

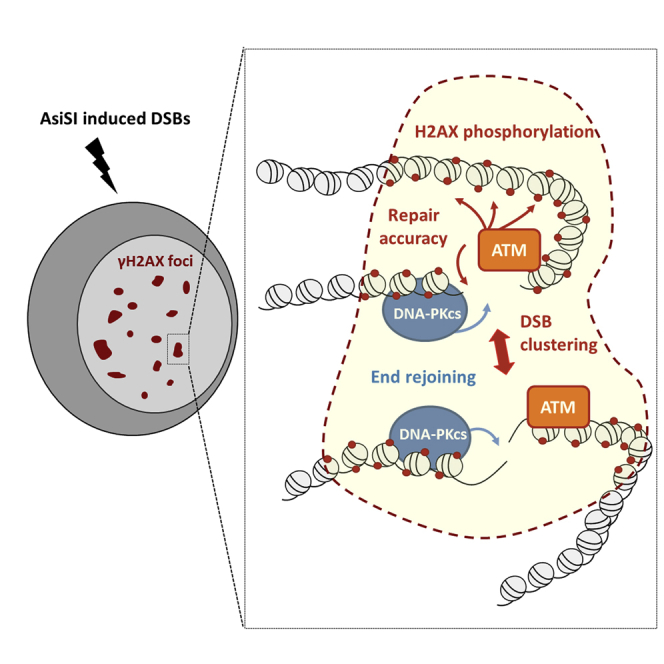
Summary
DNA double-strand breaks (DSBs) elicit the so-called DNA damage response (DDR), largely relying on ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PKcs), two members of the PI3K-like kinase family, whose respective functions during the sequential steps of the DDR remains controversial. Using the DIvA system (DSB inducible via AsiSI) combined with high-resolution mapping and advanced microscopy, we uncovered that both ATM and DNA-PKcs spread in cis on a confined region surrounding DSBs, independently of the pathway used for repair. However, once recruited, these kinases exhibit non-overlapping functions on end joining and γH2AX domain establishment. More specifically, we found that ATM is required to ensure the association of multiple DSBs within “repair foci.” Our results suggest that ATM acts not only on chromatin marks but also on higher-order chromatin organization to ensure repair accuracy and survival.
Keywords: DSB repair, chromatin, PI 3-kinases, γH2AX, ChIP-chip
Graphical Abstract
Highlights
-
•
Both ATM and DNA-PKcs are recruited at AsiSI-induced DSBs
-
•
Once recruited, both kinases exhibit complementary and non-redundant functions
-
•
DNA-PKcs activity is required for end joining at all AsiSI-induced DSBs
-
•
ATM activity promotes repair accuracy, H2AX phosphorylation, and DSB clustering
By inducing multiple annotated DNA double-strand breaks in the human genome, Caron et al. show that two DNA damage response kinases, ATM and DNA-PKcs, are co-recruited at DSBs but exhibit non-redundant functions in promoting end joining, repair accuracy, H2AX phosphorylation, and DSB clustering.
Introduction
Among the various DNA damage types, DNA double-strand breaks (DSBs) are the most deleterious since they can lead to various mutations and chromosomal rearrangements linked to tumor initiation and progression. DSBs can both arise during development as part of scheduled processes, such as V(D)J and immunoglobulin class-switch recombination, and be generated by environmental stresses, such as pollutants and irradiation. DSBs are mainly repaired by two distinct pathways: homologous recombination (HR), involving extensive resection and utilizing an intact copy of the damaged locus, and non-homologous end joining (NHEJ), in which the two broken ends are able to be joined with no or minimal homology (reviewed in Deriano and Roth, 2013, Jasin and Rothstein, 2013). Defects in either repair pathway results in genome instability and can be lethal at very early developmental stages.
DSB detection rapidly elicits the so-called DNA damage response (DDR), which largely relies on the activity of the phosphatidylinositol 3-kinase (PI3K)-related kinases DNA-PKcs, ATM, and ATR (reviewed in Sirbu and Cortez, 2013). All three of these kinases have been found to be mutated in human disorders associated with genome instability: severe combined immunodeficiency (DNA-PKcs), ataxia telangiectasia (ATM), and Seckel syndrome (ATR). While DNA-PKcs and ATM have a function restricted to the DSB response, ATR is activated following a wider range of damage types, especially those occurring during DNA replication. These kinases are rapidly recruited and activated at DSBs through direct interactions with the Ku heterodimer (DNA-PKcs), the MRN complex (ATM), and ATRIP (ATR) (Falck et al., 2005). Once recruited, they have been proposed to participate in repair on three different levels (Sirbu and Cortez, 2013).
