

Abstract
Electron microscopy is used in biological research to study the ultrastructure at high resolution to obtain information on specific cellular processes. Serial block face-scanning electron microscopy is a relatively novel electron microscopy imaging technique that allows three-dimensional characterization of the ultrastructure in both tissues and cells by measuring volumes of thousands of cubic micrometres yet at nanometre-scale resolution. In the scanning electron microscope, repeatedly an image is acquired followed by the removal of a thin layer resin embedded biological material by either a microtome or a focused ion beam. In this way, each recorded image contains novel structural information which can be used for three-dimensional analysis.
Here, we explore focused ion beam facilitated serial block face-scanning electron microscopy to study the endothelial cell–specific storage organelles, the Weibel–Palade bodies, during their biogenesis at the Golgi apparatus. Weibel–Palade bodies predominantly contain the coagulation protein Von Willebrand factor which is secreted by the cell upon vascular damage. Using focused ion beam facilitated serial block face-scanning electron microscopy we show that the technique has the sensitivity to clearly reveal subcellular details like mitochondrial cristae and small vesicles with a diameter of about 50 nm. Also, we reveal numerous associations between Weibel–Palade bodies and Golgi stacks which became conceivable in large-scale three-dimensional data. We demonstrate that serial block face-scanning electron microscopy is a promising tool that offers an alternative for electron tomography to study subcellular organelle interactions in the context of a complete cell.
Keywords: Endothelial cells, FIB, Golgi apparatus, SBF-SEM, Weibel–Palade body
Introduction
Microscopy on biological samples is a powerful tool to localize specific proteins or organelles to study their behaviour, their morphological properties and their associations with other cell components. Transmission electron microscopy (TEM) can reveal cellular structures at the highest resolution but is often limited in showing ‘the bigger picture’ in the third dimension as the material has to be sectioned into 100–200 nm thick slices to allow imaging. Examining serial TEM sections in combination with the collection of serial electron tomograms can partially overcome this limitation (Hoffpauir et al., 2007; Barcena & Koster, 2009). However, both techniques are laborious and time consuming because of the required time for acquisition, processing and handling. Serial sectioning and serial electron tomography are therefore practically applicable for limited volumes in the order of tens of cubic micrometres. Studies focusing on cell–cell contacts in tissue, or studies to characterize the three-dimensional (3D) organization of subcellular organelles, are therefore not particularly suitable to be approached using TEM. A relatively novel way to study the relationships between cells or organelles is serial block face-scanning electron microscopy (SBF-SEM)(Knott & Genoud, 2013). In this imaging modality, the Scanning Electron Microscope (SEM) images the block face of resin-embedded material every time a microtome or focused ion beam (FIB) has removed of a thin slice of material. As SBF-SEM can be performed automatically, large volumes of resin-embedded material can be imaged which potentially can reveal novel features which stay unnoticed in two-dimensional TEM sections. The differences between SBF-SEMs using an integrated microtome and those that use a FIB, are mainly related to the attainable slice thickness and to the surface area that is removed from the block: the microtome can section down to 25 nm and removes a slice from the entire block surface whereas the FIB operates locally on a selected area and can remove slices as thin as 3 nm (Knott & Genoud, 2013). Currently, SBF-SEM is a particular popular technique to image cell–cell interactions in tissues. Because of its technical capabilities, we set out to explore whether the technique could also be effectively used to study subcellular organelles. In this paper, we describe the application of SBF-SEM to study Weibel–Palade bodies (WPBs) in endothelial cells (Weibel & Palade, 1964).
WPBs are storage organelles found in endothelial cells. These cells line blood vessels and form a barrier between the blood and the underlying tissue (Weibel & Palade, 1964). The WPBs were serendipitously discovered by TEM and were described as rod- or cigar-shaped organelles of 0.1–0.3 μm in diameter and 1–5 μm in length. WPBs typically display densely packed tubules (Weibel & Palade, 1964; Valentijn et al., 2011), which are formed by the haemostatic protein Von Willebrand factor (VWF). VWF is secreted by endothelial cells upon vascular damage, inflammation or hypoxia and initiates the first steps in haemostasis (Valentijn et al., 2011).
