

Abstract
The cellular REDOX regulatory systems play a central role in maintaining REDOX homeostasis that is crucial for cell integrity, survival, and proliferation. To date, a substantial amount of data has demonstrated that cancer cells typically undergo increasing oxidative stress as the tumor develops, upregulating these important antioxidant systems in order to survive, proliferate, and metastasize under these extreme oxidative stress conditions. Since a large number of chemotherapeutic agents currently used in the clinic rely on the induction of ROS overload or change of ROS quality to kill the tumor, the cancer cell REDOX adaptation represents a significant obstacle to conventional chemotherapy. In this review we will first examine the different factors that contribute to the enhanced oxidative stress generally observed within the tumor microenvironment. We will then make a comprehensive assessment of the current literature regarding the main antioxidant proteins and systems that have been shown to be positively associated with tumor progression and chemoresistance. Finally we will make an analysis of commonly used chemotherapeutic drugs that induce ROS. The current knowledge of cancer cell REDOX adaptation raises the issue of developing novel and more effective therapies for these tumors that are usually resistant to conventional ROS inducing chemotherapy.
1. Introduction
Reactive oxygen species (ROS) are radical and nonradical oxygen-containing chemical molecules that show different degrees of reactivity. ROS include biologically important molecules such as superoxide anion (O2 •−), hydroxyl radical (•OH) and hydrogen peroxide (H2O2). For a long time ROS have been tagged as harmful oxidants; however it is currently well established that the ROS molecule H2O2 constitutes an important second messenger in cell signaling transduction. This is due to its high diffusion rate across membranes particularly through the use of aquaporins and its ability to target reactive/REDOX sensitive cysteine residues in proteins [1–6]. Activation of NADPH oxidases (NOX) through the binding of growth factors, cytokines, neurotransmitters, and hormones to their receptors leads to the inducible production of O2 •− that is subsequently converted into H2O2, by the enzyme superoxide dismutase (SOD). To date, H2O2-dependent signaling has been shown to play a key role in the regulation of many cellular processes including differentiation, proliferation, migration, and apoptosis [3, 7–9]. Due to their reactive properties these species contribute, on different levels, to protein oxidation, lipid peroxidation, and/or DNA damage that can ultimately result in cell death or tumorigenesis (in case of DNA mutagenesis) [10, 11]. Consequently cells have several antioxidant systems to inactivate ROS and to recycle oxidized molecules; these include catalase, SOD, glutathione peroxidases, and thioredoxin peroxidases. The main cellular antioxidant systems responsible for recycling REDOX sensitive proteins are the thioredoxin (Trx) and glutathione (GSH) systems that reduce active cysteine residues present in ROS scavenging proteins (reestablishing their antioxidant function) and other proteins, whose functions are regulated by the oxidative status of key reactive cysteine residues (e.g., protein tyrosine phosphatases (PTPs), transcription factors, and the phosphatase and tensin homolog, PTEN) (Figure 1).
Figure 1.
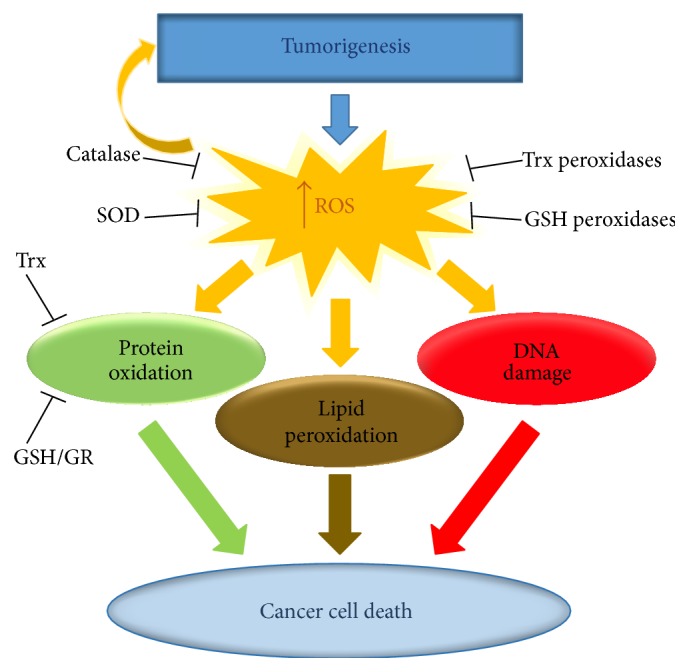
The cellular antioxidant systems. Tumor progression induces increasing oxidative stress. Cells have several antioxidant systems to directly inactivate ROS (e.g., Trx peroxidases, GSH peroxidases, catalase, and SOD) as well as REDOX regulatory systems that recycle/reactivate the ROS scavenging proteins and other REDOX sensitive proteins (e.g., PTPs, PTEN, and transcription factors).
Several studies have shown an important role for ROS in tumor development [12, 13]. Cancer cells generally display elevated ROS compared to normal counterparts that give them a proliferative advantage and promote malignant progression [14]. In the tumor site the hypoxic cancer cells (in a low oxygen environment) typically show even higher levels of ROS compared to nonhypoxic cancer cells [15]. To deal with the increasing oxidative stress experienced as the tumor progresses, cancer cells upregulate the cellular antioxidant systems. This allows the cancer cells to maintain low to moderate levels of ROS (important for the activation of proliferative signaling pathways) while avoiding high levels of ROS that have damaging effects and can induce cell death [16–18].
A large number of currently used chemotherapeutic agents kill cancer cells in part through the generation of ROS. These drugs are less toxic to normal cells that have lower endogenous levels of ROS. Consequently, the upregulation of antioxidant proteins within the cancer cells will render them more resistant to chemotherapy [17–20].
In this review we will make a comprehensive examination of the current literature regarding the redox regulatory systems that become upregulated in cancer and their role in promoting tumor progression and resistance to chemotherapy. A better understanding of the molecular mechanisms used by cancer cells to adapt and survive to oxidative stress may also allow the development of more efficient chemotherapeutic treatments.
2. Sources of ROS in Cancer
The existing high levels of ROS typically observed in cancer cells are the result of accumulation of intrinsic and/or environmental factors.
2.1. Intrinsic Sources of ROS
Several factors within the tumor site contribute to the generation of high levels of ROS in the cancer cells. The more relevant factors include hypoxia (low levels of oxygen), enhanced cellular metabolic activity, mitochondrial dysfunction, increased growth factor receptor mediated signaling transduction, oncogene activity, increased activity of oxidases, lipoxygenases, cyclooxygenases and thymidine phosphorylase, and the crosstalk between cancer cells and immune cells recruited to the tumor site (Figure 2) [21, 22].
Figure 2.

Sources of ROS in cancer. A number of intrinsic and extrinsic factors contribute to oxidative stress within the tumor as illustrated in the figure.
Within the tumor mass, it has been well established that hypoxic cancer cells generally have higher levels of ROS compared to nonhypoxic counterparts. Hypoxia contributes to the production of ROS through different mechanisms. The major regulator of the hypoxic response is the transcription factor Hypoxia Inducible Factor (HIF). The regulation of the alpha subunit of this transcription factor (HIF-α) is dependent on intracellular levels of oxygen. HIF-α regulation is mediated by the action of prolyl hydroxylases (PHDs), enzymes that in the presence of normal concentrations of oxygen (normoxia) are able to hydroxylate HIF-α at two prolyl residues allowing the binding of the protein von Hippel-Lindau (pVHL). VHL in its turn recruits E3 ubiquitin ligases which target HIF-1α for proteasomal degradation [23–25]. Low levels of oxygen lead to the inhibition of PHDs and subsequent stabilization and accumulation of the HIF-α subunit. HIF-α translocates to the nucleus and binds to the HIF-1β subunit and cofactors such as CBP/p300 inducing the transcription of more than one hundred genes involved in promoting angiogenesis (formation of new blood vessels from preexisting blood vessels), glycolysis, epithelial-mesenchymal transition (EMT), proliferation, invasion, and recruitment of inflammatory cells to the tumor site [26, 27].
Hypoxia induced glycolysis and subsequent inhibition of oxidative phosphorylation at the mitochondrial membrane leads to an increase in ROS levels mediated by the mitochondrial complex III [28–30]. The hypoxic response also promotes the elevation of intracellular ROS via HIF dependent transcriptional activation of genes that encode for growth factors and their receptors. Binding of these growth factors to their receptors at the surface of cancer cells triggers signaling pathways that induce the activation of NADPH oxidases (NOX). NOX are responsible for the production of O2 •−, which is then converted into H2O2.
HIF-1α and HIF-2α stabilization are also dependent on ROS. Gao et al. showed for the first time a decrease in tumor growth in mice treated with the antioxidant N-acetyl cysteine (NAC) that was traced to a redox mediated reduction in the levels of HIF [31]. In this way ROS is not only a by-product of hypoxia, but it also contributes to the hypoxic response by stabilizing its main regulator and thus creating a positive feedback loop. A number of studies have shown that the ROS produced in the mitochondria during hypoxia promote the oxidation/inactivation of prolyl hydroxylases (PHDs) thus stabilizing HIF-1α and HIF-2α [30, 32]. Furthermore the enhanced activation of NOX observed during hypoxia promotes NF-kβ dependent HIF-1α transcription [33].
In cancer cells (as in normal cells), the mitochondria is thought to be the largest contributor to intracellular ROS that are generated as by-products of oxidative phosphorylation. The mitochondrial electron transport chain is composed of complexes I–IV and ATP synthase in the inner mitochondrial membrane. Complexes I and II oxidize NADH and FADH2, respectively, and transfer the resulting electrons to ubiquinol, which carries it to complex III. Within complex III the electrons are shuttled across the inner mitochondrial membrane to cytochrome c, which carries them to complex IV. The flow of electrons throughout the respiratory chain culminates at complex IV with the reduction of molecular oxygen to water. The incremental release of energy resulting from electron transfer is used to pump protons (hydrogen ions) into the intermembrane space generating a proton gradient, the dissipation of which is used by ATP synthase, to power the phosphorylation of ADP to ATP. Throughout this process, a small percentage of molecular oxygen can also undergo a one-electron reduction generating O2 •− [27, 34]. O2 •− is produced in complexes I and III of the electron transport chain and released to the intermembrane space (approximately 80%) or to the mitochondrial matrix (approximately 20%) [35, 36]. O2 •− can be converted into H2O2 that is able to cross the membranes and translocate to the cytoplasm and subsequently into the nucleus [1, 2]. In these different cellular compartments H2O2 can oxidize several cellular components, including proteins, lipids, and DNA (in the nucleus and mitochondria). Although peroxisomes are cellular organelles that are involved in ROS scavenging (via catalase mediated conversion of H2O2 into O2 and H2O) they also contribute to intracellular ROS production through the β-oxidation of fatty acids and by flavin oxidase activity [37]. The endoplasmic reticulum (ER) also contributes to the elevation of intracellular ROS levels via protein oxidation (disulphide bond formation) that occurs during the process of protein folding [38].
NOX constitute another important source of ROS in cancer cells. The NOX family consists of seven members, namely, NOX 1–5 and the dual oxidases DUOX 1-2. NOX are composed of transmembrane and cytoplasmic subunits that exert distinct functions (regulatory/catalytic). The catalytic subunit of all NOX isoforms contains six transmembrane domains in the N-terminal half, four conserved histidine residues located in the third and fifth transmembrane helices which coordinate two hemes, and a flavoprotein and an NADPH-binding domain in the cytosolic C-terminal region [39]. The NOX catalytic subunit transfers an electron from intracellular NADPH across the cytoplasmic membrane to oxygen via FAD and the two heme groups, with the second heme group being responsible for reducing extracellular molecular oxygen to produce O2 •−. The O2 •− can be rapidly converted into H2O2 either spontaneously or through the action of SOD, which crosses the plasma membrane into the cytoplasm [39]. NOX are activated by various signaling proteins, including growth factors, cytokines, hormones, and neurotransmitters that are typically overexpressed within the tumor microenvironment leading to exacerbated activation of NOX and consequently increased levels of ROS within the cancer cells [3, 8].