First of all, both ATM and DNA-PKcs play a direct role in repair at the break site in a manner that largely depends on their kinase activity. DNA-PKcs is a core component of the NHEJ machinery that allows both synapsis of DNA ends and the stable recruitment of the XRCC4/DNA Ligase 4 complex, required for end joining (Calsou et al., 2003). Consequently, its impairment leads to ends rejoining defects as measured by pulse field gel electrophoresis (PGFE) (Beamish et al., 2000). In contrast, ATM is dispensable for repair of most DSBs arising after irradiation, but required for efficient repair of DSBs induced in heterochromatin (Beucher et al., 2009, Goodarzi et al., 2008) or with blocked DNA ends (Álvarez-Quilón et al., 2014).
Second, upon activation, PI3K-like kinases elicit checkpoint activation by phosphorylating a large number of substrates that either remain at the break site and thus play a direct role in signal amplification or diffuse from the break and mediate signal transduction that eventually leads to cell cycle arrest (Sirbu and Cortez, 2013).
Finally, these DSB-activated kinases trigger a profound remodeling of the chromatin structure at the vicinity of the break. One of their main substrates is the H2AX histone variant, incorporated in roughly one-tenth of nucleosomes (although its distribution is slightly biased toward gene rich regions [Iacovoni et al., 2010, Seo et al., 2012]). Phosphorylation of H2AX on serine S139, referred to as γ-H2AX (Rogakou et al., 1998), is one of the earliest events that takes place at DSBs (for review, see Scully and Xie, 2013). Remarkably, γ-H2AX spreads on neighboring chromatin to form megabase-wide chromatin domains (Iacovoni et al., 2010, Savic et al., 2009), whose function is still unclear, although it has been suggested to act as a platform to recruit additional repair, signaling, and chromatin modifying enzymes. However, while H2AX null mice exhibit a range of phenotypes consistent with DSB coping defects (such as tumor susceptibility, sterility, and immunodeficiency; for review, see Scully and Xie [2013]), they are viable, indicating that DSB repair can occur without H2AX phosphorylation. At a molecular level, γ-H2AX serves as an anchor for MDC1, believed to mediate all γ-H2AX-dependent functions. Although not required for their initial recruitment, the γ-H2AX/MDC1 module is necessary for the accumulation and retention of DNA repair factors at DSBs (for review, see Scully and Xie, 2013), leading to the hypothesis that γ-H2AX could be involved in forming “repair foci” that concentrate repair factors and thus potentiate repair. Whether these repair foci contain one or more DSBs is still a matter of debate (Aten et al., 2004, Jakob et al., 2009b, Krawczyk et al., 2012, Soutoglou et al., 2007). Finally, several studies have pointed out a role for γ-H2AX in HR repair (Sonoda et al., 2007, Xie et al., 2004) and in homology search (Renkawitz et al., 2013).
Tremendous efforts have been made to identify both specific and/or overlapping functions of these DSB-activated PI3K-like kinases using genetics as well as specific chemical inhibition (e.g., see Callén et al., 2009, Riballo et al., 2004). However, their relative contributions are still unclear given that they share many common substrates, including γ-H2AX, and operate at different levels of the DDR, from break detection to signal transduction. These studies and their interpretation have been further complicated by the use of different DSB-induction methods, including high or low doses, global or localized radiation, genotoxic drugs (some of which lead to the generation of DSBs only during DNA replication), and controlled restriction enzymes inducing one (I-SceI; Zn-FokI) or multiple (I-PpoI, AsiSI) clean DSBs at different positions throughout the genome.
We recently reported the genome-wide distribution of γ-H2AX, XRCC4 (an NHEJ component), and RAD51 (involved in HR) around more than 100 DSBs throughout the human genome using the DIvA system (DSB inducible via AsiSI) expressing an inducible AsiSI restriction enzyme. These studies revealed that both repair and signaling depend on the localization of the DSB on the genome and the underlying chromatin context (Aymard et al., 2014, Iacovoni et al., 2010), with HR being promoted at transcriptionally active loci on the genome.
Here, we used the abovementioned DIvA system in order to clarify the function of ATM and DNA-PKcs in the repair and signaling of clean DSBs induced at different genomic locations and engaged in different repair pathways. Our study revealed that both ATM and DNA-PKcs are essential for survival and recruited at all DSBs, independently of their propensity to be repaired by HR or not. However, we clearly show that, once recruited, they exhibit non-redundant functions regarding signaling and repair. At the repair level, DNA-PKcs is absolutely required for end joining at all investigated DSBs, while ATM is dispensable although promoting repair fidelity. By contrast, ATM is required for γH2AX establishment at all breaks, while DNA-PKcs is dispensable for DSB-induced chromatin signaling. We also found that multiple AsiSI-induced DSBs are able to associate within repair foci, in a manner that strictly depends on ATM, but not DNA-PKcs, activity. Our study sheds light on the respective roles of ATM and DNA-PKcs regarding end joining, γH2AX domain establishment, and repair foci formation.