As endothelial cells have to respond on demand, a sufficient storage pool of WPBs is formed in advance. Biogenesis of WPBs occurs at the Golgi apparatus and is possibly induced by VWF itself. It has been shown that expression of full-length VWF in nonendothelial cells can result in the formation of so called ‘pseudo-WPBs’ (Voorberg et al., 1993; Michaux et al., 2003). However, recent data suggest that the Golgi is also heavily involved in controlling WPB content and WPB size (Ferraro et al., 2014). Studying the morphology of forming WPBs by TEM is still providing novel information but is limited in providing the large-scale 3D information as only thin sections of cells can be studied at once. Here, we explore FIB facilitated SBF-SEM to determine whether the technique provides the resolution and the sensitivity to study subcellular interactions such as WPB biogenesis at the Golgi apparatus.
Materials and methods
Cell culture
Human umbilical vein endothelial cells obtained from Lonza (Walkersville, MD, USA) were cultured in endothelial growth medium-2 (EGM-2 bullet kit, Lonza). Cell culture petri dishes and Thermanox™ cover slips (Thermo Scientific, Hudson, NH, USA) were coated with 1% gelatin in Phosphate Buffered Saline (PBS) prior to cell seeding. Cells were grown for about 7 days till confluent.
Transmission electron microscopy
TEM samples were prepared as previously described (Valentijn et al., 2008). Briefly, the cells were fixed in 2% glutaraldehyde in 0.1 M cacodylate buffer pH 7.4. Cells were postfixed with 1% osmium tetroxide. Cells were dehydrated using a gradient series of ethanol and were embedded in Epon LX-112. The resin was polymerized for 48 h at 60°C. Ultrathin sections of 100–120 nm thickness were prepared and placed on copper Electron Microscopy (EM) grids. The sections were poststained with 7% uranyl acetate and Reynold’s lead citrate. TEM images were acquired using a Tecnai 12 (FEI company, Eindhoven, the Netherlands) TEM at 120 kV equipped with an Eagle 4k × 4k CCD camera (FEI company).
Focused ion beam-SEM
Samples for FIB-SEM imaging were prepared as previously described (Holcomb et al., 2013). In short, cells were fixed for 1 h in 1.5% glutaraldehyde in 0.1 M cacodylate buffer pH 7.4. Cells were placed on ice for 1 h in 2% osmium tetroxide with 1.5% potassium ferrocyanide in 0.1 M cacodylate buffer. A 1% thiocarbohydrazide solution was prepared fresh in water and filtered through a 0.22 μm Millipore syringe filter before use. It was placed on the cells for 20 min at room temperature. Then, the cells were incubated in 2% osmium tetroxide in water for 30 min. Cells were kept overnight in 1% aqueous uranyl acetate at 4°C. Next day, Walton’s lead aspartate was freshly prepared, filtered through a 0.22 μm Millipore syringe and placed on the cells for 30 min at 60°C. Dehydration was performed using a series of ethanol incubations. The cells were then placed in ice-cold 100% anhydrous acetone. Durcupan Araldite casting resin M (Durcupan ACM) was infiltrated into the cells using 25%, 50% and 75% Durcupan solutions in acetone. Cells were placed in 100% Durcupan resin overnight. Excess resin was removed and resin-filled BEEM capsules were placed on top of the cells. The resin was polymerized at 60°C for 48–72 h.
The polymerized resin capsules were removed from the cover slips. The monolayer of cells is retained on the surface of the resin block. A 1-mm slice was cut from the resin block using a saw. The block was placed on a SEM stub using carbon tape. Silver paint was applied on the sides of the block to electrically ground the sample. Additionally, a thin layer of iridium was sputtered on top of samples using an Emitech K575X coater (Quorum Technologies Ltd., Laughton, UK).
Imaging was performed using an Auriga CrossBeam (Carl Zeiss Microscopy GmbH, Munich, Germany) SEM at an acceleration voltage of 1.7 kV using an image aperture of 30 μm (high current; beam current: ∼220 pA). The signals from the InLens secondary electron detector and the energy selected backscattered electron detector were mixed because it provided the best signal-to-noise ratio. The grid voltage of the detector was set at 1108 V. After every acquired image, a 10 nm slice was removed using the FIB. A milling current of 4 nA was used at an acceleration voltage of 30 kV.