Nowadays, it is well established that, in addition to the cancer cells, the tumor mass is constituted by innate immune cells (including macrophages, neutrophils, mast cells, myeloid-derived suppressor cells, dendritic cells, and natural killer cells); adaptive immune cells (T and B lymphocytes); and cells from the surrounding stroma that are recruited to the tumor site (consisting of fibroblasts, endothelial cells, and pericytes) [40]. These noncancerous cells constitute the tumor microenvironment and play a key role in tumorigenesis. Tumor associated cells are capable of inducing the generation of ROS in cancer cells through the secretion of various growth factors and other signaling proteins leading to the activation of NOX in the cancer cell. Neutrophils and macrophages can produce a rapid burst of O2 •− (primarily mediated via NOX) leading to the subsequent generation of H2O2 that can diffuse through the cytoplasmic membrane and into the neighboring cancer cells [35, 41]. Tumor associated macrophages (TAMs) can also produce nitric oxide within the tumor site through the activation of nitric oxide synthase 2 (NOX2). Nitric oxide reacts with superoxide to produce peroxynitrite (ONOO-) that can either be protonated to peroxynitrous acid and then spontaneously decompose into nitrogen dioxide (NO2) and •OH (highly reactive/oxidizing) or react with carbon dioxide (CO2), yielding nitrosoperoxy carboxylate (ONOOCO2) which decomposes into carbonate radicals (CO3 •−) and NO2 [42, 43].
2.2. Environmental Sources of ROS
Environmental sources of ROS can also significantly contribute to tumorigenesis. Ionizing radiation is the most studied source of environmental ROS and has been shown to play a role in all stages of carcinogenesis from cancer initiation to tumor progression. Ionizing radiation generates ROS through the radiolysis of water molecules and also by secondary reactions that can persist and diffuse within the cells [44].
Other environmental factors such as tobacco components, xenobiotics, chlorinated compounds, metal ions, barbiturates, and phorbol esters can generate ROS in the cells by metabolism to primary radical intermediates or by activating endogenous sources of ROS (Figure 2) [45].
3. Cancer REDOX Adaptation
To balance the beneficial effects of low to moderate levels of ROS, which induce proliferative signaling pathways, and avoid the harmful oxidant effects of high levels of ROS that can damage proteins, lipids, and DNA, cancer cells undergo REDOX adaptation. Increasing evidence has demonstrated a crucial role for the cellular antioxidant systems in supporting tumor initiation, progression, and chemoresistance. Bellow we summarize what is currently known regarding the main antioxidant proteins/systems involved in cancer (Figure 3).
Figure 3.

Antioxidant systems in cancer. Cancer cells undergo REDOX adaptation to survive and proliferate in an environment with increasing oxidative stress. Regulation of ROS levels by the cellular antioxidant systems is crucial to maintain a proliferative and mutagenic phenotype (associated with low/moderate levels of ROS) and avoid apoptosis or senescence (associated with high levels of ROS).
3.1. GSH System
The GSH and Trx systems are the main antioxidant systems responsible for the reduction of redox sensitive proteins in the cells.
The tripeptide GSH is the most abundant nonenzymatic antioxidant in the cell. GSH is a multifunctional antioxidant being able to (i) function as a cofactor of several oxidative stress detoxifying enzymes, for example, glutathione peroxidase (GPx), glutathione transferase, and others; (ii) directly scavenge ROS, such as hydroxyl radical and singlet oxygen; (iii) detoxify hydrogen peroxide and lipid peroxides through the catalytic activity of glutathione peroxidase; (iv) regenerate/reduce vitamins C and E; and (v) react with oxidized sulphenic acid and thiyl radical groups in proteins to form S-glutathionylated proteins (protein-SSG), protecting these proteins from further oxidation. S-Glutathionylated proteins can then be further reduced by the glutathione cycle through the action of glutathione reductase and small proteins such as glutaredoxin and thioredoxin [46–48].
It is well known that GSH metabolism is modified in a vast number of cancers. High levels of GSH have been reported in many types of cancer including breast, melanoma, and liver. Furthermore a direct correlation between GSH concentration and cellular proliferation and metastasis has also been established [18, 47, 49]. It is important to keep in mind that the GSH system has many components and that a slight alteration in one of them may have an instant result, disrupting the balance of cellular antioxidant response and REDOX homeostasis. Various modifications within the GSH system have been observed in cancer: higher levels of GSH-related enzymes, such as γ-glutamylcysteine ligase (GCL), γ-glutamyl-transpeptidase (GGT), and glutathione-S-transferases (GST), have been reported, as well as higher expression of GSH-transporting pumps [50–52]. Decreased GSH/GSSG ratio has also been found in advanced cancer patients; this can be explained by an increased generation of H2O2 that readily oxidizes GSH, turning it into GSSG [48].
The GSH system is a major contributor to chemotherapy resistance. This system participates not only in the cellular antioxidant defense, but also in drug-resistance metabolic processes including the detoxification and efflux of xenobiotics. The GSH system also participates in DNA repair processes as well as in the inactivation of the cancer cell proapoptotic pathway [18, 51, 53, 54]. GSH is able to directly interact with cisplatin and trisenox, inactivating these chemotherapeutics [55, 56]. For all these reasons, significant effort has been engaged at depleting cellular GSH levels to sensitize tumors to the cytotoxic effects of chemotherapeutic drugs.
Recently, a study showed that GSH plays a crucial role in tumor initiation [57]. Using the oncogene-induced murine model of mammary cancer (MMTV-PyMT) it was shown that a 75% depletion in the levels of GSH in these mice led to the formation of fewer tumors that progressed more slowly than those in mice with normal GSH levels [57].
3.2. TRX System
Thioredoxins (Trx) are small proteins (with a molecular weight of approximately 12 KDa) that repair oxidized proteins. Trx possess a conserved Cys-Gly-Pro-Cys redox catalytic site that is able to reduce oxidized thiols (reactive cysteine residues) in target proteins. Trx reductases (TrxR) recycle the oxidized Trx through the transfer of reducing equivalents from NADPH to the oxidized catalytic site of Trx (Trx-S2) reducing it to a dithiol (Trx-(SH)2) [9, 58]. Amongst all members of the thioredoxin system, Trx1 and TrxR1 have emerged as critical redox regulators and as potential therapeutic targets for many types of human cancers [59].
Trx are major protein disulphide reductases in the cell, being able to catalyze the reduction of disulphide bonds in proteins orders of magnitude faster than GSH [60]. Trx are involved in multiple redox-dependent signaling pathways in cancer by regulating redox-sensitive transcription factors (e.g., activator protein 1 (AP-1), NF-κB, and p53); signaling proteins (e.g., protein tyrosine phosphatases, PTPs, and PTEN); and other antioxidant proteins involved in the regulation of these signaling cascades (e.g., peroxiredoxins). Reduced Trx has been shown to bind to and inhibit apoptosis signal-regulated kinase 1 (ASK1) [61]. ASK1 activates the c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase pathways leading to apoptosis [62, 63]. Furthermore, Trx also play an important role controlling cancer cell growth through the supply of reducing equivalents for DNA synthesis [64].
Elevated levels of Trx have been reported in multiple types of cancers including cervical, liver, gastric, lung, and colorectal cancers [48, 64]. Many studies have also revealed a positive correlation between elevated levels of Trx in human tumors and resistance to chemotherapy [65–67]. Although a large number of reports have established a key role for the Trx system in cancer, a recent study seems to indicate that the Trx system is essential for tumor progression, but not initiation, where the GSH system plays a more prominent role [57].
3.3. SOD
Superoxide dismutase (SOD) is responsible for the dismutation of O2 •− into oxygen (O2) and H2O2 (that is less reactive than O2 •−), thus providing an antioxidant defense [47]. In humans there are three isoforms of SOD: cytosolic Cu, Zn-SOD, mitochondrial Mn-SOD, and extracellular SOD (EC-SOD) [68]. Of these isoforms, the mitochondrial Mn-SOD has been more associated with cancer by functioning as a mitochondrial ROS switch.
A large number of reports demonstrated the elevation of Mn-SOD in tumors (e.g., lung, esophageal, gastric, head and neck, prostate, and colon) compared to matched normal tissues and elucidated that the enhanced expression of Mn-SOD resulted in increased cancer cell invasiveness, growth, survival, and metastatic behavior [69–75]. Overexpression of Mn-SOD is likely a compensatory mechanism to intrinsic ROS stress, especially in cancer cells with mitochondrial dysfunction and consequently increased levels of O2 •−. In this situation enhanced expression of Mn-SOD might help the cancer cell to counteract superoxide stress and thus promote tumor progression. A potential mechanism for Mn-SOD induced metastasis is the H2O2-dependent increase in matrix metalloproteases expression [76, 77]. Interestingly, other reports demonstrated low activity of Mn-SOD in a wide range of cancers including cervical, breast, prostate, lung, and liver [78–82]. These studies also showed that overexpression of Mn-SOD in cancer cells led to decreased proliferation, anchorage-dependent growth, and invasiveness. However, these tumor suppressive properties of Mn-SOD were mainly observed in vitro, in experimental systems where cancer cells were artificial transfected with Mn-SOD-expressing vectors to induce high expression of this enzyme [78, 83]. Under these conditions, the cancer cell redox homeostasis can be significantly disturbed, leading to inhibition of cancer cell proliferation. However, the physiological relevance of such tumor-suppression function of Mn-SOD is still unclear in cancer cells in vivo. Also, Mn-SOD cancer cell growth suppression can be modulated by diminishing the levels of carcinogen-inducing O2 •− [84–86] and sensitization of cancer cells to cell death induced by different ROS-generating agents [87]. This dual effect of SOD acting as either a tumor promoter or a tumor suppressor should be addressed taking into account the nature of the ROS molecules generated in low or high SOD expressing cancer cells. O2 •− (which is elevated in tumors with low SOD activity) is a reductant of iron, which subsequently generates •OH by transferring the electron to H2O2 [88]. The highly reactive •OH can lead to enhanced mutagenesis via DNA oxidation. On the other hand, increased SOD activity will lead to enhanced levels of H2O2 that oxidizes reactive cysteine residues in proteins with high specificity, constituting an important second messenger in a wide variety of signaling pathways that activate proliferation, invasiveness, and metastasis. Furthermore, increased levels of O2 •− (associated with low levels of SOD) might be more beneficial to cancer cells in the early stages of tumor development where oxidative stress within the tumor mass is still low; increased levels of H2O2 (associated with high levels of SOD) might have a more prominent role at later stages of tumor progression, lowering unspecific damage of cellular components, while increasing H2O2 dependent signaling pathways. However there is still a lot of work to be done to investigate these hypotheses.
3.4. Catalase
Catalase (CAT) was originally regarded as a monofunctional peroxisomal enzyme that efficiently catalyzes the conversion of H2O2 into water and O2. This notion has evolved over the years and catalase is now considered a multifunctional enzyme that exhibits not only classic catalase activity, but also peroxidase [89, 90] and oxidase functions [91]. Moreover, catalase activity is not limited to the degradation of H2O2, as this enzyme also degrades peroxynitrite in an enzymatic reaction that, similarly to its classical reaction with H2O2, involves the formation of compound I (CATFeIV=O•+) [92, 93]. In addition, compound I of catalase can oxidize NO (CATFeIV=O•+ + 2•NO + H2O → CATFeIII + 2H+ + 2NO2 −) [94, 95], whereas native ferricatalase (CATFeIII) is reversibly inhibited by NO [96]. Thus, catalase has the potential to execute a central modulatory function at the cross point between H2O2 and NO/peroxynitrite-mediated signaling pathways.
Decreased CAT activity has been reported in cancer [69]. This low CAT activity leads to high levels of H2O2 and creates an intracellular environment favorable to DNA mutagenesis and to the activation of H2O2-dependent signaling pathways that typically trigger cell proliferation, invasion and metastatic phenotypes, consequently promoting tumor progression [35, 97, 98].
However, malignant cells may acquire membrane-associated catalase. Cancer cells exhibit enhanced NOX1-dependent superoxide anion generation [93, 99–102]. Various studies have shown that the overexpression of membrane-associated catalase on the outer surface of tumor cells has a protective role against apoptosis induced intercellular ROS signaling [93, 101–103]. Catalase-mediated protection from intercellular ROS signaling was found in all human cancer cell lines studied so far (more than 70 cell lines) [104], indicating that this might be an important mechanism for tumor survival. Efficient protection of tumor cells by membrane-associated catalase is not in contradiction to the finding that, in general, tumor cells have less catalase than normal tissue, as the surface of the tumor cell with its high local concentration of catalase represents a small proportion of the total cellular mass [105].
