracing evidence is missing, it is suggested that Kras-associated ADR progresses to PanIN lesions [47], in part, because the expression of key signaling pathways in ADR cells is mimicking expression detected in PanIN lesions [48]. Moreover, in mouse models with expression of mutant Kras in the pancreas lesions of acinar to ductal reprogramming precede PanIN development [43, 48]. In addition, PanINs in mice and humans are usually associated with areas of ADM/ADR [48].
4.2. Acinar Cells Give Rise to PanINs
In contrast to mouse models where mutant Kras is activated during pancreatic embryogenesis [49], mutant Kras expressed in mature acinar cells leads to PanIN formation but is insufficient to cause PDA [36–38, 46]. However, when combined with chronic pancreatitis, which is a risk factor for the development of PDA [50], mutant Kras (Kras G12V) was able to accelerate precursor formation with progression to cancer [37]. This suggests that inflammation can induce synergistic protumorigenic changes in acinar cells that cooperate with mutant Kras. Pancreatitis experiments in wild-type animals have shown that these changes could comprise reactivation of developmental factors that have been shown to be important for Kras-mediated neoplastic transformation of pancreatic acinar cells which will be discussed in detail below [42, 43].
The above mentioned studies have shown that adult pancreatic acinar cells are capable of forming PanIN lesions in the context of mutant Kras. It is also important to note that ductal cells in CK19 promoter-based expression of mutant Kras were able, albeit at much lower frequency, to form PanIN lesions [51]. In a study by Kopp et al. it was investigated by comparative recombination in adult pancreatic duct/CACs and acinar cells using tamoxifen inducible Sox9CreER and Ptf1a CreER-mediated recombination of mutant Kras [5]. Interestingly, it was found that pancreatic ductal cells including CACs were virtually incapable of transforming into PanIN lesions, whereas acinar cells readily transformed into PanIN lesions with a >100-fold greater efficiency as compared to ductal cells. Scientists in the field have speculated extensively about the possibility of CACs being the cell of origin of PanIN and PDAC. However, the compartment of CACs is still characterized insufficiently and, hence, the genetic tools to address this question appropriately are missing. Taken together, the most recent findings suggest that this is a very unlikely scenario and strongly support the model that at least in the murine Kras model acinar cells serve as the origin for PanIN lesions. This also prompts the question of which factors are important for acinar cells to transform into ADR/PanIN. Previous studies have taught us that recapitulation of developmental factors is occurring during acinar transformation. One of these is the embryonic transcription factor Sox9 that is expressed in pancreatic progenitor cells and becomes restricted to ductal cells in the adult organ [52]. Kopp et al. investigated if this factor not only is a marker of acinar cells undergoing transformation but also plays an essential function in this process [5]. To test this, Sox9 was deleted from acinar cells expressing oncogenic Kras, which resulted in a complete blockage of PanIN formation (Figure 1). Vice versa, overexpression of Sox9 in the context of oncogenic Kras dramatically catalyzed preneoplastic transformation. Mechanistically, it was shown that ectopic expression of Sox9 alone in acinar cells can erode cell integrity evidenced by downregulation of markers of acinar differentiation, such as Mist1, and concomitant upregulation of the ductal factor CK19 [5]. Interestingly, previous studies have shown that combining mutant Kras with a deletion of Mist1 also accelerates formation of PanIN lesions in the pancreas [53], which further highlights the relevance of a stable acinar differentiation state as a barrier for Kras-mediated transformation (Figure 1).
Figure 1.