Image processing and visualization
To refine the alignment of the image stack as produced by the instrument, first the support layer was localized within each image. Next, the images were pairwise aligned by weighted phase correlation. The Gaussian weight window was set at an offset to the support layer and the tapering was chosen such that the 3-sigma boundary was well within the cell monolayer. The pairwise shift was determined by a parabolic fit centred around the location of maximum correlation value. All image processing was implemented using MATLAB® and the DIPimage toolbox (Hendriks et al., 1999). Segmentation of subcellular structures in the aligned image stack was performed in Amira® (Visualization Sciences Group, FEI company). To load the data set, the images were cropped and binned two times in x and y. The structures were segmented by using the threshold option on selected regions or by manual drawing. Additionally, the segmented volumes were smoothened.
Results
SBF-SEM on a single cell layer reveals most of the major cell structures
To explore whether we could use SBF-SEM for the analysis of WPBs forming at the Golgi apparatus, we first wanted to establish to which extent details can be resolved by SEM in chemically fixed cells. Performing SBF-SEM on single cell layers can be more challenging as the underlying resin has the tendency to charge by the lack of conducting material. We experienced that the level of detail required for the identification of WPB was best imaged with a SBF-SEM system that uses a FIB to remove material. In a FIB-SEM system, the sample is tilted in the SEM chamber to alternately image the sample from the top by SEM, and mill material away with the FIB from the side (Fig.1A). To open up the material and to start subsequent imaging and FIB milling, a trench has to be milled by the FIB (Figs.1A, B). As shown in Figure1(B), the human umbilical vein endothelial cells appeared as a very thin line just beneath the resin block surface.
Figure 1.
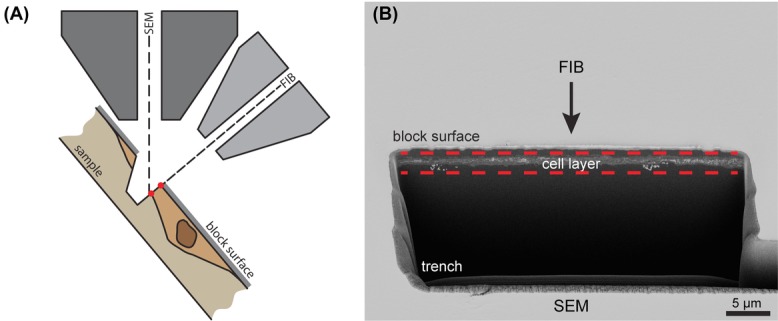
Focused ion beam milling for serial block face (SBF)-scanning electron microscopy (SEM). Overview of the imaging setup and the sample. (A) Schematic representation showing the beam line of the electrons of the scanning electron microscope and the ions of the focused ion beam (FIB) with respect to the sample. The sample is tilted such that the electron beam can image from the top whereas the FIB can remove material from the side. A trench is created by the FIB to make the sample accessible for SBF imaging. (B) Low magnification overview imaged by the SEM showing the trench created for SBF-SEM imaging. Between the dashed lines, the monolayer of cell material is observed. Scale bar is 5 μm.
When zooming in on the exposed cells, many intracellular organelles were clearly revealed when acquiring several image stacks. Apart from the nucleus, we most often observed the abundant membrane stacks of the Golgi apparatus (Fig.2A), the wide spreading network of the endoplasmic reticulum (Fig.2B), the lysosomes (Fig.2B) and the mitochondria (Fig.2C). As shown in Figure2(C), the internal cristae within the mitochondria were clearly visible. In addition to this, we could also distinguish centrosomes (Fig.2D-D’), occasional microtubules (arrowheads, Fig.2E) and caveolae (arrowheads, Fig.2F) which are tiny 50–100 nm vesicles that are formed at the plasma membrane of endothelial cells for transcytosis. Common cellular structures that were not detected in the SBF-SEM stacks were mainly fine proteinaceous components such as actin filaments, clathrin coats and ribosomes.