3.5. Peroxiredoxins
Peroxiredoxins (Prdx) are thioredoxin peroxidases that are divided into three classes: typical 2-cysteine peroxiredoxins (PrxI–IV), atypical 2-cysteine peroxiredoxins (PrxV), and 1-cysteine peroxiredoxins (PrxVI) [50]. Although their individual roles in cellular redox regulation and antioxidant protection are distinct, they all catalyze the reduction of H2O2, organic hydroperoxides, and peroxynitrite, to balance intracellular ROS levels [106, 107]. Due to their widespread distribution within the cell, Prdx are thought to constitute broad-range cellular antioxidant defenders. However, recent studies have demonstrated that typical 2-cysteine peroxiredoxins (Prdx I–IV) play key cell signaling regulatory roles by either interacting directly with specific REDOX-sensitive signaling protein(s) or by being located in the vicinity of REDOX-dependent signaling cascades (lipid rafts) and buffering ROS generated by NOX [108]. The 2-Cys Prx have been implicated in the regulation of diverse cellular processes, including proliferation, migration, apoptosis, and metabolism [108].
Overexpression of all known Prdx has been reported in many types of cancers including prostate, breast, lung, thyroid, and mesothelioma and is associated with cancer cell survival and tumor progression [109–112]. The ROS buffering and signaling regulatory (prosurvival) functions of Prdx have been suggested to play a role in tumor chemoresistance [106, 107, 112–114].
3.6. APE1/Ref-1
The Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) is a key enzyme that in addition to its DNA base excision repair function it also exerts important cellular functions in the REDOX control of multiple transcription factors involved in cancer progression, including NF-κB, STAT3, AP-1, HIF-1, and p53 [115–118]. All of these transcription factors possess reactive cysteine residue(s) within their DNA binding domain that can be readily oxidized/inactivated by cellular ROS. APE1/Ref-1 is able to reduce these cysteine residue(s) through the exchange of a proton from one or two of its redox cysteine residues (Cys65, Cys93, Cys99, or Cys138). This restores the DNA binding capability of the transcription factors and subsequently promotes transcription of their target genes. Oxidized APE1/Ref-1 is then recycled/reduced by the Trx system [119].
APE1/Ref-1 is highly expressed in many cancers (e.g., breast, lung, liver, and gliomas) [120–122]. APE1/Ref-1 role in cancer progression is likely due to its ability to increase DNA repair and to activate antiapoptotic, inflammatory, and growth-promoting transcription factors [121]. APE1/Ref-1 has also been shown to be associated with tumor resistance to both ionizing radiation and chemotherapy [123–125]. Consequently, APE/Ref-1 has emerged as a promising therapeutic target for cancer treatment [115, 120, 126].
3.7. Transcription Factors
Transcription factors can indirectly counteract ROS in cancer. The transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) binds to antioxidant response elements (ARE) in the regulatory regions of target genes and induces the transcription/expression of antioxidant enzymes [e.g., NAD(P)H:quinone oxidoreductase 1 (NQO1); SOD; catalase; heme oxygenase-1 (HO-1), TrxR, GST, GSH-synthetizing enzymes, and UDP-glucuronosyltransferases (UGTs)] [127–129]. NRF2 is considered to be the master regulator of intracellular antioxidant responses. NRF2 is regulated by its binding partner KEAP1 that targets NRF2 to proteasomal degradation [130]. It has been shown that hyperactive and oncogenic phosphoinositide 3-kinase- (PI3K-)AKT signaling which is commonly observed in a large number of cancer cells activate NRF2 [46]. NRF2 mutations have also been found in several types of tumors such as skin, esophageal, lung, ovarian, and breast cancers [131]. Generally these mutations are observed in the KEAP1-binding domain of NRF2 preventing KEAP1-mediated degradation of this transcription factor [131, 132]. Inactivating mutations in KEAP1 have also been identified [133]. All of these mutations lead to the constitutive stabilization of the NRF2 protein in the nucleus, providing strong evidence for a role for NRF2 in tumorigenesis. High expression of antioxidant NRF2 target genes has been linked to chemoresistance. For instance, HO-1 seems to be an important effector of NRF-2 induced chemoresistance in myeloid leukemias and inhibition of either NRF2 or HO-1 sensitizes the tumor to therapy [127]. It was also described that NRF2 enhances the cellular antioxidant capacity to counteract ROS mediated stress in cancer cells overexpressing oncogenic KRas [20].
Forkhead box O (FOXO) transcription factors belong to the large group of forkhead transcription factors that are all characterized by a conserved DNA binding domain termed the forkhead box [134]. FOXO activate the transcription of genes that encode for antioxidant proteins, including Mn-SOD, catalase, and sestrin 3 [135]. Although FOXO is generally considered to be a tumor suppressor and FOXO inactivation has been reported in a number of cancers [136, 137], recent studies have indicated that these transcription factors might also have protumorigenic functions. It has been shown that activated FOXO transcription factors support the survival of Acute Myeloid Leukemia (AML) cells [138]. FOXO transcription factors are inactivated by the PI3K-AKT pathway. Phosphorylation of FOXO by AKT leads to FOXO translocation from the nucleus into the cytoplasm, where they cannot exert their transcriptional function [134]. Interestingly, it has been observed that the FOXO genes are involved in chromosomal translocations that lead to alveolar rhabdomyosarcoma and acute lymphoblastic leukemia (ALL) [139]. Specifically, the paired box 3- (PAX3-) FOXO1 translocation is found in approximately 60% of all alveolar rhabdomyosarcoma tumors [140, 141]. The PAX3-FOXO1 fusion protein is insensitive to AKT inactivation, meaning that AKT cannot inhibit PAX-FOXO1 translocation to the nucleus or regulate its transcriptional activity [142]. However the contribution of FOXO transcriptional activity within the fusion protein is still fairly unknown and increased activity of PAX3 has been reported [141].
3.8. Dietary Antioxidant Compounds
Dietary antioxidants are nonenzymatic compounds that, although less specific compared to enzymatic antioxidants, appear to be important in cellular responses to oxidative stress including during tumorigenesis. For instance, vitamin C (ascorbic acid) is a water-soluble antioxidant which is mostly present in the cell in its reduced form, ascorbate, which acts as a reductant and enzyme cofactor and directly reacts with O2 •−, •OH, and various lipid hydroperoxides. Vitamin E (α-tocopherol) is a fat-soluble antioxidant, which acts as a free radical scavenger by converting free radicals into tocopheryl radicals, thus lowering their radical damaging abilities. Selenium is a nonmetal element that forms part of antioxidant selenoproteins such as glutathione peroxidase and thioredoxin reductase. Vitamin A (β-carotene) is a known fat-soluble antioxidant and its antioxidant property derives from its ability to quench singlet oxygen and trap peroxyl radicals. Deficiency of vitamin A causes oxidative stress, inhibiting cell repair function and causing cell damage [46, 47].
Much debate has focused on the consumption of antioxidant supplements by cancer patients undergoing chemotherapy due to concerns that the antioxidants may interfere with the mechanism of action of ROS generating therapeutic agents and subsequently decrease their efficacy [143, 144]. In this way, antioxidant supplementation might help the cancer cells to achieve redox homeostasis during tumor progression (generally accompanied by increasing oxidative stress). A recently published research using B-Raf and K-Ras induced lung cancer mouse models has demonstrated that the dietary antioxidant compound, vitamin E, can actually promote tumor progression [145]. This study further showed that vitamin E increased cell proliferation by decreasing ROS, DNA damage, and p53 expression in both mouse and human lung cancer cells [145].
Interestingly vitamin C has also been shown to have a prooxidant function. Vitamin C can reduce catalytic metals within the cells that in turn react with oxygen, producing O2 •− that subsequently dismutates into H2O2. The prooxidant function of vitamin C is observed at pharmacological doses (millimolar concentrations). It is currently accepted that the anticancer effect of pharmacological doses of ascorbate is mediated by the generation of high concentrations of extracellular H2O2 that diffuses through the plasma membrane causing DNA damage and triggering proapoptotic signaling pathways which will ultimately lead to cell death [146, 147]. A recent study investigated the synergistic action of pharmacological doses of vitamin C in combination with chemotherapeutic drugs used in the clinic for the treatment of ovarian cancer. These results showed that vitamin C significantly increased the efficacy of carboplatin and/or paclitaxel using a tumorigenic mouse model, where ovarian cancer cells were intraperitoneally injected in the mice [147]. A small pilot phase 1/2a clinical trial was also conducted in patients newly diagnosed with stage III/IV ovarian cancer [147]. Reduction of chemotherapy-associated toxicity was observed in patients receiving intravenous vitamin C in addition to carboplatin and paclitaxel treatment. A tendency for enhanced overall survival and longer time to relapse in patients receiving vitamin C in combination with carboplatin and paclitaxel compared to patients receiving only carboplatin and paclitaxel was observed; however these results were not statistically significant. A larger clinical trial might be useful to further clarify these results. The use of ascorbate in cancer therapy is still a matter of debate as preclinical studies have shown a large variation in ascorbate sensitivity even within cancers of the same type. This may be the result of intrinsic differences between cancer cells from diverse origins (e.g., antioxidant defenses) or the tumor microenvironment in the case of organ-specific cancers, which are presently poorly understood. The fact that vitamin C can function as an antioxidant (at low doses) or a prooxidant (at high doses) is most likely at the basis of these contradictory results and dosage as well as other factors (e.g., intrinsic antioxidant defenses, expression of sodium-dependent vitamin C transporters, activation of signaling pathway(s), etc.) should be taken into account when considering the use of this compound in combination with ROS producing chemotherapeutics.
3.9. Others
3.9.1. Annexin A2
Annexin A2 belongs to the family of annexins, which are commonly described as calcium-dependent phospholipid-binding proteins [9]. Annexin A2 is a multifunctional protein that has been shown to be overexpressed in a large number of cancers (e.g., breast, liver, gastric, pancreatic, lung, gliomas, colorectal, and ovarian) and to be positively associated with metastasis and resistance to chemotherapy [9, 148–151]. It was demonstrated that annexin A2 is unique among the vertebrate annexins in that it possesses an N-terminal reactive cysteine residue, Cys-8, which is reversibly oxidized by H2O2 inactivating this ROS molecule and generating H2O and O2. Oxidized annexin A2 is then recycled/reduced by the Trx system. Thus one molecule of annexin A2 has the ability to degrade several molecules of H2O2 [9, 58]. The role of the antioxidant function of annexin A2 in tumorigenesis was investigated using a mouse animal model. This study showed that the growth of tumors derived from annexin A2 depleted cancer cells was significantly impaired compared to tumors derived from control cancer cells (expressing normal levels of annexin A2). However, the growth impairment of the annexin A2-depleted tumors was rescued by the administration of the antioxidant compound, N-acetyl cysteine (NAC), in the mice during tumor formation [9, 58]. These results indicate that annexin A2 REDOX regulatory function plays a significant role in promoting tumor growth. It was also demonstrated that annexin A2 depleted cancer cells were significantly more sensitive to death induced by the ROS generating chemotherapeutic agents, etoposide, doxorubicin, and tamoxifen [58], elucidating for the first time a molecular mechanism by which annexin A2 provides resistance to chemotherapy, by functioning as an antioxidant protein. More recently it has been shown that a fraction of cellular annexin A2 translocates into the nucleus in response to different oxidative stress stimuli, including X-ray radiation, Gamma-radiation, UV radiation, etoposide, chromium VI, and H2O2 [9, 152, 153]. Nuclear annexin A2 was shown to protect cellular DNA from oxidative damage [153].
3.9.2. DJ-1
DJ-1 is a multifunctional protein that has been shown to also have antioxidant functions. DJ-1 contains three reactive cysteine residues, namely, Cys46, Cys53, and Cys106, being Cys 106 the most susceptible to oxidation. In fact, Cys 106 is essential for DJ-1 cytoprotective function against oxidative stress and mutation of this cysteine residue abolishes all functions of DJ-1 [154–158]. The antioxidant functions attributed to DJ-1 include the upregulation of intracellular glutathione synthesis via increasing glutamate cysteine ligase enzyme [159]; inhibition of oxidative stress induced apoptosis via direct binding of its Cys 106 residue to ASK1 [156, 160]; stabilization of NRF2 transcription factor that regulates the expression of many antioxidant genes (as described above) by preventing the binding of NRF2 to its inhibitor KEAP1 [161]. A recent study using cardiac cells showed that DJ-1 was able to inhibit ischemia/reperfusion-induced ROS generation, via upregulation of antioxidant enzymes [162]. These results suggest that DJ-1 might also trigger a similar antioxidant response in hypoxic cancer cells. However this has not yet been investigated.