Results
ATM and DNA-PKcs Are Necessary for Cell Survival after AsiSI-Induced DSBs
In order to evaluate the sensitivity of human cells upon induction of a few hundred clean DSBs dispersed throughout the genome, we developed a clonogenic assay using AID-DIvA cells, an improved version of our DIvA model (Aymard et al., 2014). This cell line stably expresses a construct carrying AsiSI-ER fused to an auxin inducible degron (AID), which triggers the rapid degradation of the restriction enzyme upon auxin addition and thus enables repair of AsiSI-induced DSBs. As shown in Figure 1A, 4OHT treatment reduced clonogenic survival to about 20%, whereas auxin addition rescued cell survival to about 60%. Notably, both ATM and DNA-PKcs inhibition, using the highly potent and selective inhibitors KU55933 (ATMi) or Nu7441 (DNAPKi), respectively, led to a significant decrease in clonogenic survival (Figure 1A), whereas these two inhibitors only mildly affected survival in the absence of DSB induction (Figure S1A). Combining both inhibitors mimicked the effect observed with the DNA-PKcs inhibitor alone (Figure S1B). This indicates that both kinases are essential for cell survival after induction of AsiSI-induced DSBs.
Figure 1.

Function of ATM and DNA-PKcs in Repair Kinetics and Repair Accuracy at AsiSI-Induced DSBs
(A) Clonogenic assays in AID-DIvA cells after 4OHT treatment (4 hr), followed by auxin treatment (4 hr) in the presence of ATM inhibitor (KU55933), in the presence of DNA-PKcs inhibitor (Nu7441), or without inhibitor as indicated. Colonies were counted 10 days after 4OHT/auxin treatments. Average and SEM of biological replicates are shown (n = 3).
(B) Cleavage assay (Chailleux et al., 2014) in AID-DIvA cells treated with 4OHT (4 hr) followed by auxin (4 hr), in the absence or presence of ATM or DNA-PKcs inhibitors, as indicated. Immunoprecipitated DNA was analyzed close to four DSBs, either RAD51 unbound (indicated in blue, upper panels) or RAD51 bound (indicated in red, lower panel). The percentage of sites that remain broken for each DSB after the indicated time of auxin treatment are presented. Average and SEM (n = 3, technical replicates) of a representative experiment are shown (out of three independent experiments).
(C) Cleavage assay in AID-DIvA cells untreated (−4OHT) and treated with 4OHT (+4OHT) followed by auxin addition in the absence (+OHT+auxin) or presence of the ATM inhibitor (+4OHT+auxin+ATMi) and an additional round of 4OHT treatment (+4OHT+auxin+4OHT and +4OHT+auxin+ATMi+4OHT). Immunoprecipitated DNA in each condition was analyzed by quantitative PCR at two AsiSI-induced DSBs (HR-prone DSB-III in red and non-HR-prone DSB3 in blue). Normalized pull-down efficiencies from a representative experiment are shown.
DNA-PKcs, but Not ATM, Is Required for AsiSI-Induced DSB Repair
We next thought to directly evaluate the function of these two kinases on the repair of various AsiSI-induced DSBs. Indeed, we found recently that distinct AsiSI-induced DSBs are not repaired equivalently, and we identified a subset of DSBs, localized in transcriptionally active, H3K36me3-rich chromatin, that are “HR-prone,” i.e., preferentially recruit RAD51, undergo resection, and rely on HR for efficient repair. On the other hand, AsiSI-induced DSBs localized either in intergenic regions or inactive genes could not recruit RAD51 and relied on XRCC4 for repair (Aymard et al., 2014).
In order to assay the respective roles of ATM and DNA-PKcs in repair at those HR- and non-HR-prone DSBs, we used a previously described cleavage assay that allows one to measure repair kinetics at selected AsiSI-induced DSBs (Aymard et al., 2014, Chailleux et al., 2014). Interestingly, while DNA-PKcs inhibition led to a severe repair defect, AsiSI-induced DSBs were efficiently religated upon ATM inhibition (Figure 1B). The combination of both inhibitors led to a repair deficiency similar to the one observed upon DNA-PKcs inhibition (Figure S1C). Notably, the effects of DNA-PKcs and ATM inhibition were identical on non-HR-prone or HR-prone DSBs (Figure 1B, compare the three top panels to the bottom panel). Altogether these data indicate that AsiSI-induced DSB repair depends on DNA-PKcs, but not on ATM activity, and that the function of these kinases is independent of the pathway used for repair and on the genomic location of the break.
Since ATM inhibition led to a severe survival defect (Figure 1A), we tested the accuracy of DSB repair in the presence of the ATM inhibitor. Faithful repair of an AsiSI-induced DSB leads to the reconstitution of the restriction site, thus available for a new round of enzymatic cleavage. We therefore measured the ability of AsiSI-ER to re-cleave sites once repair has occurred upon auxin addition either in the presence or absence of the ATM inhibitor. As described above, ATM inhibition during the repair step did not impede the re-joining of a non-HR prone (DSB-3; Figure 1C, top panel) or of an HR-prone DSB (DSB-III; Figure 1C, bottom panel) (compare yellow and blue bars). However, we observed that an additional round of 4OHT treatment led to reduced AsiSI cleavage when DSB repair was performed in the presence of the ATM inhibitor compared to control (Figure 1C; compare brown and purple bars). Again, both HR and non-HR-prone DSBs behaved similarly. This indicates that ATM inhibition reduces repair accuracy at AsiSI-induced DSBs.