Figure 2.
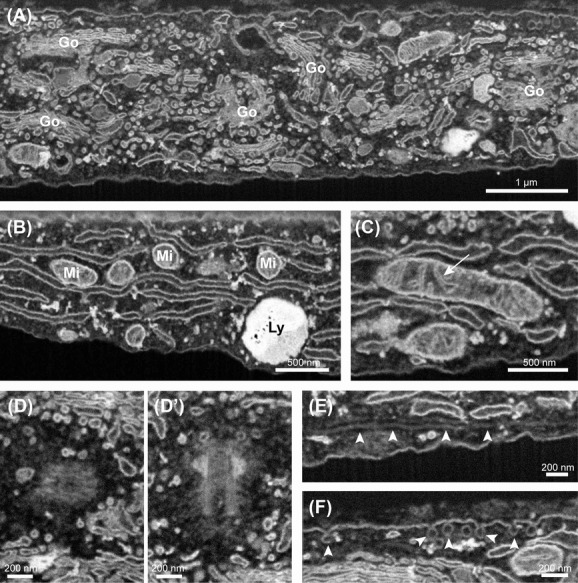
Organelles and subcellular structures by serial block face (SBF)-scanning electron microscopy (SEM). Overview of the subcellular structures and cell organelles in endothelial cells observed by SBF-SEM. (A) The extensive membrane stacks of the Golgi apparatus (Go). (B) Membrane network of the endoplasmic reticulum. In addition, also a lysosome (Ly) and parts of mitochondria (Mi) are observed. (C) Mitochondrion in which the internal cristae are clearly resolved (arrow). (D and D’) Pair of centrosomes. (D) Centrosome from the top. (D’) Centrosome from the side. (E) Microtubule (arrowheads) running just beneath the plasma membrane. (F) Caveolae at the plasma membrane. Scale bar panel (A) is 1 μm. Scale bar panel (B) and (C) is 500 nm. Scale bar panels (D)–(F) is 200 nm.
Because of the severe fixation and staining procedure that was used to prepare the specimen for SBF-SEM imaging, some of these structures may be perturbed and not be visible.
WPB imaging by SBF-SEM
In our search to WPBs in the acquired image stacks, we experienced that longitudinally sectioned WPBs were observed less frequently. Further analysis of the data indicated that most WPBs were visualized in a cross-sectional orientation (Fig.3A). The elongated shape of these WPBs was revealed when visualizing the 3D volume in a different orientation and upon segmentation (Supplementary video S1).
Figure 3.
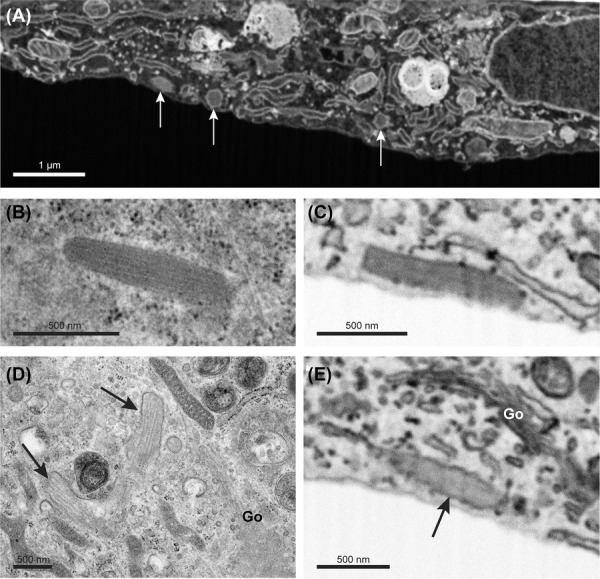
Weibel–Palade bodies imaged by SEM and TEM. Weibel–Palade bodies (WPBs) imaged by SEM and TEM in endothelial cells. (A) SBF-SEM slice showing cross-sectioned WPBs (arrows). (B) Longitudinal-sectioned WPB imaged by TEM displaying internal striations of Von Willebrand factor tubules. (C) Inverted SEM image of a longitudinal-sectioned WPB that displays a uniform stained interior. (D) Immature WPBs (arrows) near the Golgi apparatus (Go) imaged by TEM. Separate VWF tubules are clearly visible. (E) Inverted SEM image of an immature WPB near the Golgi apparatus (Go). Interior is less electron dense than the WPB in panel C but the VWF tubules are not resolved. Scale bar panel (A) is 1 μm. Scale bar panels (B)–(E) is 500 nm.