DJ-1 has been shown to be upregulated in many human cancer types (e.g., lung, prostate, endometrial, and bladder), which correlated with cancer cell proliferation, tumor survival, and chemoresistance [59, 163–165].
4. ROS Inducing Chemotherapy
Typically, cancer cells exhibit higher levels of endogenous ROS compared to normal cells. For this reason a commonly used strategy for killing cancer cells consists of using ionizing radiation and/or chemotherapeutic drugs that induce the generation of these oxidants to levels that are capable of triggering apoptosis once ROS levels reach or exceed a certain threshold within the cell. The rationale for this approach relies on the fact that since cancer cells already have high levels of endogenous ROS prior to treatment they should reach this apoptotic threshold faster/easier compared to normal cells [166, 167].
Accordingly, oxidative stress has been recognized as a tumor specific target for the design of chemotherapeutic agents. A large number of chemotherapeutic drugs capable of inducing oxidative stress, by interfering with various antioxidant/redox regulatory proteins and/or ROS inducing pathways, are currently being used in the clinic (Table 1) [168].
Table 1.
Chemotherapeutic drugs commonly used in the clinic capable of inducing ROS.
| Drug name | Tumor type | Mechanism of action for ROS induction | References |
|---|---|---|---|
| Actinomycin D | Sarcomas; Wilms' tumor; testicular; melanoma; neuroblastoma; germ cell; retinoblastoma; choriocarcinoma | Inhibition of Bcl-2 | [179] |
|
| |||
| Bleomycin | Melanoma; Hodgkin's and non-Hodgkin's lymphomas; testicular; head and neck; cervical; malignant pleural effusions | Formation of Fe(II)-bleomycin-DNA complex that is oxidized by O2 | [185, 187] |
|
| |||
| Busulfan | Chronic myeloid leukemia | GSH depletion and NOX activation | [182, 183] |
|
| |||
| Carmustine | Brain; Hodgkin's and non-Hodgkin's lymphoma; melanoma; myeloma | GSH depletion via inhibition of GR | [169] |
|
| |||
| Cisplatin | Ovarian; colon; testicular; germ cell; bladder; lung; head and neck | Increased expression of p47phox subunit of NOX | [17, 188] |
|
| |||
| DMAT | Prostate | Inhibition of CK2 activity | [173] |
|
| |||
| Doxorubicin | Hodgkin's and non-Hodgkin's lymphoma; leukemia; breast; gastric; neuroblastoma; ovarian; lung; soft tissue and bone sarcomas; thyroid; bladder | p53-dependent transcription of cytochrome oxidase 2; FOXO3-dependent transcription of Noxa and BIM; quinone metabolism | [190] |
|
| |||
| Etoposide | Lymphomas; leukemias; neuroblastoma; breast; lung; testicular; gastric | FOXO3 dependent transcription of Noxa and BIM | [188, 190] |
|
| |||
| 5-Fluorouracil | Gastric; colon; gynecological; breast; head and neck; lung; skin | p53-dependent transcription of ROMO 1 | [188, 189] |
|
| |||
| Gemcitabine | Pancreatic; lung; in combination with other drugs: breast, bladder, and ovarian | Activation of AKT and ERK 1/2 which leads to upregulation of CXCR4 | [198] |
|
| |||
| Mitomycin C | Colon; breast; head and neck; bladder; cervical; gastric; pancreatic; liver | Inhibition of Bcl-2 | [180, 202] |
|
| |||
| Paclitaxel | Ovarian; breast; non-small cell lung carcinoma; Kaposi's sarcoma | Activation of Rac1 subunit of NOX; disruption of the mitochondrial membrane | [177, 178] |
|
| |||
| Tamoxifen | Breast | Inhibition of CK2 activity | [175] |
The chemotherapeutic agent carmustine interferes with the GSH antioxidant system by inhibiting glutathione reductase activity. GSH is an abundant cellular antioxidant that plays a key role in cellular REDOX homeostasis as already described in this review. Inhibition of glutathione reductase leads to the build-up of oxidized glutathione (GSSG) that can no longer exert its antioxidant function; as a consequence a significant accumulation of ROS within the cell is observed, driving caspase-3 activation and apoptosis [166, 169, 170].
Other chemotherapeutic drugs function through ROS-independent as well as ROS-inducing mechanisms. This is the case for the estrogen receptor inhibitor, tamoxifen, and for 2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) that inhibit the catalytic activity of protein cyclin kinase 2 (CK2). Under normal conditions, CK2 phosphorylates the apoptosis repressor with caspase recruitment domain (ARC) and Bid (proapoptotic member of the Bcl-2 family) proteins. Phosphorylated ARC localizes to the mitochondria, inhibiting caspase 8, while Bid phosphorylation by CK2 protects it from being cleaved by caspase 8. In conjunction phosphorylation of ARC and Bid by CK2 contributes to cell survival via inhibition of apoptosis. However, when CK2 is inhibited by tamoxifen or DMAT, it is not able to phosphorylate neither ARC nor Bid proteins. This will lead to caspase 8 activation, which will cleave Bid promoting its translocation to the mitochondrial membrane with the subsequent release of cytochrome C and activation of additional caspases, ultimately triggering apoptosis [171–173]. Disruption of the mitochondrial membrane by Bid will also lead to the release of ROS into the cytosol. CK2 inhibition also mediates NOX activation and the subsequent generation of ROS (O2 •− and H2O2). This occurs because CK2 can phosphorylate the cytosolic subunit of NOX, p47phox, negatively regulating NOX activity. Inhibition of CK2 by tamoxifen or DMAT will thus enhance NOX activity and induce the elevation of intracellular ROS which will also contribute to cell death induced by these chemotherapeutic agents [174–176].
Paclitaxel (also known as taxol) is another drug that is able to enhance NOX activity. This chemotherapeutic is a microtubule stabilizing agent that interferes with the normal breakdown of spindle microtubules during cell division, inhibiting mitosis and inducing apoptosis [177]. Paclitaxel has been shown to induce the translocation of Rac1 (regulatory subunit of NOX) from the cytosol to the plasma membrane. The increase of Rac 1 in the plasma membrane promoted the activation of NOX and led to the production of O2 •−, which was subsequently converted to H2O2 either spontaneously or by SOD [178]. This study showed that paclitaxel displayed a significant cytotoxic bystander effect in neighboring cancer cells [178], suggesting that this effect might play a substantial role in the antitumor activity of this drug. It is noteworthy that paclitaxel has limited ability to reach cancer cells that are distant from the vasculature. The fact that H2O2 can diffuse reasonably far from its site of production and can use aquaporins to enter the cells suggest that this mechanism might contribute significantly to paclitaxel induced cancer cytotoxicity.
The chemotherapeutic drugs actinomycin D and mitomycin C also activate the mitochondria-dependent apoptotic pathway. This occurs via inactivation of the antiapoptotic protein, Bcl-2. Bcl-2 forms heterodimers with the proapoptotic Bcl-2 family members: Bax, Bak, Bid, BIM, PUMA, and BAD sequestering these proteins and inhibiting apoptosis. Inactivation of Bcl-2 by actinomycin D or mitomycin C will elicit the release of the proapoptotic Bcl-2 family members causing mitochondrial membrane potential collapse and cytochrome C release from the mitochondria into the cytosol, subsequently increasing intracellular ROS levels. In the cytosol, cytochrome C induces apoptosis through the activation (via cleavage) of caspase-3 and caspase-9. Excessive ROS generation also contributes to cell death induced by these chemotherapeutic drugs [179–181].
A wide variety of DNA damaging (genotoxic) chemotherapeutic agents have been shown to induce cancer cell death in part through the increase of intracellular ROS levels. This is the case of busulfan, an alkylating agent that causes significant DNA damage by promoting the crosslinking between DNA molecules and also between DNA and proteins. Since these crosslinks can potentially originate DNA strand breaks it was hypothesized that busulfan most likely activates the DNA-damage response p53 signaling pathway to induce senescence or apoptosis. Interestingly, a p53 independent pathway has been reported for busulfan induced senescence. It was demonstrated that busulfan is metabolized through conjugation with GSH. This reaction is catalyzed by GST. This leads to depletion of intracellular levels of GSH, followed by a continuous increase in ROS production via NOX activation, which in turn activate the ERK and p38 MAPK signaling pathways, inducing cell senescence [182–184].
Bleomycin (BLM) is a chelator and a DNA damaging agent that is known to produce single- and double-strand breaks in the DNA as well as the release of free bases. BLM binds to DNA via its bithiazole and N-terminal moieties and interacts with Fe(II) via its ferrous binding site, forming an Fe(II)-BLM-DNA complex. BLM induces an increase in intracellular levels of ROS due to oxidation of the Fe(II)-BLM-DNA complex by O2 that leads to the generation of O2 •− and •OH [185–187].
Other genotoxic chemotherapeutics, such as doxorubicin and 5-Fluorouracil, trigger a p53-dependent proapoptotic pathway in order to kill cancer cells. The tumor suppressor p53 is involved in the maintenance of DNA integrity and under normal conditions it is maintained at low levels by the E3 ubiquitin ligase MDM2. Doxorubicin and 5-fluorouracil induce the stabilization of p53 and the subsequent transcription of p53 regulated genes including cytochrome oxidase 2, a component of the cellular respiratory chain that has been shown to generate ROS [188]. Another molecular mechanism by which 5-Fluorouracil induces the generation of ROS is by p53-dependent transcription of the ROS modulator 1 (ROMO1) gene. ROMO 1 protein localizes at the mitochondrial membrane and induces mitochondrial ROS generation [188, 189].
Doxorubicin also induces cancer cell apoptosis in a p53-independent way. Both doxorubicin and etoposide genotoxic chemotherapeutics promote the accumulation and nuclear translocation of the forkhead transcription factor, FOXO3, which activates the transcription of the proapoptotic genes Noxa and BIM. This promotes the collapse of the mitochondrial membrane with the subsequent release of cytochrome C and ROS into the cytosol ultimately inducing apoptosis [190–192]. Another mechanism by which doxorubicin produces ROS involves the addition of one-electron to the quinone moiety of doxorubicin resulting in the formation of a semiquinone that quickly regenerates into a quinone by reducing O2 to O2 •− and H2O2. The one-electron redox cycling of doxorubicin is accompanied by a release of iron from intracellular stores; binding of doxorubicin with iron results in formation of 3 : 1 drug-iron complexes that convert O2 •− and H2O2 into more potent •OH [193].
Doxorubicin is known to produce serious side effects, the most dangerous of which being cardiomyopathy which has been closely associated with abnormalities in mitochondrial functions including defects in the respiratory chain/oxidative phosphorylation system, decreased ATP production, mitochondrial DNA damage, modulation of mitochondrial sirtuin activity, and ROS generation [193–196]. For this reason it is important to take into account that using ROS to kill cancer cells might also have serious consequences for the patient that need to be taken into serious consideration before administration of these chemotherapeutic compounds.
The chemotherapeutic drug, cisplatin, is an alkylating agent that causes DNA crosslinking ultimately leading to p53-dependent apoptosis. It has been shown that DNA damage induced by cisplatin triggers a p53-dependent upregulation of ROS that activate p38 and JNK signaling pathways inducing apoptosis in a wide variety of cancer cell lines [188, 197].
The chemotherapeutic gemcitabine (2′-2′ difluorodeoxy cytidine, dFdC) is a prodrug that is phosphorylated by deoxycytidine kinase within the cell to form the active compounds gemcitabine diphosphate (dFdCDP) and gemcitabine triphosphate (dFdCTP). The active forms of gemcitabine incorporate into the DNA, causing cell cycle arrest and subsequent apoptosis [198]. It has also been shown that gemcitabine induces ROS as an additional anticancer mechanism [199]. Interestingly a report has shown that gemcitabine induced ROS activate the proproliferative and prosurvival MAPKS, ERK1/2, and AKT. The activation of these pathways leads to the enhanced nuclear accumulation of NF-kB and HIF-1α transcription factors which induce the transcription and subsequent expression of CXCR4 protein [198, 200]. These authors also demonstrated that increased expression of CXCR4 enhanced cancer cell invasion and migration to CXCL12 chemokine rich environments, namely, the stroma and blood vessels, promoting in this way chemoresistance due to escape from the tumor site [198]. For this reason, ROS concentrations within the cell should be considered during chemotherapy treatment since survival and metastatic mechanisms can be induced. It is crucial that ROS concentrations are increased until a threshold is reached where the cellular balance is tipped to activate apoptotic pathway(s) to stop tumor progression [176, 200]. Although high levels of ROS induced by chemotherapeutics can have dangerous side effects (as seen for doxorubicin), low dosages can also promote tumor growth instead of death. It becomes clear that finding the optimal dosage for the use of these chemotherapeutic agents is crucial for an effective cancer therapy.