Activated ATM and DNA-PKcs Are Recruited to the Vicinity of DSBs
To further investigate the functions of both kinases in DSB repair, we next analyzed their distribution around multiple DSBs induced on chromosome 1 and 6 by chromatin immunoprecipitation followed by hybridization on tiling arrays (ChIP-chip). To this end, 4OHT-treated or untreated DIvA cells were subjected to ChIP using antibodies directed against the activated forms of these kinases (phosphorylated ATM-Ser1981 and phosphorylated DNA-PKcs-S2056, two autophosphorylation events indicative of kinase activation) (Figures S2A–S2C). Inspection of ChIP-chip profiles indicated that both P-ATM and P-DNA-PKcs were detected in the vicinity of AsiSI-DSBs upon 4OHT treatment (see two examples, Figure 2A). Averaged P-ATM and P-DNA-PKcs profiles on all DSBs induced on chromosome 1 and 6 (24 sites; Iacovoni et al., 2010) showed a significant recruitment of both kinases within a ∼5-kb window around DSBs (Figures 2B and 2C). However, while P-ATM and P-DNA-PKcs enrichment was restricted to the vicinity of the breaks for most of the DSBs, we could identify one DSB leading to a large spreading of both kinases on the surrounding chromatin (Figure S2D). Although the reasons for such a spreading are presently not clear, this indicated that the distribution of P-ATM and P-DNA-PKcs depends on the chromatin and/or the genomic context of the DSB. Of note, ATM was also dispensable for repair at this specific DSB (Figure S2E).
Figure 2.

Recruitment of P-ATM and P-DNA-PKcs at AsiSI-Induced DSBs
(A) ChIP-chip analyses in DIvA cells after 4OHT treatment (4 hr), using anti P-ATM S1981 (left panels) or anti-P-DNA-PKcs S2056 (right panels) antibodies. Profiles of both activated kinases before (in black) and after (in red) 4OHT treatment are shown at two AsiSI-induced DSBs (indicated by black arrows).
(B) Averaged signal for P-ATM S1981 (top) and P-DNA-PKcs S2056 (bottom) over 40-kb windows and centered at the AsiSI site are shown either before (black) or after (red) 4OHT treatment.
(C) Box plot representing the distribution of the averaged P-ATM S1981 (top) and P-DNA-PKcs S2056 (bottom) signals, calculated on 2-kb windows around each of the 24 AsiSI sites of chr1/6, with or without the indicated 4OHT treatment. ∗∗p < 0.01, ∗∗∗∗p < 0.001 (paired Student’s t test).
(D) Averaged P-ATM S1981 signal (x axis) calculated on 2-kb windows at each AsiSI site plotted against the averaged P-DNA-PKcs S2056 signal (y axis) calculated over the same window. r = 0.62 (Pearson).
(E) Heatmap representation of P-ATM S1981 (left panels) and P-DNA-PKcs S2056 (right panels) signal across all AsiSI-induced DSBs on chromosomes 1 and 6 (sorted by P-ATM increasing signal over a 20-kb window).
Importantly, both kinases showed a similar profile and were found to be similarly enriched at each AsiSI-induced DSB on chromosome 1 and 6. Accordingly, the average enrichment of P-ATM over a 2-kb window correlated with the averaged enrichment of P-DNA-PKcs calculated on the same window (r = 0.62, Pearson) (Figure 2D). Heatmap representation of our ChIP-chip results, where each individual cleaved AsiSI site is represented, confirmed the propensity of a DSB to recruit P-DNA-PKcs when recruiting P-ATM and failed to reveal sites solely and specifically occupied by either P-DNA-PKcs or P-ATM (Figure 2E).
Altogether, our ChIP-chip data indicate that (1) both activated kinases are similarly recruited at DSBs and (2) they are mostly localized to the immediate proximity of the break, in sharp contrast to the megabase-wide spreading of γ-H2AX.