When observed by TEM, the internal structure of longitudinally sectioned WPB typically reveals striations of VWF tubules (Fig.3B). However, when we examined WPBs in the SEM data, the tubules were hardly resolved and appeared as a uniformly stained mass as shown in the inverted SEM image depicted in Figure3(C). Revealing the VWF tubules is essential to study WPB formation and maturation as the size and the amount of tubules provides information on the advancement of the WPB during its biogenesis. As shown in Figure3(D), TEM on immature WPBs near the membranes of the Golgi apparatus clearly reveals the initial loose packaging of the VWF tubules. During the formation process, these immature WPBs will further increase in size and are finally condensed to create tightly packed mature WPBs. The WPBs observed around Golgi stacks by SBF-SEM appeared, similar to those imaged by TEM, less electron dense but unfortunately did not reveal the VWF tubules (Fig.3E). A more tailored approach to the image acquisition as well as in sample preparation will probably be required to resolve such kind of detail.
Analysis of the Golgi apparatus and surrounding WPBs reveals close relationships
To obtain insight into the cellular spatial organization of forming WPBs around the Golgi, a stack of almost 1600 images was collected at 25 000× magnification. We imaged an area of about 11.4 μm by 8.5 μm with a milling thickness of 10 nm which resulted in a voxel resolution of 3.7 × 3.7 × 10 nm. In total, a volume of about 16 μm depth was imaged and removed from the block surface in a time span of approximately 50 h which corresponds to an acquisition speed of about 32 images per hour. This was achieved using a FIB milling time of about 2 s and a SEM acquisition time of about 2 min per image. To analyse and segment the volume in Amira®, the data set was aligned by phase correlation with a Gaussian window function. In addition, the data were cropped and binned two times in x and y.
Figure4(A) shows a 3D representation of the imaged volume containing cellular material. In this volume, we segmented the Golgi apparatus and the WPBs (Figs.4B–C and Supplementary video S2) to reveal the distribution of the Golgi as well as its associations with WPBs. In addition, we also modelled the nucleus and cell membrane to show the Golgi and WPBs in the context of the cell. From the model we can observe that the WPBs that are in close proximity to a Golgi stack appear to concentrate in some specific region. Several of these WPBs even reveal tight associations and possible connections with Golgi stacks (WPBs are indicated in red in Figs.4B, C, see also Supplementary video S2). Interestingly, most of the modelled WPBs are already of remarkable size suggesting that advanced immature WPBs remain associated with the Golgi membrane. In addition, the segmentation of the cell membrane emphasizes the flatness of the endothelial cell, a characteristic that is often overlooked and underestimated when flat-embedded endothelial cells are studied by conventional TEM.
Figure 4.

Serial block face (SBF)-scanning electron microscopy (SEM) reveals Weibel–Palade bodies in close association with the Golgi. Analysis and modelling of a large SBF-SEM stack to study WPBs in relation to the Golgi apparatus in endothelial cells. (A) Three-dimensional overview of the data set. The data set consist of almost 1600 images and was acquired at 25 000× magnification using a 10 nm slice thickness. For processing, the data were binned two times in x and y which resulted in a voxel resolution of 7.4 × 7.4 × 10 nm. (B) Segmentation of the volume reveals the large size of the Golgi apparatus (green) and the WPBs (red) that are in close association with the Golgi. In addition, we segmented the peripheral WPBs (yellow) and the nucleus (blue). The inserts show one of the WPBs that was found in close relationship with the Golgi. Scale bar is 500 nm. To have a better view on the ultrastructure, the data were rotated with respect to the orientation in which the data were acquired for panels (A) and (B). (C) Segmentation of the cell membrane additionally reveals the confined and flat morphology of the endothelial cell. Scale bar is 1 μm.