All of the chemotherapeutics described above (summarized in Table 1) generate ROS that will contribute to tumor cell death. This is because excessive ROS can induce significant DNA damage, protein oxidation, organelles, and/or membranes oxidation tipping the cellular balance away from the proliferative advantages of low/moderate ROS levels to drive apoptosis in the presence of high ROS levels [201].
5. Conclusions and Future Directions
Increasing evidence has established that the cellular antioxidant systems play a key role not only in regulating (normal) cellular redox homeostasis, but also in protecting cancer cells from the increasing oxidative stress that they are subjected to during tumor progression. A large number of antioxidant proteins have been shown to be upregulated in cancer and to promote resistance to chemotherapy. These include not only proteins of the GSH and Trx systems that play a major role in recycling/reducing REDOX sensitive proteins, whose function is regulated by oxidation/reduction of key reactive cysteine residues, but also ROS scavenging proteins and transcription factors that induce the cellular antioxidant response. Novel antioxidant proteins, such as annexin A2 and DJ-1, have also been identified more recently that contribute significantly to tumorigenesis. In summary, since it was established that tumors are typically under substantial oxidative stress and as such have to adapt to survive in this extreme environment; increasing interest has been drawn to identifying the antioxidant/redox regulatory proteins involved in the tumor REDOX adaptation and in understanding the molecular mechanisms by which these proteins promote tumor progression and resistance to chemotherapy. Taking into account that many chemotherapeutic drugs currently in use in the clinic rely to varying degrees on ROS overload to kill the cancer cells, the tumor REDOX adaptation presents a major obstacle for the efficacy of these therapies. One approach to overcome this problem could be to deplete the cancer cell from REDOX regulatory potential through the downregulation of antioxidant protein(s) and peptides that have been shown to play crucial roles for tumor survival and growth in combination with chemotherapeutics that induce ROS.
Acknowledgments
Patrícia Alexandra Madureira has received funding from the European Union Seventh Framework Programme (FP7/2007–2013) under Grant agreement no. PCOFUND-GA-2009-246542 and from the Foundation for Science and Technology of Portugal and is a recipient of a Bolsa de Cientista Convidado (BCC) (ref. SFRH/BCC/105831/2014) from the Foundation for Science and Technology of Portugal, FCT Research Center Grant UID/BIM/04773/2013 CBMR 1334. The authors would like to thank Dr. Richard Hill for his critical review of the paper. The authors would like to thank Manuel Borges for his help with generating Figure 3.
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Bienert G. P., Møller A. L. B., Kristiansen K. A., et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. The Journal of Biological Chemistry. 2007;282(2):1183–1192. doi: 10.1074/jbc.m603761200. [DOI] [PubMed] [Google Scholar]
- 2.Bienert G. P., Schjoerring J. K., Jahn T. P. Membrane transport of hydrogen peroxide. Biochimica et Biophysica Acta—Biomembranes. 2006;1758(8):994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Rhee S. G. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 4.Marchissio M. J., Francés D. E. A., Carnovale C. E., Marinelli R. A. Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicology and Applied Pharmacology. 2012;264(2):246–254. doi: 10.1016/j.taap.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 5.Bertolotti M., Bestetti S., García-Manteiga J. M., et al. Tyrosine Kinase signal modulation: a matter of H2O2 membrane permeability? Antioxidants and Redox Signaling. 2013;19(13):1447–1451. doi: 10.1089/ars.2013.5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller E. W., Dickinson B. C., Chang C. J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(36):15681–15686. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veal E. A., Day A. M., Morgan B. A. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26(1):1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Trachootham D., Lu W., Ogasawara M. A., Valle N. R.-D., Huang P. Redox regulation of cell survival. Antioxidants and Redox Signaling. 2008;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madureira P. A., Waisman D. M. Annexin A2: the importance of being redox sensitive. International Journal of Molecular Sciences. 2013;14(2):3568–3594. doi: 10.3390/ijms14023568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson A. L., Loeb L. A. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutation Research. 2001;477(1-2):7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 11.Rubin R., Farber J. L. Mechanisms of the killing of cultured hepatocytes by hydrogen peroxide. Archives of Biochemistry and Biophysics. 1984;228(2):450–459. doi: 10.1016/0003-9861(84)90010-9. [DOI] [PubMed] [Google Scholar]
- 12.Kumar B., Koul S., Khandrika L., Meacham R. B., Koul H. K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Research. 2008;68(6):1777–1785. doi: 10.1158/0008-5472.can-07-5259. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa K., Takenaga K., Akimoto M., et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320(5876):661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 14.Weinberg F., Chandel N. S. Reactive oxygen species-dependent signaling regulates cancer. Cellular and Molecular Life Sciences. 2009;66(23):3663–3673. doi: 10.1007/s00018-009-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell E. L., Klimova T. A., Eisenbart J., Schumacker P. T., Chandel N. S. Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Molecular and Cellular Biology. 2007;27(16):5737–5745. doi: 10.1128/MCB.02265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H. J., Chae H.-Z., Kim Y.-J., et al. Preferential elevation of Prx I and Trx expression in lung cancer cells following hypoxia and in human lung cancer tissues. Cell Biology and Toxicology. 2003;19(5):285–298. doi: 10.1023/b:cbto.0000004952.07979.3d. [DOI] [PubMed] [Google Scholar]
- 17.Li C., Thompson M. A., Tamayo A. T., et al. Over-expression of Thioredoxin-1 mediates growth, survival, and chemoresistance and is a druggable target in diffuse large B-cell lymphoma. Oncotarget. 2012;3(3):314–326. doi: 10.18632/oncotarget.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traverso N., Ricciarelli R., Nitti M., et al. Role of glutathione in cancer progression and chemoresistance. Oxidative Medicine and Cellular Longevity. 2013;2013:10. doi: 10.1155/2013/972913.972913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ooi A., Wong J.-C., Petillo D., et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20(4):511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 20.Denicola G. M., Karreth F. A., Humpton T. J., et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–110. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmström K. M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature Reviews Molecular Cell Biology. 2014;15(6):411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 22.Storz P. Reactive oxygen species in tumor progression. Frontiers in Bioscience. 2005;10(2):1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 23.Cockman M. E., Masson N., Mole D. R., et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. The Journal of Biological Chemistry. 2000;275(33):25733–25741. doi: 10.1074/jbc.m002740200. [DOI] [PubMed] [Google Scholar]
- 24.Maxwell P. H., Wlesener M. S., Chang G.-W., et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 25.Srinivas V., Zhang L.-P., Zhu X.-H., Caro J. Characterization of an oxygen/redox-dependent degradation domain of hypoxia-inducible factor alpha (HIF-alpha) proteins. Biochemical and Biophysical Research Communications. 1999;260(2):557–561. doi: 10.1006/bbrc.1999.0878. [DOI] [PubMed] [Google Scholar]
- 26.Ebert B. L., Bunn H. F. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Molecular and Cellular Biology. 1998;18(7):4089–4096. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klimova T., Chandel N. S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death and Differentiation. 2008;15(4):660–666. doi: 10.1038/sj.cdd.4402307. [DOI] [PubMed] [Google Scholar]
- 28.Catalano V., Turdo A., Di Franco S., Dieli F., Todaro M., Stassi G. Tumor and its microenvironment: a synergistic interplay. Seminars in Cancer Biology. 2013;23(6):522–532. doi: 10.1016/j.semcancer.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Bell E. L., Klimova T. A., Eisenbart J., et al. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. Journal of Cell Biology. 2007;177(6):1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandel N. S., McClintock D. S., Feliciano C. E., et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. The Journal of Biological Chemistry. 2000;275(33):25130–25138. doi: 10.1074/jbc.m001914200. [DOI] [PubMed] [Google Scholar]
- 31.Gao P., Zhang H., Dinavahi R., et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guzy R. D., Hoyos B., Robin E., et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metabolism. 2005;1(6):401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Bonello S., Zähringer C., BelAiba R. S., et al. Reactive oxygen species activate the HIF-1alpha promoter via a functional NFkappaB site. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(4):755–761. doi: 10.1161/01.ATV.0000258979.92828.bc. [DOI] [PubMed] [Google Scholar]
- 34.Murphy M. P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liou G.-Y., Storz P. Reactive oxygen species in cancer. Free Radical Research. 2010;44(5):479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brand M. D. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45(7-8):466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrader M., Fahimi H. D. Peroxisomes and oxidative stress. Biochimica et Biophysica Acta—Molecular Cell Research. 2006;1763(12):1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Malhotra J. D., Kaufman R. J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants and Redox Signaling. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 39.Paletta-Silva R., Rocco-Machado N., Meyer-Fernandes J. R. NADPH oxidase biology and the regulation of tyrosine kinase receptor signaling and cancer drug cytotoxicity. International Journal of Molecular Sciences. 2013;14(2):3683–3704. doi: 10.3390/ijms14023683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Visser K. E., Eichten A., Coussens L. M. Paradoxical roles of the immune system during cancer development. Nature Reviews Cancer. 2006;6(1):24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 41.Babior B. M. The respiratory burst oxidase. Current Opinion in Hematology. 1995;2(1):55–60. doi: 10.1097/00062752-199502010-00008. [DOI] [PubMed] [Google Scholar]
- 42.Cui S., Reichner J. S., Mateo R. B., Albina J. E. Activated murine macrophages induce apoptosis in tumor cells through nitric oxide-dependent or -independent mechanisms. Cancer Research. 1994;54(9):2462–2467. [PubMed] [Google Scholar]
- 43.Szabó C., Ischiropoulos H., Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nature Reviews Drug Discovery. 2007;6(8):662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 44.Riley P. A. Free radicals in biology: oxidative stress and the effects of ionizing radiation. International Journal of Radiation Biology. 1994;65(1):27–33. doi: 10.1080/09553009414550041. [DOI] [PubMed] [Google Scholar]
- 45.Klaunig J. E., Kamendulis L. M., Hocevar B. A. Oxidative stress and oxidative damage in carcinogenesis. Toxicologic Pathology. 2010;38(1):96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 46.Gorrini C., Harris I. S., Mak T. W. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery. 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 47.Jiang B., Xiao S., Khan M. A., Xue M. Defective antioxidant systems in cervical cancer. Tumor Biology. 2013;34(4):2003–2009. doi: 10.1007/s13277-013-0804-1. [DOI] [PubMed] [Google Scholar]
- 48.Valko M., Rhodes C. J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 49.Yeh C.-C., Hou M.-F., Wu S.-H., et al. A study of glutathione status in the blood and tissues of patients with breast cancer. Cell Biochemistry and Function. 2006;24(6):555–559. doi: 10.1002/cbf.1275. [DOI] [PubMed] [Google Scholar]