γ-H2AX Induction at AsiSI-DSBs Relies on ATM Activity
We next wondered how inhibition of these kinases would impact H2AX phosphorylation. Indeed, while the role of ATM in H2AX phosphorylation is well established, the function of DNA-PKcs toward this modification is still unclear since many studies have reported conflicting results, likely because of the various means used to induce DSBs. In addition, each kinase could be responsible for γH2AX depending on the genomic localization of the break and/or the pathway used for repair. Finally, one could also envisage that both kinases are required at different locations around the DSB, to establish a proper γH2AX domain. Thus, we used ATM and DNA-PKcs inhibitors to clarify the respective involvement of each kinase in H2AX phosphorylation around distinct DSBs located in various chromatin contexts. While western blot analysis revealed that ATM inhibition led to a major defect in global H2AX phosphorylation upon the induction of clean AsiSI-dependent DSBs, this was not the case with DNA-PKcs inhibition (Figure 3A). This result was confirmed by immunofluorescence analysis of 4OHT-treated DIvA cells. Indeed, while treatment with the ATM inhibitor led to a dramatic decrease in γH2AX foci, inhibition of DNA-PKcs did not result in any detectable impairment of the γH2AX signal (Figure 3B, see examples on the top panel and the quantification of four independent experiments on the bottom panel). We next performed γH2AX ChIP-chip experiments to investigate at higher resolution the effect of PI3K-like kinase inhibitors on γH2AX profiles around more than 20 DSBs induced on chromosomes 1 and 6. ATM inhibition led to a dramatic decrease in the γH2AX signal around DSBs. In contrast, inhibition of DNA-PKcs did not alter the γH2AX profile induced at the DSB (see three examples in Figure 3C and the average γH2AX profile for the 24 DSBs induced on chr1/6 in Figure 3D). Interestingly, we observed the same effects at all DSBs, regardless of the DSB location (Figure 3E). Altogether these data indicate that upon induction of multiple clean DSBs, ATM seems to be the main kinase responsible for H2AX phosphorylation over entire megabase domains and at all DSBs independently of their genomic location.
Figure 3.

ATM, but Not DNA-PKcs, Mediates H2AX Phosphorylation on Megabase-wide Domains around AsiSI-Induced DSBs
(A) Western blot analyses of DIvA cells treated or not with 4OHT and ATM or DNA-PKcs inhibitors as indicated and stained for γH2AX (top) and tubulin (loading control, bottom).
(B) The presence of γH2AX foci was monitored by immunofluorescence in untreated or 4OHT-induced cells (4 hr), in the presence or not of ATM or DNA-PKcs inhibitors, as indicated. The bottom panel shows quantification from five biological replicates.
(C) γH2AX ChIP-chip analyses in DIvA cells after 4OHT treatment (4 hr), in the presence or absence of ATM and DNA-PKcs inhibitors as indicated. Profiles of γH2AX are shown at three AsiSI-induced DSBs (indicated by black arrows).
(D) The averaged γH2AX signals in 4OHT-treated cells (in red) supplemented with ATM (blue) or DNA-PKcs inhibitors (gray) over a 2-Mb region flanking cleaved AsiSI sites are shown.
(E) Heatmap showing γH2AX distribution over each AsiSI cleaved site for each condition (sorted by increasing γH2AX level on a 500-kb window).
However, since a faint γH2AX signal was still detected upon ATM inhibition (Figures 3A–3E), we wondered whether DNA-PKcs activity could account for this residual H2AX phosphorylation. A combined treatment with ATM and DNA-PKcs inhibitors did not lead to a further decrease of γH2AX as detected by western blot (Figure 4A) and immunostaining (Figure 4B). Surprisingly, γH2AX ChIP-chip experiments in DIvA cells treated with both inhibitors even revealed an increased γH2AX signal at the sites of DSB, compared to the signal in cells treated with ATM inhibitor alone (see Figure 4C for a few examples and Figure 4D for the averaged profiles). This indicates that DNA-PKcs does not contribute to the phosphorylation of H2AX and rather inhibits this event after induction of clean DSBs on the human genome. It also strongly suggests that another kinase is able to phosphorylate H2AX when both ATM and DNA-PKcs activities are impaired (and to a lesser extent when only ATM activity is impaired). We thus also performed additional experiments by combining ATM and DNA-PKcs inhibitors with inhibitors directed against other PI3K family members or kinases formerly found as able to phosphorylate H2AX or to regulate the DNA damage response although in different conditions (Lu et al., 2006, Shen et al., 2013, Ward and Chen, 2001). However, neither ATR inhibitor (Toledo et al., 2011) (Figures S3A–S3E), mTOR inhibitor (Figures S4A–S4C), nor JNK inhibitor (Figures S4D and S4E) decreased the residual γH2AX signal observed upon ATMi and DNAPKi combination, indicating that a yet unidentified kinase is able to mediate H2AX phosphorylation to some extent, especially in conditions were ATM and DNA-PKcs activities are impaired.
Figure 4.

Inhibition of Both DNA-PKcs and ATM Does Not Abrogate γH2AX
(A) γH2AX (top) and tubulin (loading control, bottom) western blot analyses of DIvA cells treated with ATM and/or DNA-PKcs inhibitors as indicated.
(B) The presence of γH2AX foci was monitored by immunofluorescence in 4OHT-induced cells (4 hr), in the presence of the ATM inhibitor alone or in combination with the DNA-PKcs inhibitor, as indicated. The bottom panel shows quantification from five biological replicates.
(C) γH2AX ChIP-chip analyses in DIvA cells after 4OHT treatment (4 hr) and in the presence of the ATM inhibitor alone or in combination with DNA-PKcs inhibitor, as indicated. Profiles of γH2AX are shown at three AsiSI-induced DSBs (indicated by black arrows).
(D) The averaged γH2AX signals in 4OHT-treated cells (in red) supplemented with ATM (blue) or ATM+DNA-PKcs inhibitors (purple) over a 2-Mb region flanking cleaved AsiSI sites are shown.