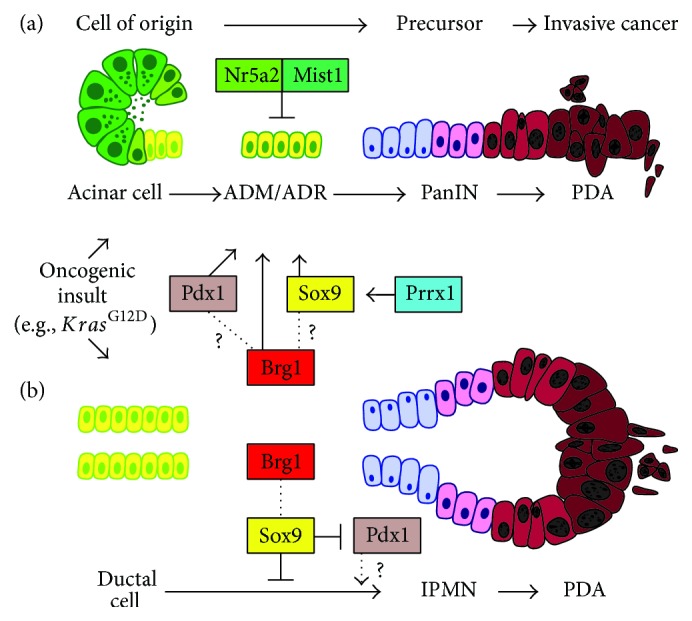
A model for developmental factors regulating acinar and duct cell transformation. (a) Acinar cells undergo acinar to ductal reprogramming (ADM/ADR) on their way to become PanINs and PDA. The ductal reprogramming of acinar cells is inhibited by factors that maintain stable acinar differentiation, such as Mist1 and Nr5a2. Instead, the transcription factors Sox9 and Pdx1 promote ductal reprogramming. Prrx1 and the chromatin remodeler Brg1 also promote ductal reprogramming, possibly by regulating other transcription factors, such as Sox9 or Pdx1. (b) Ductal cells transform into IPMN lesions by undergoing a dedifferentiation step (“ductal retrogression”) evidenced by an upregulation of the progenitor marker Pdx1. The chromatin remodeler Brg1 suppresses ductal retrogression by regulating Sox9 expression, which in turn inhibits Pdx1. This points to an opposing function of Brg1 in acinar versus duct cell transformation.
4.3. Epithelial Cell Plasticity
The notion that loss of acinar cell integrity is a prerequisite for PDA formation is accompanied by the emergence of a developmental program as mentioned above [44]. This phenomenon is not exclusive to pancreatic cancer but also present in other processes involving ADM during pancreatic regeneration in response to pancreatitis [42]. Besides the already mentioned Sox9 and Pdx1, Notch- and Shh-signaling, among others, are reactivated within the acinar compartment after challenging the gland using cerulein [42]. Interestingly, persistent overexpression of Pdx1 within the pancreatic epithelium by transgenic means leads to ADM formation suggesting that Pdx1 has a driving function in acinar cell dedifferentiation [54] (Figure 1). More recently, using an unbiased approach comparing gene expression profiles of the developing pancreas, acute pancreatitis and mutant Kras-driven carcinogenesis revealed a transcriptional program shared in these processes [55]. Of note, Prrx1 emerged as the most regulated transcription factor in this system. The Prrx1 gene encodes two major variants, Prrx1a and Prrx1b, generated by alternative splicing [56]. Remarkably, PRRX1B is upregulated in ADM as well as PanINs and induces Sox9 expression on a transcriptional level thereby fostering a ductal phenotype [55] (Figure 1). In PDA, both splice variants, Prrx1a and Prrx1b, control differentially epithelial-to-mesenchymal transition (EMT) and the reverse process, mesenchymal-to-epithelial transition (MET) (Takano, Reichert et al. unpublished data). In general, the degree of cellular plasticity is illustrated by the ability to undergo EMT/MET both being critical in embryonic development (EMT type I), tissue regeneration (EMT type II), and cancer (EMT type III) [57]. The two latter types are characterized by an inflammatory response in contrast to EMT type I observed in the embryo [58]. However, all subtypes can be identified by the expression of EMT-transcription factors (EMT-TFs), including Slug, Snail, Twist, and Zeb [59]. Interestingly, Slug has been found to cooperate with Sox9 in promoting tumorigenicity in breast cancer cells [60].