- 50.Acharya A., Das I., Chandhok D., Saha T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxidative Medicine and Cellular Longevity. 2010;3(1):23–34. doi: 10.4161/oxim.3.1.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Estrela J. M., Ortega A., Obrador E. Glutathione in cancer biology and therapy. Critical Reviews in Clinical Laboratory Sciences. 2006;43(2):143–181. doi: 10.1080/10408360500523878. [DOI] [PubMed] [Google Scholar]
- 52.O'Brien M. L., Tew K. D. Glutathione and related enzymes in multidrug resistance. European Journal of Cancer Part A. 1996;32(6):967–978. doi: 10.1016/0959-8049(96)00051-2. [DOI] [PubMed] [Google Scholar]
- 53.Calvert P., Yao K.-S., Hamilton T. C., O'Dwyer P. J. Clinical studies of reversal of drug resistance based on glutathione. Chemico-Biological Interactions. 1998;111-112:213–224. doi: 10.1016/s0009-2797(98)00008-8. [DOI] [PubMed] [Google Scholar]
- 54.Du Z.-X., Zhang H.-Y., Meng X., Guan Y., Wang H.-Q. Role of oxidative stress and intracellular glutathione in the sensitivity to apoptosis induced by proteasome inhibitor in thyroid cancer cells. BMC Cancer. 2009;9, article 56 doi: 10.1186/1471-2407-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Köberle B., Tomicic M. T., Usanova S., Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochimica et Biophysica Acta. 2010;1806(2):172–182. doi: 10.1016/j.bbcan.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 56.Ong P.-S., Chan S.-Y., Ho P. C. Differential augmentative effects of buthionine sulfoximine and ascorbic acid in As2O3-induced ovarian cancer cell death: oxidative stress-independent and -dependent cytotoxic potentiation. International Journal of Oncology. 2011;38(6):1731–1739. doi: 10.3892/ijo.2011.986. [DOI] [PubMed] [Google Scholar]
- 57.Harris I. S., Treloar A. E., Inoue S., et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 58.Madureira P. A., Hill R., Miller V. A., Giacomantonio C., Lee P. W. K., Waisman D. M. Annexin A2 is a novel cellular redox regulatory protein involved in tumorigenesis. Oncotarget. 2011;2(12):1075–1093. doi: 10.18632/oncotarget.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raninga P. V., Trapani G. D., Tonissen K. F. Cross talk between two antioxidant systems, thioredoxin and DJ-1: consequences for cancer. Oncoscience. 2014;1(1):95–110. doi: 10.18632/oncoscience.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnér E. S. J., Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. European Journal of Biochemistry. 2000;267(20):6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 61.Saitoh M., Nishitoh H., Fujii M., et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO Journal. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ichijo H., Nishida E., Irie K., et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 63.Tobiume K., Matsuzawa A., Takahashi T., et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. The EMBO Reports. 2001;2(3):222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Powis G., Mustacich D., Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radical Biology & Medicine. 2000;29(3-4):312–322. doi: 10.1016/s0891-5849(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 65.Wang J., Kobayashi M., Sakurada K., Imamura M., Moriuchi T., Hosokawa M. Possible roles of an adult T-cell leukemia (ATL)-derived factor/thioredoxin in the drug resistance of ATL to adriamycin. Blood. 1997;89(7):2480–2487. [PubMed] [Google Scholar]
- 66.Kawahara N., Tanaka T., Yokomizo A., et al. Enhanced coexpression of thioredoxin and high mobility group protein 1 genes in human hepatocellular carcinoma and the possible association with decreased sensitivity to cisplatin. Cancer Research. 1996;56(23):5330–5333. [PubMed] [Google Scholar]
- 67.Yokomizo A., Ono M., Nanri H., et al. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Research. 1995;55(19):4293–4296. [PubMed] [Google Scholar]
- 68.Landis G. N., Tower J. Superoxide dismutase evolution and life span regulation. Mechanisms of Ageing and Development. 2005;126(3):365–379. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 69.Ho J. C.-M., Zheng S., Comhair S. A. A., Farver C., Erzurum S. C. Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Research. 2001;61(23):8578–8585. [PubMed] [Google Scholar]
- 70.Hu H., Luo M.-L., Du X.-L., et al. Up-regulated manganese superoxide dismutase expression increases apoptosis resistance in human esophageal squamous cell carcinomas. Chinese Medical Journal. 2007;120(23):2092–2098. [PubMed] [Google Scholar]
- 71.Izutani R., Asano S., Imano M., Kuroda D., Kato M., Ohyanagi H. Expression of manganese superoxide dismutase in esophageal and gastric cancers. Journal of Gastroenterology. 1998;33(6):816–822. doi: 10.1007/s005350050181. [DOI] [PubMed] [Google Scholar]
- 72.Janssen A. M. L., Bosman C. B., van Duijn W., et al. Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clinical Cancer Research. 2000;6(8):3183–3192. [PubMed] [Google Scholar]
- 73.Salzman R., Kaňková K., Pácal L., Tomandl J., Horáková Z., Kostřica R. Increased activity of superoxide dismutase in advanced stages of head and neck squamous cell carcinoma with locoregional metastases. Neoplasma. 2007;54(4):321–325. [PubMed] [Google Scholar]
- 74.Malafa M., Margenthaler J., Webb B., Neitzel L., Christophersen M. MnSOD expression is increased in metastatic gastric cancer. Journal of Surgical Research. 2000;88(2):130–134. doi: 10.1006/jsre.1999.5773. [DOI] [PubMed] [Google Scholar]
- 75.Miar A., Hevia D., Muñoz-Cimadevilla H., et al. Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radical Biology and Medicine. 2015;85:45–55. doi: 10.1016/j.freeradbiomed.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 76.Nelson K. K., Ranganathan A. C., Mansouri J., et al. Elevated Sod2 activity augments matrix metalloproteinase expression: evidence for the involvement of endogenous hydrogen peroxide in regulating metastasis. Clinical Cancer Research. 2003;9(1):424–432. [PubMed] [Google Scholar]
- 77.Ranganathan A. C., Nelson K. K., Rodriguez A. M., et al. Manganese superoxide dismutase signals matrix metalloproteinase expression via H2O2-dependent ERK1/2 activation. The Journal of Biological Chemistry. 2001;276(17):14264–14270. doi: 10.1074/jbc.M100199200. [DOI] [PubMed] [Google Scholar]
- 78.Chuang T.-C., Liu J.-Y., Lin C.-T., et al. Human manganese superoxide dismutase suppresses HER2/neu-mediated breast cancer malignancy. FEBS Letters. 2007;581(23):4443–4449. doi: 10.1016/j.febslet.2007.08.021. [DOI] [PubMed] [Google Scholar]
- 79.Cullen J. J., Weydert C., Hinkhouse M. M., et al. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Research. 2003;63(6):1297–1303. [PubMed] [Google Scholar]
- 80.Hu Y., Rosen D. G., Zhou Y., et al. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. Journal of Biological Chemistry. 2005;280(47):39485–39492. doi: 10.1074/jbc.m503296200. [DOI] [PubMed] [Google Scholar]
- 81.Oberley L. W., Buettner G. R. Role of superoxide dismutase in cancer: a review. Cancer Research. 1979;39(4):1141–1149. [PubMed] [Google Scholar]
- 82.Soini Y., Vakkala M., Kahlos K., Pääkkö P., Kinnula V. MnSOD expression is less frequent in tumour cells of invasive breast carcinomas than in in situ carcinomas or non-neoplastic breast epithelial cells. The Journal of Pathology. 2001;195(2):156–162. doi: 10.1002/path.946. [DOI] [PubMed] [Google Scholar]
- 83.Behrend L., Mohr A., Dick T., Zwacka R. M. Manganese superoxide dismutase induces p53-dependent senescence in colorectal cancer cells. Molecular and Cellular Biology. 2005;25(17):7758–7769. doi: 10.1128/MCB.25.17.7758-7769.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oberley L. W. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomedicine and Pharmacotherapy. 2005;59(4):143–148. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 85.Ridnour L. A., Oberley T. D., Oberley L. W. Tumor suppressive effects of MnSOD overexpression may involve imbalance in peroxide generation versus peroxide removal. Antioxidants and Redox Signaling. 2004;6(3):501–512. doi: 10.1089/152308604773934260. [DOI] [PubMed] [Google Scholar]
- 86.Zhang Y., Smith B. J., Oberley L. W. Enzymatic activity is necessary for the tumor-suppressive effects of MnSOD. Antioxidants and Redox Signaling. 2006;8(7-8):1283–1293. doi: 10.1089/ars.2006.8.1283. [DOI] [PubMed] [Google Scholar]
- 87.Li N., Oberley T. D., Oberley L. W., Zhong W. Overexpression of manganese superoxide dismutase in DU145 human prostate carcinoma cells has multiple effects of cell phenotype. Prostate. 1998;35(3):221–233. doi: 10.1002/(sici)1097-0045(19980515)35:3lt;221::aid-pros8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 88.McCord J. M., Keele B. B., Jr., Fridovich I. An enzyme-based theory of obligate anaerobiosis: the physiological function of superoxide dismutase. Proceedings of the National Academy of Sciences of the United States of America. 1971;68(5):1024–1027. doi: 10.1073/pnas.68.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Keilin D., Hartree E. F. Catalase, peroxidase and metmyoglobin as catalysts of coupled peroxidatic reactions. Biochemical Journal. 1955;60(2):310–325. doi: 10.1042/bj0600310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kremer M. L. Peroxidatic activity of catalase. Biochimica et Biophysica Acta. 1970;198(2):199–209. doi: 10.1016/0005-2744(70)90052-5. [DOI] [PubMed] [Google Scholar]
- 91.Vetrano A. M., Heck D. E., Mariano T. M., Mishin V., Laskin D. L., Laskin J. D. Characterization of the oxidase activity in mammalian catalase. The Journal of Biological Chemistry. 2005;280(42):35372–35381. doi: 10.1074/jbc.m503991200. [DOI] [PubMed] [Google Scholar]
- 92.Gebicka L., Didik J. Catalytic scavenging of peroxynitrite by catalase. Journal of Inorganic Biochemistry. 2009;103(10):1375–1379. doi: 10.1016/j.jinorgbio.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 93.Heinzelmanna S., Bauer G. Multiple protective functions of catalase against intercellular apoptosis-inducing ROS signaling of human tumor cells. Biological Chemistry. 2010;391(6):675–693. doi: 10.1515/BC.2010.068. [DOI] [PubMed] [Google Scholar]
- 94.Brunelli L., Yermilov V., Beckman J. S. Modulation of catalase peroxidatic and catalatic activity by nitric oxide. Free Radical Biology and Medicine. 2001;30(7):709–714. doi: 10.1016/s0891-5849(00)00512-8. [DOI] [PubMed] [Google Scholar]
- 95.Wink D. A., Mitchell J. B. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radical Biology & Medicine. 1998;25(4-5):434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 96.Brown G. C. Reversible binding and inhibition of catalase by nitric oxide. European Journal of Biochemistry. 1995;232(1):188–191. doi: 10.1111/j.1432-1033.1995.tb20798.x. [DOI] [PubMed] [Google Scholar]
- 97.Castaldo S. A., Da Silva A. P., Matos A., et al. The role of CYBA (p22phox) and catalase genetic polymorphisms and their possible epistatic interaction in cervical cancer. Tumor Biology. 2015;36(2):909–414. doi: 10.1007/s13277-014-2714-2. [DOI] [PubMed] [Google Scholar]
- 98.Fruehauf J. P., Meyskens F. L., Jr. Reactive oxygen species: a breath of life or death? Clinical Cancer Research. 2007;13(3):789–794. doi: 10.1158/1078-0432.ccr-06-2082. [DOI] [PubMed] [Google Scholar]
- 99.Bittinger F., González-García J. L., Klein C. L., Brochhausen C., Offner F., Kirkpatrick C. J. Production of superoxide by human malignant melanoma cells. Melanoma Research. 1998;8(5):381–387. doi: 10.1097/00008390-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 100.López-Lázaro M. Excessive superoxide anion generation plays a key role in carcinogenesis. International Journal of Cancer. 2007;120(6):1378–1380. doi: 10.1002/ijc.22493. [DOI] [PubMed] [Google Scholar]
- 101.Bechtel W., Bauer G. Modulation of intercellular ROS signaling of human tumor cells. Anticancer Research. 2009;29(11):4559–4570. [PubMed] [Google Scholar]
- 102.Bechtel W., Bauer G. Catalase protects tumor cells from apoptosis induction by intercellular ROS signaling. Anticancer Research. 2009;29(11):4541–4557. [PubMed] [Google Scholar]
- 103.Bauer G. Reactive oxygen and nitrogen species: efficient, selective, and interactive signals during intercellular induction of apoptosis. Anticancer Research. 2000;20(6):4115–4139. [PubMed] [Google Scholar]