Discussion
SBF-SEM is an effective method to study the 3D organization of subcellular structures. We show that this technique clearly resolves the general cell components at a resolution level that shows details at the level of mitochondrial cristae and vesicles of about 50 nm in diameter. In addition, we could recognize the endothelial-specific storage organelles, the WPBs, and reveal their organization around the Golgi apparatus. Interestingly, the WPBs that were in close proximity to the Golgi seemed to cluster at specific region. Moreover, close associations between Golgi apparatus membranes and WPBs were observed that are exciting to study in further detail, especially because large WPBs were observed as well. This could suggest that the WPBs are fully formed at the Golgi.
When we compared the morphology of the WPBs that were observed by SEM to electron micrographs of WPBs imaged by TEM, we observed that the characteristic tubular interior of the WPB was not visible in the SEM data. We speculate that these tubular structures were difficult to image by the SEM due the severe fixation and staining method. In addition, the limited depth in which the electrons interact with the stained material in the resin block and the limited sensitivity of the used detectors may also have influenced the image quality. Most of the currently available staining methods were largely designed to study neuronal networks in brain tissue and are therefore mainly focusing on intense membrane staining (Tapia et al., 2012; Holcomb et al., 2013). To reveal the VWF tubules, a different staining protocol could be needed to preserve and highlight these structures as well. Another improvement can be expected by the application of high pressure freezing and freeze substitution as they improve the ultrastructural preservation of the WPBs and VWF tubules (Zenner et al., 2007). To date, only a few freeze substitution protocols have been published for SBF-SEM (Hall et al., 2012; Villinger et al., 2012).
A critical point for imaging single cell layers is the conductivity of the sample. Single cell layers tend to charge faster than blocks of embedded tissue due to the high resin content that lacks conductive material that is formed by the metals staining cellular structures. We observed significant variation between samples which were prepared similarly. Some of the samples were stable during the FIB milling and subsequent SEM imaging whereas others were a challenge to image because of apparent charging which was visible in image deformation and image shifts. We experienced that leaving the sample under vacuum for several days as well as applying an additional iridium coat on the block surface reduced charging effects. Mixing currently available resins with cell-impermeable conductive material may solve the charging issues and could be beneficial for SBF-SEM imaging of single cell layers. However, this possibility has not been explored so far.
In spite of the occasional charging issue, the obtained image quality using the available sample preparation methods combined with high-end SEMs is a revolutionary step forward in the capabilities of electron microscopy to image cells and tissue thousands of cubic micrometres in size yet at 5–20 nm resolution. Acquired SEM images as presented here, are to a large extent similar to TEM, revealing detailed structures such as the mitochondrial cristae and small 50 nm vesicles. A major advantage is that FIB facilitated SBF-SEM can image large volumes in a single data collection session which saves time when compared to electron tomography on serial sections. A second advantage compared to serial section electron tomography is that a continuous image stack can be acquired and that no information is lost during sectioning as is often seen in serial TEM sections. On the down side, SBF-SEM does not yet reveal the extent of details as resolved by electron tomography. For example, electron tomography easily reveals individual tubules of VWF in WPBs (Valentijn et al., 2008).
To localize specific areas of interest for SBF-SEM imaging, correlative light and electron microscopy (CLEM) could be beneficial. With CLEM, light microscopy is used to localize and map the fluorescently stained structures of interest in a biological sample for subsequent investigation by EM in order to reveal the morphological and structural characteristics (Mironov & Beznoussenko, 2012). Currently, several groups have shown the power of CLEM and SBF-SEM in tissue and cells (Armer et al., 2009; Murphy et al., 2011; Lucas et al., 2014,). However, to perform CLEM to localize subcellular structures in 3D by SBF-SEM, some challenges have to be met. CLEM on subcellular structures depends much more on the accuracy of the overlay between the fluorescence data and the SBF-SEM stack when compared to CLEM aiming to relocate a single cell in tissue. As also shown by Murphy et al., the fluorescent images are most often acquired in a different orientation than the orientation used to obtain the SBF-SEM stack. Therefore, much more advanced image processing is required to overlay the two data sets (Murphy et al., 2011). The use of bimodal fiducials that are visible in both imaging modalities may therefore be used to obtain an accurate overlay. For monolayers, for example, bimodal fiducials could be applied underneath the cells prior to cell seeding and on top to the cell prior to fluorescence microscopy. Also novel image processing tools are required that can overlay and display the two correlated data sets in a suitable way for analysis and annotation.