4.4. Acinar Cells in Human Pancreatic Cancer Initiation
In summary, murine studies have identified acinar cells as the origin for PanIN lesions and highlighted the role of developmental factors in this process. An open question remains if these studies in mouse models are applicable to the situation of pancreatic cancer initiation in humans. Although it is impossible to directly prove in vivo that acinar cells transform into PanINs in human pancreas, there is some evidence that this process may also be true for human pancreatic carcinogenesis. In vitro studies have shown that human pancreatic acinar cells have a comparable plasticity, as their murine equivalents, and readily transdifferentiate into duct-like cells in culture [61]. Shi et al. examined in human pancreas specimens if the initiating oncogenic Kras mutation can be detected in acinar cells that are in proximity to ADM/PanIN [62]. Although the authors could not detect Kras mutations in acinar cells and isolated areas of ADM, expectedly PanINs but also areas of adjacent ADM exhibited a high frequency of mutant Kras. This data could be interpreted that ADM adjacent to PanIN represents retrograde extensions of the latter and thus would not support an acinar origin for PanINs. However, it is also conceivable that it was impossible to detect mutant Kras in morphologically normal acinar cells, because Kras mutant acinar cells may promptly transdifferentiate into ADM in humans and Kras mutant ADM may have a high propensity to immediate formation of PanIN lesions.
The acinar origin hypothesis is also supported by genome wide association studies that identified frequent single-nucleotide polymorphisms (SNPs) in pancreatic developmental factors that are associated with pancreatic cancer. These developmental factors comprised not only PDX1 but also the important regulators of acinar differentiation HNF1A and NR5A2 [63, 64]. The role of Nr5a2 in maintaining a stable acinar differentiation state and preventing Kras-driven PanIN formation was subsequently demonstrated in studies in mice [65, 66] (Figure 1). These investigations supported the relevance of the identified SNPs in NR5A2 as pancreatic cancer susceptibility lesions and further suggest a role of acinar cells in human preneoplastic transformation.
5. Development of IPMN from Its Cellular Origin
Whereas PanINs and PanIN-PDA have been studied in great detail, in part, due to the prevalence of GEMM recapitulating PanIN-PDA formation [67, 68], the molecular properties of IPMN are less well characterized and few suitable mouse models for studying IPMN-PDA progression had been developed [69, 70]. In humans, IPMNs are macroscopically cystic lesions that develop within the pancreatic main and/or branch duct system and show a direct connection to the ducts. This fact and the ductal morphology have led to the assumption of duct cells as the cellular origin for this precursor lesion. However, lineage-tracing studies in murine models had been hampered by the lack of suitable inducible Cre-lines that allow recombination specifically in adult duct cells. Therefore, proving a ductal origin of the preneoplastic lesions in mouse models of IPMN had been impossible.
With the recent availability of such duct-specific Cre-lines, it was demonstrated in a study on a novel GEMM of IPMN that the preneoplastic lesions in this particular model derive from ductal cells [4]. In an initial experiment, it was shown that embryonic pancreas specific deletion (Ptf1a Cre) of Brg1, the catalytic subunit of the SWItch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complexes, in combination with expression of mutant Kras led to the formation of cystic neoplastic lesions in adult mice that resembled human IPMNs [4]. This mouse model faithfully recapitulated human IPMN-PDA development and mirrored the human situation with a less malignant biological behavior as compared with PanIN-PDA. Next, it was interrogated, in this model, whether the IPMN lesions were of ductal or acinar origin by using Cre-lines that allow specific recombination of the Brg1 and mutant Kras alleles in adult acinar (Ptf1a CreER) or ductal (Hnf1β-Cre ERT2) cells [4]. These experiments showed that Brg1 deletion renders adult duct cells sensitive to Kras-mediated transformation evidenced by the occurrence of duct atypia and lesions that resembled IPMNs. In contrast, Brg1 deletion in adult acinar cells in the context of mutant Kras did not lead to IPMN formation and, moreover, PanIN formation was blocked. This suggests that Brg1 plays a dual role in PDA precursor development by inhibiting duct transformation to IPMN and promoting acinar transformation to PanIN in the context of an oncogenic stimulus by mutant Kras (Figure 1).