- 104.Bauer G. Tumor cell-protective catalase as a novel target for rational therapeutic approaches based on specific intercellular ROS signaling. Anticancer Research. 2012;32(7):2599–2624. [PubMed] [Google Scholar]
- 105.Böhm B., Heinzelmann S., Motz M., Bauer G. Extracellular localization of catalase is associated with the transformed state of malignant cells. Biological Chemistry. 2015 doi: 10.1515/hsz-2014-0234. [DOI] [PubMed] [Google Scholar]
- 106.McDonald C., Muhlbauer J., Perlmutter G., Taparra K., Phelan S. A. Peroxiredoxin proteins protect MCF-7 breast cancer cells from doxorubicin-induced toxicity. International Journal of Oncology. 2014;45(1):219–226. doi: 10.3892/ijo.2014.2398. [DOI] [PubMed] [Google Scholar]
- 107.Whitaker H. C., Patel D., Howat W. J., et al. Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. British Journal of Cancer. 2013;109(4):983–993. doi: 10.1038/bjc.2013.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park J., Lee S., Lee S., Kang S. W. 2-cys peroxiredoxins: emerging hubs determining redox dependency of mammalian signaling networks. International Journal of Cell Biology. 2014;2014:10. doi: 10.1155/2014/715867.715867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ishii T., Warabi E., Yanagawa T. Novel roles of peroxiredoxins in inflammation, cancer and innate immunity. Journal of Clinical Biochemistry and Nutrition. 2012;50(2):91–105. doi: 10.3164/jcbn.11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li L., Zhang Y.-G., Chen C.-L. Anti-apoptotic role of peroxiredoxin III in cervical cancer cells. FEBS Open Bio. 2013;3:51–54. doi: 10.1016/j.fob.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park J. H., Kim Y. S., Lee H. L., et al. Expression of peroxiredoxin and thioredoxin in human lung cancer and paired normal lung. Respirology. 2006;11(3):269–275. doi: 10.1111/j.1440-1843.2006.00849.x. [DOI] [PubMed] [Google Scholar]
- 112.Kinnula V. L., Lehtonen S., Sormunen R., et al. Overexpression of peroxiredoxins I, II, III, V, and VI in malignant mesothelioma. Journal of Pathology. 2002;196(3):316–323. doi: 10.1002/path.1042. [DOI] [PubMed] [Google Scholar]
- 113.Poschmann G., Grzendowski M., Stefanski A., Bruns E., Meyer H. E., Stühler K. Redox proteomics reveal stress responsive proteins linking peroxiredoxin-1 status in glioma to chemosensitivity and oxidative stress. Biochimica et Biophysica Acta—Proteins and Proteomics. 2015;1854(6):624–631. doi: 10.1016/j.bbapap.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 114.Wang T., Diaz A. J. G., Yen Y. The role of peroxiredoxin II in chemoresistance of breast cancer cells. Breast Cancer: Targets and Therapy. 2014;6:73–80. doi: 10.2147/bctt.s61281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fishel M. L., Kelley M. R. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Molecular Aspects of Medicine. 2007;28(3-4):375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 116.Fishel M. L., Wu X., Devlin C. M., et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. The Journal of Biological Chemistry. 2015;290(5):3057–3068. doi: 10.1074/jbc.m114.621995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang Y., Wang J., Xiang D., Wang D., Xin X. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer and indentification of the therapeutic potential of APE1/Ref-1 inhibitor. International Journal of Oncology. 2009;35(5):1069–1079. [PubMed] [Google Scholar]
- 118.Kelleher Z. T., Matsumoto A., Stamler J. S., Marshall H. E. NOS2 regulation of NF-κB by S-nitrosylation of p65. The Journal of Biological Chemistry. 2007;282(42):30667–30672. doi: 10.1074/jbc.m705929200. [DOI] [PubMed] [Google Scholar]
- 119.Wei S. J., Botero A., Hirota K., et al. Thioredoxin nuclear translocation and interaction with redox factor-1 activates the activator protein-1 transcription factor in response to ionizing radiation. Cancer Research. 2000;60(23):6688–6695. [PubMed] [Google Scholar]
- 120.Kaur G., Cholia R. P., Mantha A. K., Kumar R. DNA repair and redox activities and inhibitors of apurinic/apyrimidinic endonuclease 1/redox effector factor 1 (APE1/Ref-1): a comparative analysis and their scope and limitations toward anticancer drug development. Journal of Medicinal Chemistry. 2014;57(24):10241–10256. doi: 10.1021/jm500865u. [DOI] [PubMed] [Google Scholar]
- 121.Yang Z., Yang S., Misner B. J., Liu-Smith F., Meyskens F. L. The role of APE/Ref-1 signaling pathway in hepatocellular carcinoma progression. International Journal of Oncology. 2014;45(5):1820–1828. doi: 10.3892/ijo.2014.2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bobola M. S., Blank A., Berger M. S., Stevens B. A., Silber J. R. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clinical Cancer Research. 2001;7(11):3510–3518. [PubMed] [Google Scholar]
- 123.Bobola M. S., Jankowski P. P., Gross M. E., et al. Apurinic/apyrimidinic endonuclease is inversely associated with response to radiotherapy in pediatric ependymoma. International Journal of Cancer. 2011;129(10):2370–2379. doi: 10.1002/ijc.25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xiong G. S., Sun H. L., Wu S. M., Mo J. Z. Small interfering RNA against the apurinic or apyrimidinic endonuclease enhances the sensitivity of human pancreatic cancer cells to gemcitabine in vitro. Journal of Digestive Diseases. 2010;11(4):224–230. doi: 10.1111/j.1751-2980.2010.00442.x. [DOI] [PubMed] [Google Scholar]
- 125.Minisini A. M., Loreto C., Mansutti M., et al. Topoisomerase IIα and APE/ref-1 are associated with pathologic response to primary anthracycline-based chemotherapy for breast cancer. Cancer Letters. 2005;224(1):133–139. doi: 10.1016/j.canlet.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 126.Yang S., Meyskens F. L., Jr. Apurinic/apyrimidinic endonuclease/redox effector factor-1(APE/Ref-1): a unique target for the prevention and treatment of human melanoma. Antioxidants and Redox Signaling. 2009;11(3):639–650. doi: 10.1089/ars.2008.2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gañán-Gómez I., Wei Y., Yang H., Boyano-Adánez M. C., García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radical Biology and Medicine. 2013;65:750–764. doi: 10.1016/j.freeradbiomed.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 128.Sakurai A., Nishimoto M., Himeno S., et al. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. Journal of Cellular Physiology. 2005;203(3):529–537. doi: 10.1002/jcp.20246. [DOI] [PubMed] [Google Scholar]
- 129.Ramprasath T., Selvam G. S. Potential impact of genetic variants in Nrf2 regulated antioxidant genes and risk prediction of diabetes and associated cardiac complications. Current Medicinal Chemistry. 2013;20(37):4680–4693. doi: 10.2174/09298673113209990154. [DOI] [PubMed] [Google Scholar]
- 130.Kansanen E., Kuosmanen S. M., Leinonen H., Levonenn A.-L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biology. 2013;1(1):45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim Y. R., Oh J. E., Kim M. S., et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. Journal of Pathology. 2010;220(4):446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- 132.Shibata T., Ohta T., Tong K. I., et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(36):13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hayes J. D., McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends in Biochemical Sciences. 2009;34(4):176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 134.Sunters A., Madureira P. A., Pomeranz K. M., et al. Paclitaxel-induced nuclear translocation of FOXO3a in breast cancer cells is mediated by c-Jun NH2-terminal kinase and Akt. Cancer Research. 2006;66(1):212–220. doi: 10.1158/0008-5472.can-05-1997. [DOI] [PubMed] [Google Scholar]
- 135.Vurusaner B., Poli G., Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radical Biology and Medicine. 2012;52(1):7–18. doi: 10.1016/j.freeradbiomed.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 136.Ciechomska I., Pyrzynska B., Kazmierczak P., Kaminska B. Inhibition of Akt kinase signalling and activation of Forkhead are indispensable for upregulation of FasL expression in apoptosis of glioma cells. Oncogene. 2003;22(48):7617–7627. doi: 10.1038/sj.onc.1207137. [DOI] [PubMed] [Google Scholar]
- 137.Modur V., Nagarajan R., Evers B. M., Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. The Journal of Biological Chemistry. 2002;277(49):47928–47937. doi: 10.1074/jbc.m207509200. [DOI] [PubMed] [Google Scholar]
- 138.Sykes S. M., Lane S. W., Bullinger L., et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146(5):697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Burgering B. M. T., Medema R. H. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. Journal of Leukocyte Biology. 2003;73(6):689–701. doi: 10.1189/jlb.1202629. [DOI] [PubMed] [Google Scholar]
- 140.Linardic C. M. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Letters. 2008;270(1):10–18. doi: 10.1016/j.canlet.2008.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Olanich M. E., Barr F. G. A call to ARMS: targeting the PAX3-FOXO1 gene in alveolar rhabdomyosarcoma. Expert Opinion on Therapeutic Targets. 2013;17(5):607–623. doi: 10.1517/14728222.2013.772136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.del Peso L., González V. M., Hernández R., Barr F. G., Núñez G. Regulation of the forkhead transcription factor FKHR, but not the PAX3-FKHR fusion protein, by the serine/threonine kinase Akt. Oncogene. 1999;18(51):7328–7333. doi: 10.1038/sj.onc.1203159. [DOI] [PubMed] [Google Scholar]
- 143.Block K. I., Koch A. C., Mead M. N., Tothy P. K., Newman R. A., Gyllenhaal C. Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of the evidence from randomized controlled trials. International Journal of Cancer. 2008;123(6):1227–1239. doi: 10.1002/ijc.23754. [DOI] [PubMed] [Google Scholar]
- 144.Goodman M., Bostick R. M., Kucuk O., Jones D. P. Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radical Biology and Medicine. 2011;51(5):1068–1084. doi: 10.1016/j.freeradbiomed.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 145.Sayin V. I., Ibrahim M. X., Larsson E., Nilsson J. A., Lindahl P., Bergo M. O. Antioxidants accelerate lung cancer progression in mice. Science Translational Medicine. 2014;6(221) doi: 10.1126/scitranslmed.3007653.221ra15 [DOI] [PubMed] [Google Scholar]
- 146.McConnell M. J., Herst P. M. Ascorbate combination therapy: new tool in the anticancer toolbox? Science Translational Medicine. 2014;6(222) doi: 10.1126/scitranslmed.3008488.222fs6 [DOI] [PubMed] [Google Scholar]
- 147.Ma Y., Chapman J., Levine M., Polireddy K., Drisko J., Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Science Translational Medicine. 2014;6(222) doi: 10.1126/scitranslmed.3007154.222ra18 [DOI] [PubMed] [Google Scholar]
- 148.Luo C., Liu Q., Zhang P., Li M., Chen Z., Liu Y. Prognostic significance of annexin II expression in non-small cell lung cancer. Clinical and Translational Oncology. 2013;15(11):938–946. doi: 10.1007/s12094-013-1028-y. [DOI] [PubMed] [Google Scholar]
- 149.Xu X. H., Pan W., Kang L., Feng H., Song Y. Association of annexin A2 with cancer development (review) Oncology Reports. 2015;33(5):2121–2128. doi: 10.3892/or.2015.3837. [DOI] [PubMed] [Google Scholar]
- 150.Zhang Z. D., Li Y., Fan L. Q., Zhao Q., Tan B. B., Zhao X. F. Annexin A2 is implicated in multi-drug-resistance in gastric cancer through p38MAPK and AKT pathway. Neoplasma. 2014;61(6):627–637. doi: 10.4149/neo_2014_078. [DOI] [PubMed] [Google Scholar]
- 151.Madureira P. A., Surette A. P., Phipps K. D., Taboski M. A. S., Miller V. A., Waisman D. M. The role of the annexin A2 heterotetramer in vascular fibrinolysis. Blood. 2011;118(18):4789–4797. doi: 10.1182/blood-2011-06-334672. [DOI] [PubMed] [Google Scholar]
- 152.Waters K. M., Stenoien D. L., Sowa M. B., et al. Annexin A2 modulates radiation-sensitive transcriptional programming and cell fate. Radiation Research. 2013;179(1):53–61. doi: 10.1667/rr3056.1. [DOI] [PubMed] [Google Scholar]