In our journey to explore SBF-SEM for the imaging of WPBs, we experienced that SBF-SEM is a suitable tool to further investigate the more detailed morphological characteristics of the WPBs formation at the Golgi apparatus. To optimally use SBF-SEM for this purpose, optimization of the fixation and staining protocol is needed to elucidate the VWF tubules as well. So far we were able to show the WPB distribution around the Golgi in a large 3D volume. In addition, we could reveal that some of these WPBs were in close proximity or even connected to the Golgi membrane. Apart from WPB biogenesis, SBF-SEM will also be an effective approach to study the 3D organization upon WPB exocytosis. However, to efficiently study these biological processes in a targeted way, the combination with a CLEM method will be needed to pinpoint the structures of interest using fluorescence.
Acknowledgments
The authors would like to thank Roman Koning for help with Amira. This work was financially supported by the Netherlands Organization for Scientific Research (NWO TOP, grant no. 91209006).
Authors’ contributions
M.J.M. designed and performed the experiments, analysed data, made the figures and wrote the manuscript; F.G.A.F. analysed data and reviewed the manuscript; H.Z. performed experiments and reviewed the manuscript; J.E. and A.J. provided input on the experimental design and reviewed the manuscript. All authors consented with the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Supporting Information
Disclaimer: Supplementary materials have been peer-reviewed but not copyedited.
Video S1. Three-dimensional visualization of cross-sectioned Weibel–Palade bodies in endothelial cells. The data set was acquired at 25 000× magnification using a 10 nm slice thickness. For processing, the data were binned two times in x and y which resulted in a voxel resolution of 7.4 × 7.4 × 10 nm.
Video S2. Three-dimensional visualization of the Golgi apparatus with associating Weibel–Palade bodies in endothelial cells. The data set consist of almost 1600 images and was acquired at 25 000× magnification using a 10 nm slice thickness. For processing, the data were binned two times in x and y that resulted in a voxel resolution of 7.4 × 7.4 × 10 nm.
References
- Armer HE, Mariggi G, Png KM, Genoud C, Monteith AG, Bushby AJ, Gerhardt H. Collinson LM. Imaging transient blood vessel fusion events in zebrafish by correlative volume electron microscopy. PLoS One. 2009;4:e7716. doi: 10.1371/journal.pone.0007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcena M. Koster AJ. Electron tomography in life science. Semin. Cell Dev. Biol. 2009;20:920–930. doi: 10.1016/j.semcdb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro F, Kriston-Vizi J, Metcalf DJ, et al. A two-tier Golgi-based control of organelle size underpins the functional plasticity of endothelial cells. Dev. Cell. 2014;29:292–304. doi: 10.1016/j.devcel.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Hartwieg E. Nguyen KC. Modern electron microscopy methods for C. elegans. Methods Cell Biol. 2012;107:93–149. doi: 10.1016/B978-0-12-394620-1.00004-7. [DOI] [PubMed] [Google Scholar]
- Hendriks CL, Van Vliet L, Rieger B, van Kempen G. van Ginkel M. 1999. DIPimage: A Scientific Image Processing Toolbox for MATLAB. Quantitative Imaging Group, Faculty of Applied Sciences, Delft University of Technology, Delft, the Netherlands.