This study gave the first experimental evidence that ductal cells can serve as the origin for IPMN lesions and highlighted the relevance of chromatin remodeling in this process. In a subsequent work, Roy et al. investigated the mechanistic basis for this observation [71]. It was found that combination of mutant Kras and loss of Brg1 in pancreatic ducts led to a dedifferentiation of mature ductal cells that was termed “ductal retrogression.” Ductal retrogression was characterized by a reduced expression of markers of mature ductal differentiation and an upregulation of pancreatic progenitor factors such as Pdx1. One of the downregulated ductal markers was Sox9. Mechanistically it was shown that ectopic expression of Sox9 in the context of combined mutant Kras/Brg1 loss prevented ductal retrogression and IPMN formation (Figure 1). The upregulation of Pdx1 during ductal retrogression mirrors the expression pattern in pancreatic acinar cells during injury or Kras-mediated dedifferentiation. This points to Pdx1 as a unique factor in transformation of both acinar and ductal cells. Therefore, future studies need to explore a functional role of Pdx1 in this process and whether inhibition of this progenitor factor can prevent the formation of both PanIN and IPMN.
6. Conclusions with Translational Aspects
In contrast to some other cancers, embryonic signaling pathways like TGFβ, Wnt-β-catenin, and Hedgehog alone are not sufficient for the initiation of PDA [44]. Although the expression of a primitive ductal program can be launched, an oncogenic insult, most frequently mutated and constitutive active Kras, is required to drive pancreatic cancer progression. Apparently, acinar cells display the highest degree of cellular plasticity in order to adopt an undifferentiated progenitor state upon inflammatory or oncogenic stimuli. At the same time, it is becoming more and more clear that, although morphologically relatively uniform, PDA represents an extremely complex disease. In the field of pancreatic cancer research, we are lagging behind in terms of subtype identification compared to other solid cancer entities, for example, breast cancer. The cell of origin or precursor type leading to PDA has significant impact towards the prognosis of PDA patients. This suggests that not only the genetic makeup of a given cancer cell but its primordial lineage plays an important role. Our knowledge has been fueled to a large extent by mouse models of pancreatic cancer but in order to address the complexity of this disease, human model systems are needed. For example, Kim et al. used an elegant induced-pluripotency approach to reprogram human PDA cells in order to recapitulate human disease progression [72]. Another study by Boj et al. suggests that an organoidculture system established from surgery specimen or endoscopic biopsy material grown in a three-dimensional matrix might be a useful tool to address this complexity [73]. In addition, this model system harbors the opportunity to test personalized therapies for pancreatic cancer patients, potentially even in real time.
In general, mouse models have significantly contributed to our understanding of virtually all aspects of PDA biology. However, so far we were not able to make use of this knowledge in order to improve PDA patients' care. The recent advances in pancreatic cancer treatment have been generated by not molecular defined strategies but rather pragmatic approaches using extremely toxic chemotherapeutic regimens [74] or increasing the delivery of established cytotoxic drugs [75].
The genetic heterogeneity of PDA and distinct oncogenic susceptibilities of defined compartments within the gland make this disease the opposite of what clinicians call a “chameleon”; one disease with many faces, PDA, instead, might represent numerous diseases with the same appearance. Thus, tailored therapies taking the mutational landscape of the respective tumor into account need to be developed and an even more profound understanding of the cellular plasticity and the regulating genetic factors in pancreatic carcinogenesis is required.