ATM, but Not DNA-PKcs, Is Required for Coalescence of AsiSI-Induced DSBs into Large Repair Foci
The fact that AsiSI induces a constant and known number of DSBs, which in addition are always located at the same genomic locations in all cells, prompted us to analyze γH2AX foci structure and distribution by microscopy in order to also identify the function of both kinases in the global organization of damaged chromatin. Interestingly, in normal, 4OHT-treated cells, γH2AX immunostaining revealed a number of foci largely reduced compared to the number of γH2AX domains as depicted linearly on chromosomes by ChIP-seq (Aymard et al., 2014) (roughly 80 foci per cell compared to about 300 AsiSI-induced DSBs; Figure S5A), suggesting that these γH2AX foci may contain more than one γH2AX domain and thus more than a single DSB. Although still controversial, previous studies have demonstrated that DSBs induced by γ or laser (α-particles) irradiation are able to cluster (Aten et al., 2004, Krawczyk et al., 2012). In order to investigate whether AsiSI-induced DSBs may also exhibit clustering, we developed a DIvA cell line that expresses the 53BP1 repair protein fused to GFP, to follow DSB movement within the nuclear space by live cell imaging using a spinning disk confocal laser microscope. 53BP1-GFP was efficiently recruited at AsiSI-induced DSBs within the first hour of 4OHT treatment (Movies S1 and S2). Strikingly, 53BP1-induced foci were highly dynamic and could undergo several cycles of association and dissociation within bigger foci (Figure S5C; Movies S3, S4, and S5), which confirms that multiple AsiSI-induced breaks are able to associate together within repair centers.
To get more insights into the structure of AsiSI-induced γH2AX foci in normal conditions and upon kinase inhibition, we further used high-resolution microscopy. In 4OHT-treated cells, γH2AX foci appeared to be composed of small substructures (Figure 5A, left panel), likely reflecting individual DSBs. To analyze the spatial distribution of these substructures, we performed a statistical test, using the Icy spatial analysis plug-in that describes the distribution of individual dots within the nucleus (Lagache et al., 2013). This plug-in, based on the Ripley’s K function, statistically assesses the presence of clusters by comparing the values of the K function to its critical quantiles under spatial randomness. Clustering is statistically significant when the K function crosses the upper quantile. Notably, this test demonstrated that individual γH2AX dots clustered in 4OHT-treated DIvA cells (Figure 5B, left panel). Interestingly, while DNA-PKcs inhibition did not alter the distribution of γH2AX foci in response to 4OHT (Figure 5A, right panel), inhibition of ATM led to a dramatic dispersion of γH2AX foci in the nucleus (Figure 5A, middle panel). Ripley K function describing the spatial distribution of γH2AX dots in each condition indicated that ATM inhibitor treatment led to a random distribution of γH2AX dots within the nucleus (Figure 5B, middle panel), while DNA-PKcs inhibition did not compromise foci clustering (Figure 5B, right panel). Altogether, these data favor a model in which AsiSI-induced DSBs are able to cluster within repair centers in an ATM-dependent, but DNA-PKcs-independent, manner.
Figure 5.

Clustering of AsiSI-Induced DSBs Depends on ATM Activity
(A) γH2AX staining in 4OHT-treated DIvA cells, in the presence of ATM or DNA-PKcs inhibitors, as indicated.
(B) Averaged Ripley function (y axis) depending on cluster size (x axis) illustrating the spatial distribution of γH2AX spots identified in 4OHT-treated DIvA cells in the presence of ATM or DNA-PKcs inhibitors, as indicated.
(C) Magnification of a 4OHT-treated cell, stained with γH2AX (red) and XRCC4 (green) in the presence of ATM or DNA-PKcs inhibitors, as indicated.
(D) Number of γH2AX foci detected in cells treated (red) or not (blue) with ATM inhibitor, at increasing doses of etoposide. Average ± SEM of the number of foci from at least three independent experiments is shown (top panel). Representative images of γH2AX foci (green) and DAPI counterstain (blue) in cells treated with 10 μM etoposide in the absence (−ATMi) or presence (+ATMi) of 10 μM ATM inhibitor are shown on the bottom panel.
To confirm that each individual substructure observed within a large γH2AX focus represents a single DSB, we also performed immunostaining against XRCC4, a DSB repair protein that accumulates at the exact break point (on roughly 500 bp, according to our recent XRCC4 ChIP-seq mapping [Aymard et al., 2014]). To detect XRCC4 by immunofluorescence in damaged 4OHT-treated cells, we applied a recently described protocol (Britton et al., 2013) that permits detection of NHEJ repair proteins in ionizing radiation-induced nuclear foci (IRIF) (which was, until recently, impossible with standard staining procedures) (Figure S5B). In normal conditions, we could clearly identify γH2AX foci containing two or more XRCC4 foci, showing that, indeed, several individual DSBs are found within a single repair focus (Figures 5C and S5D). This association of multiple XRCC4 foci within a γH2AX focus was lost upon ATM inhibition, confirming that ATM is required for DSB clustering in human cells.