- 153.Madureira P. A., Hill R., Lee P. W. K., Waisman D. M. Genotoxic agents promote the nuclear accumulation of annexin A2: role of annexin A2 in mitigating DNA damage. PLoS ONE. 2012;7(11) doi: 10.1371/journal.pone.0050591.e50591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Canet-Avilés R. M., Wilson M. A., Miller D. W., et al. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(24):9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kinumi T., Kimata J., Taira T., Ariga H., Niki E. Cysteine-106 of DJ-1 is the most sensitive cysteine residue to hydrogen peroxide-mediated oxidation in vivo in human umbilical vein endothelial cells. Biochemical and Biophysical Research Communications. 2004;317(3):722–728. doi: 10.1016/j.bbrc.2004.03.110. [DOI] [PubMed] [Google Scholar]
- 156.Im J.-Y., Lee K.-W., Junn E., Mouradian M. M. DJ-1 protects against oxidative damage by regulating the thioredoxin/ASK1 complex. Neuroscience Research. 2010;67(3):203–208. doi: 10.1016/j.neures.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim Y.-C., Kitaura H., Taira T., Iguchi-Ariga S. M. M., Ariga H. Oxidation of DJ-1-dependent cell transformation through direct binding of DJ-1 to PTEN. International Journal of Oncology. 2009;35(6):1331–1341. doi: 10.3892/ijo-00000451. [DOI] [PubMed] [Google Scholar]
- 158.Shendelman S., Jonason A., Martinat C., Leete T., Abeliovich A. DJ-1 is a redox-dependent molecular chaperone that inhibits α-synuclein aggregate formation. PLoS Biology. 2004;2(11) doi: 10.1371/journal.pbio.0020362.e362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou W., Freed C. R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T α-synuclein toxicity. The Journal of Biological Chemistry. 2005;280(52):43150–43158. doi: 10.1074/jbc.m507124200. [DOI] [PubMed] [Google Scholar]
- 160.Waak J., Weber S. S., Görner K., et al. Oxidizable residues mediating protein stability and cytoprotective interaction of DJ-1 with apoptosis signal-regulating kinase 1. The Journal of Biological Chemistry. 2009;284(21):14245–14257. doi: 10.1074/jbc.m806902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Clements C. M., McNally R. S., Conti B. J., Mak T. W., Ting J. P.-Y. DJ-1, a cancer- and Parkinson's disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(41):15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yu H.-H., Xu Q., Chen H.-P., et al. Stable overexpression of DJ-1 protects H9c2 cells against oxidative stress under a hypoxia condition. Cell Biochemistry and Function. 2013;31(8):643–651. doi: 10.1002/cbf.2949. [DOI] [PubMed] [Google Scholar]
- 163.Kumar P., Nandi S., Tan T. Z., et al. Highly sensitive and specific novel biomarkers for the diagnosis of transitional bladder carcinoma. Oncotarget. 2015;6(15):13539–13549. doi: 10.18632/oncotarget.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ismail I. A., Abdel Shakor A. B., Hong S. H. DJ-1 protects breast cancer cells against 2′-benzoyloxycinnamaldehyde-induced oxidative stress independent of Nrf2. Journal of Cellular Physiology. 2015;230(9):2262–2269. doi: 10.1002/jcp.24957. [DOI] [PubMed] [Google Scholar]
- 165.Zhu H., Liao S.-D., Shi J.-J., et al. DJ-1 mediates the resistance of cancer cells to dihydroartemisinin through reactive oxygen species removal. Free Radical Biology and Medicine. 2014;71:121–132. doi: 10.1016/j.freeradbiomed.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 166.Dall'Acqua S., Linardi M. A., Bortolozzi R., et al. Natural daucane esters induces apoptosis in leukaemic cells through ROS production. Phytochemistry. 2014;108:147–156. doi: 10.1016/j.phytochem.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 167.Klaunig J. E., Kamendulis L. M. The role of oxidative stress in carcinogenesis. Annual Review of Pharmacology and Toxicology. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 168.Fernandes A. P., Gandin V. Selenium compounds as therapeutic agents in cancer. Biochimica et Biophysica Acta—General Subjects. 1850;1850(8):1642–1660. doi: 10.1016/j.bbagen.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 169.An J. M., Kim S. S., Rhie J. H., Shin D. M., Seo S. R., Seo J. T. Carmustine induces ERK- and JNK-dependent cell death of neuronally-differentiated PC12 cells via generation of reactive oxygen species. Toxicology in Vitro. 2011;25(7):1359–1365. doi: 10.1016/j.tiv.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 170.Roy S. S., Chakraborty P., Bhattacharya S. Intervention in cyclophosphamide induced oxidative stress and DNA damage by a flavonyl-thiazolidinedione based organoselenocyanate and evaluation of its efficacy during adjuvant therapy in tumor bearing mice. European Journal of Medicinal Chemistry. 2014;73:195–209. doi: 10.1016/j.ejmech.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 171.Billen L. P., Shamas-Din A., Andrews D. W. Bid: a Bax-like BH3 protein. Oncogene. 2008;27(1) supplement 1:S93–S104. doi: 10.1038/onc.2009.47. [DOI] [PubMed] [Google Scholar]
- 172.Litchfield D. W. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochemical Journal. 2003;369(1):1–15. doi: 10.1042/bj20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Schneider C. C., Hessenauer A., Götz C., Montenarh M. DMAT, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncology Reports. 2009;21(6):1593–1597. doi: 10.3892/or_00000392. [DOI] [PubMed] [Google Scholar]
- 174.Jeon S. M., Lee S.-J., Kwon T. K., Kim K.-J., Bae Y.-S. NADPH oxidase is involved in protein kinase CKII down-regulation-mediated senescence through elevation of the level of reactive oxygen species in human colon cancer cells. FEBS Letters. 2010;584(14):3137–3142. doi: 10.1016/j.febslet.2010.05.054. [DOI] [PubMed] [Google Scholar]
- 175.Lee Y.-H., Kang B. S., Bae Y.-S. Premature senescence in human breast cancer and colon cancer cells by tamoxifen-mediated reactive oxygen species generation. Life Sciences. 2014;97(2):116–122. doi: 10.1016/j.lfs.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 176.Lee R. L., Westendorf J., Gold M. R. Differential role of reactive oxygen species in the activation of mitogen-activated protein kinases and akt by key receptors on b-lymphocytes: CD40, the b cell antigen receptor, and CXCR4. Journal of Cell Communication and Signaling. 2007;1(1):33–43. doi: 10.1007/s12079-007-0006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alexandre J., Batteux F., Nicco C., et al. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. International Journal of Cancer. 2006;119(1):41–48. doi: 10.1002/ijc.21685. [DOI] [PubMed] [Google Scholar]
- 178.Alexandre J., Hu Y., Lu W., Pelicano H., Huang P. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Research. 2007;67(8):3512–3517. doi: 10.1158/0008-5472.can-06-3914. [DOI] [PubMed] [Google Scholar]
- 179.Jin C., Wu S., Lu X., et al. Conditioned medium from actinomycin D-treated apoptotic cells induces mitochondria-dependent apoptosis in bystander cells. Toxicology Letters. 2012;211(1):45–53. doi: 10.1016/j.toxlet.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 180.Yan C., Kong D., Ge D., et al. Mitomycin C induces apoptosis in rheumatoid arthritis fibroblast-like synoviocytes via a mitochondrial-mediated pathway. Cellular Physiology and Biochemistry. 2015;35(3):1125–1136. doi: 10.1159/000373938. [DOI] [PubMed] [Google Scholar]
- 181.Wang Y., Li X., Wang X., et al. Ginsenoside Rd attenuates myocardial ischemia/reperfusion injury via Akt/GSK-3β signaling and inhibition of the mitochondria-dependent apoptotic pathway. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0070956.e70956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Korge P., Calmettes G., Weiss J. N. Increased reactive oxygen species production during reductive stress: the roles of mitochondrial glutathione and thioredoxin reductases. Biochimica et Biophysica Acta—Bioenergetics. 2015;1847(6-7):514–525. doi: 10.1016/j.bbabio.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Probin V., Wang Y., Zhou D. Busulfan-induced senescence is dependent on ROS production upstream of the MAPK pathway. Free Radical Biology and Medicine. 2007;42(12):1858–1865. doi: 10.1016/j.freeradbiomed.2007.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Townsend D. M., Tew K. D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22(47):7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sausville E. A., Stein R. W., Peisach J., Horwitz S. B. Properties and products of the degradation of DNA by bleomycin and iron(II) Biochemistry. 1978;17(14):2746–2754. doi: 10.1021/bi00607a008. [DOI] [PubMed] [Google Scholar]
- 186.Ekimoto H., Kuramochi H., Takahashi K., Matsuda A., Umezawa H. Kinetics of the reaction of bleomycin-Fe(II)-O2 complex with DNA. The Journal of Antibiotics. 1980;33(4):426–434. doi: 10.7164/antibiotics.33.426. [DOI] [PubMed] [Google Scholar]
- 187.Tobwala S., Fan W., Stoeger T., Ercal N. N-acetylcysteine amide, a thiol antioxidant, prevents bleomycin-induced toxicity in human alveolar basal epithelial cells (A549) Free Radical Research. 2013;47(9):740–749. doi: 10.3109/10715762.2013.819974. [DOI] [PubMed] [Google Scholar]
- 188.Bragado P., Armesilla A., Silva A., Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis. 2007;12(9):1733–1742. doi: 10.1007/s10495-007-0082-8. [DOI] [PubMed] [Google Scholar]
- 189.Chung Y. M., Kim J. S., Yoo Y. D. A novel protein, Romo1, induces ROS production in the mitochondria. Biochemical and Biophysical Research Communications. 2006;347(3):649–655. doi: 10.1016/j.bbrc.2006.06.140. [DOI] [PubMed] [Google Scholar]
- 190.Hagenbuchner J., Kuznetsov A., Hermann M., Hausott B., Obexer P., Ausserlechner M. J. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. Journal of Cell Science. 2012;125(part 5):1191–1203. doi: 10.1242/jcs.092098. [DOI] [PubMed] [Google Scholar]
- 191.Karpinich N. O., Tafani M., Schneider T., Russo M. A., Farber J. L. The course of etoposide-induced apoptosis in Jurkat cells lacking p53 and Bax. Journal of Cellular Physiology. 2006;208(1):55–63. doi: 10.1002/jcp.20638. [DOI] [PubMed] [Google Scholar]
- 192.Wang J.-X., Kaieda S., Ameri S., et al. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(28):10281–10286. doi: 10.1073/pnas.1404182111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacological Reviews. 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 194.Olson R. D., Mushlin P. S. Doxorubicin cardiotoxicity: analysis of prevailing hypotheses. The FASEB Journal. 1990;4(13):3076–3086. [PubMed] [Google Scholar]
- 195.Tokarska-Schlattner M., Zaugg M., Zuppinger C., Wallimann T., Schlattner U. New insights into doxorubicin-induced cardiotoxicity: the critical role of cellular energetics. Journal of Molecular and Cellular Cardiology. 2006;41(3):389–405. doi: 10.1016/j.yjmcc.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 196.Verma M., Shulga N., Pastorino J. G. Sirtuin-4 modulates sensitivity to induction of the mitochondrial permeability transition pore. Biochimica et Biophysica Acta. 2013;1827(1):38–49. doi: 10.1016/j.bbabio.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 197.Chen J., Solomides C., Parekh H., Simpkins F., Simpkins H. Cisplatin resistance in human cervical, ovarian and lung cancer cells. Cancer Chemotherapy and Pharmacology. 2015;75(6):1217–1227. doi: 10.1007/s00280-015-2739-2. [DOI] [PubMed] [Google Scholar]
- 198.Arora S., Bhardwaj A., Singh S., et al. An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor κB- and hypoxia-inducible factor 1α-mediated up-regulation of CXCR4. The Journal of Biological Chemistry. 2013;288(29):21197–21207. doi: 10.1074/jbc.m113.484576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Donadelli M., Costanzo C., Beghelli S., et al. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochimica et Biophysica Acta. 2007;1773(7):1095–1106. doi: 10.1016/j.bbamcr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 200.Chetram M. A., Hinton C. V. ROS-mediated regulation of CXCR4 in cancer. Frontiers in Biology. 2013;8(3):273–278. doi: 10.1007/s11515-012-1204-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gajek A., Denel M., Bukowska B., Rogalska A., Marczak A. Pro-apoptotic activity of new analog of anthracyclines—WP 631 in advanced ovarian cancer cell line. Toxicology in Vitro. 2014;28(2):273–281. doi: 10.1016/j.tiv.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 202.Koedrith P., Seo Y. R. Enhancement of the efficacy of mitomycin C-mediated apoptosis in human colon cancer cells with RNAi-based thioredoxin reductase 1 deficiency. Experimental and Therapeutic Medicine. 2011;2(5):873–878. doi: 10.3892/etm.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]