Acknowledgments
The authors express their gratitude to Ben Z. Stanger (University of Pennsylvania) for commenting on the developmental section of this review. This work was supported by the National Pancreas Foundation (Maximilian Reichert), the German Cancer Aid Foundation (Max-Eder Program, Deutsche Krebshilfe 111273 to Maximilian Reichert), and the AGA-Actavis Research Award in Pancreatic Disorders (Maximilian Reichert). Guido von Figura was supported by the Deutsche Forschungsgemeinschaft (FI 1719/1-1 and FI 1719/2-1). Alexander Kleger was supported by the Deutsche Forschungsgemeinschaft (KL 2544/1-1) and the Baden-Württemberg Stiftung (Eliteprogramme for Postdocs). Alexander Kleger is an Elser-Kröner-Fresenius-Memorial Fellow.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Virchow R. Krankheitswesen und Krankheitsursachen. Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 1880;79(2):185–228. doi: 10.1007/BF01881823. [DOI] [Google Scholar]
- 2.Waddell N., Pajic M., Patch A.-M., et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witkiewicz A. K., McMillan E. A., Balaji U., et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nature Communications. 2015;6, article 6744 doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Figura G., Fukuda A., Roy N., et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nature Cell Biology. 2014;16(3):255–267. doi: 10.1038/ncb2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kopp J. L., von Figura G., Mayes E., et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(6):737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stanger B. Z., Stiles B., Lauwers G. Y., et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8(3):185–195. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Gu G., Dubauskaite J., Melton D. A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 8.Spence J. R., Lange A. W., Lin S.-C. J., et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Developmental Cell. 2009;17(1):62–74. doi: 10.1016/j.devcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsson J., Carlsson L., Edlund T., Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 10.Offield M. F., Jetton T. L., Labosky P. A., et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 11.Pan F. C., Wright C. Pancreas organogenesis: from bud to plexus to gland. Developmental Dynamics. 2011;240(3):530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 12.Stanger B. Z., Tanaka A. J., Melton D. A. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature. 2007;445(7130):886–891. doi: 10.1038/nature05537. [DOI] [PubMed] [Google Scholar]
- 13.Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R. J., Wright C. V. E. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nature Genetics. 2002;32(1):128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Q., Law A. C., Rajagopal J., Anderson W. J., Gray P. A., Melton D. A. A multipotent progenitor domain guides pancreatic organogenesis. Developmental Cell. 2007;13(1):103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 15.Kopp J. L., Dubois C. L., Schaffer A. E., et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138(4):653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solar M., Cardalda C., Houbracken I., et al. Pancreatic exocrine duct cells give rise to insulin-producing β cells during embryogenesis but not after birth. Developmental Cell. 2009;17(6):849–860. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Gao T., McKenna B., Li C., et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metabolism. 2014;19(2):259–271. doi: 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaffer A. E., Freude K. K., Nelson S. B., Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Developmental Cell. 2010;18(6):1022–1029. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masui T., Swift G. H., Hale M. A., Meredith D. M., Johnson J. E., MacDonald R. J. Transcriptional autoregulation controls pancreatic Ptf1a expression during development and adulthood. Molecular and Cellular Biology. 2008;28(17):5458–5468. doi: 10.1128/mcb.00549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reichert M., Rustgi A. K. Pancreatic ductal cells in development, regeneration, and neoplasia. The Journal of Clinical Investigation. 2011;121(12):4572–4578. doi: 10.1172/jci57131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel R., Ma J., Zou Z., Jemal A. Cancer statistics, 2014. CA: A Cancer Journal for Clinicians. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 22.Ryan D. P., Hong T. S., Bardeesy N. Pancreatic adenocarcinoma. The New England Journal of Medicine. 2014;371(11):1039–1049. doi: 10.1056/nejmra1404198. [DOI] [PubMed] [Google Scholar]