In order to investigate whether our finding on the function of ATM on AsiSI-induced DSBs clustering could be generalized to other types of DSBs, we also analyzed the number of γH2AX foci in mouse embryonic fibroblasts, in response to increasing doses of etoposide. As expected, ATMi treatment led to a decrease of the γH2AX signal (data not shown) but also led to an increase in the number of γH2AX foci (Figure 5E) at each dose investigated, which suggested that ATM-mediated association in repair centers is a general feature of DSBs.
Discussion
In this study, by using the DIvA cell line, which permits the induction of multiple annotated DSBs throughout the human genome, we showed that the major DDR PI3K-like kinases ATM and DNA-PKcs have complementary and non-overlapping functions in survival, repair, and γH2AX establishment in the context of clean DSBs. We found that both kinases are recruited and activated to a similar extent at all investigated DSBs, in a manner that appears to be independent of the pathway used for subsequent repair. While DNA-PKcs activity is required for repair and survival, it is dispensable for γH2AX domain establishment. By contrast, ATM is essential for cell survival and γH2AX phosphorylation but DSBs can be religated upon ATM inhibition, although in a less accurate manner.
Notably, these functions of DDR kinases at AsiSI-induced DSBs are independent of the pathway used to repair these breaks since both HR-prone and non-HR-prone DSBs behaved similarly. Finally, we found that multiple AsiSI-induced DSBs can associate within repair centers in an ATM-dependent manner. Altogether our study uncouples the induction of γH2AX domains from the repair process itself and clarifies the function of both kinases in the DDR in response to clean DSBs generated at different locations on the genome.
ATM Is Dispensable for DNA Ends Rejoining, but Is Required for Accurate Repair of Clean, Cohesive DSBs
Our data, in agreement with previous reports (Zhao et al., 2006), indicate that inhibition of DNA-PKcs activity severely impaired repair and cell survival upon induction of clean and easily repairable DSBs, though our assay would not discriminate whether repair is drastically delayed or totally inhibited. DNA-PKcs is a core component of the NHEJ machinery (Calsou et al., 2003, DeFazio et al., 2002), and its inhibition likely compromises XRCC4/DNA Ligase 4 dependent end joining. Furthermore, inhibition of DNA-PKcs activity interferes with DNA-PKcs autophosphorylation, an event required for its dissociation from DNA ends (Merkle et al., 2002). This lack of dissociation likely blocks both resection (Shibata et al., 2011, Zhou and Paull, 2013) and the potential ability to switch to an alternative pathway (either Alt-NHEJ or HR), thus leading to a severe repair defect (Chan et al., 2002, Cui et al., 2005). Interestingly, DNA-PKcs inhibition altered repair at both non-HR-prone DSBs as well as HR-prone DSBs, as identified in our previous study (Aymard et al., 2014). Together with our recent finding that XRCC4 is recruited at both HR-prone and non-HR-prone AsiSI-DSBs (Aymard et al., 2014), as is P-DNA-PKcs (this study), this favors the idea that the NHEJ machinery will first attempt to repair all DSBs (Shibata et al., 2011) and that blocking DNA-PKcs activity can inhibit the subsequent use of the HR machinery at HR-prone DSBs. In agreement with a role in DNA end joining, P-DNA-PKcs distribution was mostly restricted to the vicinity of DSBs (this study; Chandler et al., 2014), apart from few exceptions (Figure S2; Chandler et al., 2014) where it exhibited considerable spreading. Whether those specific DSBs undergo a specific repair pathway remains to be investigated.
While DNA-PKcs activity is essential for repair, our data indicate that ATM activity is dispensable for end rejoining at either HR or non-HR-prone DSBs, i.e., at DSBs induced in active or inactive genes. Since all AsiSI-induced DSBs occur in euchromatin, this is consistent with a recent study indicating that ATM activity is also not required for repair of etoposide-induced DSBs in TDP2 proficient cells (also mainly occurring within euchromatin), (Álvarez-Quilón et al., 2014). By contrast, repair of DSBs induced in heterochromatin is ATM dependent. At those DSBs, ATM would be required for chromatin remodeling permitting the loading and processing of repair machineries (Beucher et al., 2009, Goodarzi et al., 2008, Noon et al., 2010, Shibata et al., 2010). In addition, a recent report identified ATM as required for macroH2A1 loading and chromatin condensation, which are events shown to be necessary for BRCA1 retention at DSB and HR repair (Khurana et al., 2014). Altogether these studies suggest that the main function of ATM might reside in its ability to modify the surrounding chromatin in order to facilitate repair, rather than in the repair reaction itself.