- Hoffpauir BK, Pope BA. Spirou GA. Serial sectioning and electron microscopy of large tissue volumes for 3D analysis and reconstruction: a case study of the calyx of Held. Nat. Protoc. 2007;2:9–22. doi: 10.1038/nprot.2007.9. [DOI] [PubMed] [Google Scholar]
- Holcomb PS, Hoffpauir BK, Hoyson MC, et al. Synaptic inputs compete during rapid formation of the calyx of Held: a new model system for neural development. J. Neurosci. 2013;33:12954–12969. doi: 10.1523/JNEUROSCI.1087-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G. Genoud C. Is EM dead. J. Cell Sci. 2013;126:4545–4552. doi: 10.1242/jcs.124123. [DOI] [PubMed] [Google Scholar]
- Lucas MS, Guenthert M, Gasser P, Lucas F. Wepf R. Correlative 3D imaging: CLSM and FIB-SEM tomography using high-pressure frozen, freeze-substituted biological samples. Methods Mol. Biol. 2014;1117:593–616. doi: 10.1007/978-1-62703-776-1_26. [DOI] [PubMed] [Google Scholar]
- Michaux G, Hewlett LJ, Messenger SL, Goodeve AC, Peake IR, Daly ME. Cutler DF. Analysis of intracellular storage and regulated secretion of 3 von Willebrand disease-causing variants of von Willebrand factor. Blood. 2003;102:2452–2458. doi: 10.1182/blood-2003-02-0599. [DOI] [PubMed] [Google Scholar]
- Mironov AA. Beznoussenko GV. Correlative light-electron microscopy a potent tool for the imaging of rare or unique cellular and tissue events and structures. Methods Enzymol. 2012;504:201–219. doi: 10.1016/B978-0-12-391857-4.00010-0. [DOI] [PubMed] [Google Scholar]
- Murphy GE, Narayan K, Lowekamp BC, Hartnell LM, Heymann JA, Fu J. Subramaniam S. Correlative 3D imaging of whole mammalian cells with light and electron microscopy. J. Struct. Biol. 2011;176:268–278. doi: 10.1016/j.jsb.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia JC, Kasthuri N, Hayworth KJ, Schalek R, Lichtman JW, Smith SJ. Buchanan J. High-contrast en bloc staining of neuronal tissue for field emission scanning electron microscopy. Nat. Protoc. 2012;7:193–206. doi: 10.1038/nprot.2011.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn KM, Valentijn JA, Jansen KA. Koster AJ. A new look at Weibel-Palade body structure in endothelial cells using electron tomography. J. Struct. Biol. 2008;161:447–458. doi: 10.1016/j.jsb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Valentijn KM, Sadler JE, Valentijn JA, Voorberg J. Eikenboom J. Functional architecture of Weibel-Palade bodies. Blood. 2011;117:5033–5043. doi: 10.1182/blood-2010-09-267492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villinger C, Gregorius H, Kranz C, et al. FIB/SEM tomography with TEM-like resolution for 3D imaging of high-pressure frozen cells. Histochem. Cell Biol. 2012;138:549–556. doi: 10.1007/s00418-012-1020-6. [DOI] [PubMed] [Google Scholar]
- Voorberg J, Fontijn R, Calafat J, Janssen H, van Mourik JA. Pannekoek H. Biogenesis of von Willebrand factor-containing organelles in heterologous transfected CV-1 cells. EMBO J. 1993;12:749–758. doi: 10.1002/j.1460-2075.1993.tb05709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER. Palade GE. New cytoplasmic components in arterial endothelia. J. Cell Biol. 1964;23:101–112. doi: 10.1083/jcb.23.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenner HL, Collinson LM, Michaux G. Cutler DF. High-pressure freezing provides insights into Weibel-Palade body biogenesis. J. Cell Sci. 2007;120:2117–2125. doi: 10.1242/jcs.007781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Three-dimensional visualization of cross-sectioned Weibel–Palade bodies in endothelial cells. The data set was acquired at 25 000× magnification using a 10 nm slice thickness. For processing, the data were binned two times in x and y which resulted in a voxel resolution of 7.4 × 7.4 × 10 nm.
Video S2. Three-dimensional visualization of the Golgi apparatus with associating Weibel–Palade bodies in endothelial cells. The data set consist of almost 1600 images and was acquired at 25 000× magnification using a 10 nm slice thickness. For processing, the data were binned two times in x and y that resulted in a voxel resolution of 7.4 × 7.4 × 10 nm.