- 23.Maitra A., Fukushima N., Takaori K., Hruban R. H. Precursors to invasive pancreatic cancer. Advances in Anatomic Pathology. 2005;12(2):81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 24.Gnoni A., Licchetta A., Scarpa A., et al. Carcinogenesis of pancreatic adenocarcinoma: precursor lesions. International Journal of Molecular Sciences. 2013;14(10):19731–19762. doi: 10.3390/ijms141019731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka M., Fernández-Del Castillo C., Adsay V., et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12(3):183–197. doi: 10.1016/j.pan.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Matthaei H., Norris A. L., Tsiatis A. C., et al. Clinicopathological characteristics and molecular analyses of multifocal intraductal papillary mucinous neoplasms of the pancreas. Annals of Surgery. 2012;255(2):326–333. doi: 10.1097/sla.0b013e3182378a18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poultsides G. A., Reddy S., Cameron J. L., et al. Histopathologic basis for the favorable survival after resection of intraductal papillary mucinous neoplasm-associated invasive adenocarcinoma of the pancreas. Annals of Surgery. 2010;251(3):470–476. doi: 10.1097/sla.0b013e3181cf8a19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mino-Kenudson M., Fernández-del Castillo C., Baba Y., et al. Prognosis of invasive intraductal papillary mucinous neoplasm depends on histological and precursor epithelial subtypes. Gut. 2011;60(12):1712–1720. doi: 10.1136/gut.2010.232272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanda M., Matthaei H., Wu J., et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142(4):730.e9–733.e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amato E., Molin M. D., Mafficini A., et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. The Journal of Pathology. 2014;233(3):217–227. doi: 10.1002/path.4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schönleben F., Qiu W., Bruckman K. C., et al. BRAF and KRAS gene mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/IPMC) of the pancreas. Cancer Letters. 2007;249(2):242–248. doi: 10.1016/j.canlet.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furukawa T., Kuboki Y., Tanji E., et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Scientific Reports. 2011;1, article 161 doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu J., Jiao Y., Dal Molin M., et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(52):21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J., Matthaei H., Maitra A., et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Science Translational Medicine. 2011;3(92) doi: 10.1126/scitranslmed.3002543.92ra66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao S. Y. Intraductal papillary mucinous neoplasm of the pancreas: an update. Scientifica. 2012;2012:20. doi: 10.6064/2012/893632.893632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De La O J.-P., Emerson L. L., Goodman J. L., et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(48):18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerra C., Schuhmacher A. J., Cañamero M., et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Habbe N., Shi G., Meguid R. A., et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(48):18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elsasser H. P., Adler G., Kern H. F. Time course and cellular source of pancreatic regeneration following acute pancreatitis in the rat. Pancreas. 1986;1(5):421–429. doi: 10.1097/00006676-198609000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Willemer S., Elsasser H. P., Kern H. F., Adler G. Tubular complexes in cerulein- and oleic acid-induced pancreatitis in rats: glycoconjugate pattern, immunocytochemical, and ultrastructural findings. Pancreas. 1987;2(6):669–675. doi: 10.1097/00006676-198711000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Fendrich V., Esni F., Garay M. V. R., et al. Hedgehog signaling is required for effective regeneration of exocrine pancreas. Gastroenterology. 2008;135(2):621–e8. doi: 10.1053/j.gastro.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jensen J. N., Cameron E., Garay M. V. R., Starkey T. W., Gianani R., Jensen J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128(3):728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 43.Morris J. P. T., IV, Cano D. A., Sekine S., Wang S. C., Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. The Journal of Clinical Investigation. 2010;120(2):508–520. doi: 10.1172/jci40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris J. P., Wang S. C., Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nature Reviews. Cancer. 2010;10(10):683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strobel O., Dor Y., Alsina J., et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. 2007;133(6):1999–2009. doi: 10.1053/j.gastro.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morris J. P., IV, Cano D. A., Sekine S., Wang S. C., Hebrok M. β-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. Journal of Clinical Investigation. 2010;120(2):508–520. doi: 10.1172/jci40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murtaugh L. C. Pathogenesis of pancreatic cancer: lessons from animal models. Toxicologic Pathology. 2014;42(1):217–228. doi: 10.1177/0192623313508250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu L., Shi G., Schmidt C. M., Hruban R. H., Konieczny S. F. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. The American Journal of Pathology. 2007;171(1):263–273. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hingorani S. R., Petricoin E. F., III, Maitra A., et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 50.Lowenfels A. B., Maisonneuve P., Cavallini G., et al. Pancreatitis and the risk of pancreatic cancer. The New England Journal of Medicine. 1993;328(20):1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 51.Ray K. C., Bell K. M., Yan J., et al. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0016786.e16786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seymour P. A., Freude K. K., Tran M. N., et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(6):1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi G., Zhu L., Sun Y., et al. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology. 2009;136(4):1368–1378. doi: 10.1053/j.gastro.2008.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyatsuka T., Kaneto H., Shiraiwa T., et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes & Development. 2006;20(11):1435–1440. doi: 10.1101/gad.1412806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reichert M., Takano S., Von Burstin J., et al. The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes & Development. 2013;27(3):288–300. doi: 10.1101/gad.204453.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Norris R. A., Kern M. J. The identification of Prx1 transcription regulatory domains provides a mechanism for unequal compensation by the Prx1 and Prx2 loci. Journal of Biological Chemistry. 2001;276(29):26829–26837. doi: 10.1074/jbc.m100239200. [DOI] [PubMed] [Google Scholar]
- 57.Kalluri R., Weinberg R. A. The basics of epithelial-mesenchymal transition. The Journal of Clinical Investigation. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nieto M. A. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159) doi: 10.1126/science.1234850.1234850 [DOI] [PubMed] [Google Scholar]
- 59.Brabletz T. To differentiate or not—routes towards metastasis. Nature Reviews. Cancer. 2012;12(6):425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- 60.Guo W., Keckesova Z., Donaher J. L., et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148(5):1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houbracken I., de Waele E., Lardon J., et al. Lineage tracing evidence for transdifferentiation of acinar to duct cells and plasticity of human pancreas. Gastroenterology. 2011;141(2):731.e4–741.e4. doi: 10.1053/j.gastro.2011.04.050. [DOI] [PubMed] [Google Scholar]
- 62.Shi C., Hong S.-M., Lim P., et al. KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin. Molecular Cancer Research. 2009;7(2):230–236. doi: 10.1158/1541-7786.mcr-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li D., Duell E. J., Yu K., et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis. 2012;33(7):1384–1390. doi: 10.1093/carcin/bgs151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Petersen G. M., Amundadottir L., Fuchs C. S., et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nature Genetics. 2010;42(3):224–228. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flandez M., Cendrowski J., Cañamero M., et al. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut. 2014;63(4):647–655. doi: 10.1136/gutjnl-2012-304381. [DOI] [PubMed] [Google Scholar]
- 66.von Figura G., Morris J. P., IV, Wright C. V. E., Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63(4):656–664. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hezel A. F., Kimmelman A. C., Stanger B. Z., Bardeesy N., Depinho R. A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes & Development. 2006;20(10):1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 68.Mazur P. K., Siveke J. T. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut. 2012;61(10):1488–1500. doi: 10.1136/gutjnl-2011-300756. [DOI] [PubMed] [Google Scholar]
- 69.Siveke J. T., Einwächter H., Sipos B., Lubeseder-Martellato C., Klöppel G., Schmid R. M. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007;12(3):266–279. doi: 10.1016/j.ccr.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 70.Bardeesy N., Cheng K.-H., Berger J. H., et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes and Development. 2006;20(22):3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roy N., Malik S., Villanueva K. E., et al. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes & Development. 2015;29(6):658–671. doi: 10.1101/gad.256628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim J., Hoffman J. P., Alpaugh R. K., et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Reports. 2013;3(6):2088–2099. doi: 10.1016/j.celrep.2013.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boj S. F., Hwang C.-I., Baker L. A., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1-2):324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conroy T., Desseigne F., Ychou M., et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England Journal of Medicine. 2011;364(19):1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 75.Von Hoff D. D., Ervin T., Arena F. P., et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England Journal of Medicine. 2013;369(18):1691–1703. doi: 10.1056/nejmoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]