Interestingly, we found that, although effective, repair of clean DSBs is less accurate upon inhibition of ATM activity. This is in contrast with a recent finding that ATM inactivation did not alter the fidelity of unblocked DSBs end rejoining on plasmids in vivo (Álvarez-Quilón et al., 2014). Thus, ATM may be required to ensure fidelity of DSB repair in a chromosomal context only. Interestingly, ATM is also required to limit the use of distal ends to repair two chromosomal I-SceI tandem DSBs (Bennardo and Stark, 2010). This role of ATM in the promotion of faithful repair may be linked to the etiology of AT patients, who show abnormal rates of tumor apparition and progression, as well as severe neurodegenerative features.
ATM Is Responsible for H2AX Phosphorylation around Clean DSBs
While the involvement of ATM in H2AX phosphorylation in response to DSBs is undebated, the function of DNA-PKcs toward γH2AX remains controversial. Indeed, some studies suggested a predominant role for ATM in H2AX phosphorylation (Burma et al., 2001, Savic et al., 2009), while others reported that ATM and DNA-PKcs could largely substitute each other in that respect (e.g., Stiff et al., 2004, Wang et al., 2005). These apparent discrepancies likely arise from the variety of damages induced by X-rays or γ rays. Our study demonstrates that induction of clean DSBs throughout the human genome elicits γH2AX formation in an ATM-dependent, but DNA-PKcs-independent, manner at all DSBs, independently of their genomic location and their propensity to be repaired by HR or not. Notably, as already reported (Burma et al., 2001, Savic et al., 2009), we observed residual γH2AX levels upon ATM inhibition. However, in contrast with the observation made on the Igκ locus (Savic et al., 2009), this phosphorylation was DNA-PKcs independent, as indicated by global as well as high-resolution analyses of γH2AX levels and distribution upon simultaneous inhibition of both kinases. This apparent contradiction likely arises from the fact that DSB repair and signaling strongly depend on the genomic context where the break occurs (Clouaire and Legube, 2015). This residual signal was also not lost upon ATR, JNK, or mTOR inhibition, indicating that yet unidentified kinase(s) contribute to H2AX phosphorylation after clean DSBs induction. Further high-resolution studies would help to identify such enzyme(s).
Importantly, ATM was the major kinase responsible for H2AX phosphorylation over the entire domain. The mechanism for γH2AX spreading is still unknown, but it could result from (1) the spreading of the kinase itself in cis, (2) the local diffusion of the kinase within repair foci, or (3) dynamic chromatin fibers encountering ATM bound to DNA ends. Our ChIP-chip mapping of activated ATM revealed that, in contrast to γH2AX, P-ATM is distributed on a restricted domain around most DSBs, excluding diffusion of the activated kinase in cis as a main mechanism for γH2AX spreading. Interestingly, in yeast, the ATM counterpart Tel1 is able to phosphorylate H2A in trans (Lee et al., 2014). In addition, we previously reported that the distribution of γH2AX is strongly influenced by preexisting high-order chromatin structure (Caron et al., 2012). These data support a model in which ATM, bound to DNA ends, would be able to phosphorylate nucleosomal H2AX brought within proximity of a DSB through local motions of the surrounding chromatin. Interestingly, upon dual inhibition of both ATM and DNA-PKcs, the residual γH2AX signal was observed on a more restricted area covering approximately 200 kb (Figure 4C), indicating that the ability to promote megabase-wide γH2AX spreading is specific to ATM and is not shared by unidentified backup kinase(s) operating in this context.
Function of ATM and DNA-PKcs in DSB Mobility and Cluster Formation
Whether DSBs are relocated in close proximity after their induction is still a matter of debate. In yeast, DSBs were reported to be highly mobile and to coalesce (Lisby et al., 2001). In mammals, conflicting results have been obtained regarding the mobility and potential clustering of radiation and nuclease-induced DSBs (Aten et al., 2004, Becker et al., 2014, Jakob et al., 2009a, Krawczyk et al., 2006, Krawczyk et al., 2012, Kruhlak et al., 2006, Soutoglou et al., 2007). In addition, a recent study indicated that damaged telomeres in ALT cells undergo clustering in an HR-machinery-dependent manner (Cho et al., 2014).
The DIvA system allows not only precise knowledge of the position of all AsiSI-induced DSBs but also their exact number within each cell. This permits comparison of the number of domains linearly depicted on the genome by ChIP-seq, with the number of γH2AX foci detected by microscopy. We found that AsiSI-induced, clean DSBs are dynamic and can frequently coalesce within larger foci. However, DSB coalescence only occurred between spatially proximal 53BP1 foci. While a single enzymatically induced DSB has limited mobility (Soutoglou et al., 2007), it was shown that I-SceI-induced DSBs would frequently pair and that this motion of broken chromatin is a critical step in the biogenesis of translocation on the human genome (Roukos et al., 2013). Furthermore, such translocation events occurred mainly with linearly or spatially proximal chromatin (Roukos et al., 2013, Zhang et al., 2012). In summary, our data, as well as the recent literature, clearly support a model where multiple DSBs induced within spatial proximity of each other frequently associate within repair foci. Given that chromosome mobility depends on the chromatin context, it is highly likely that such observed DSB associations will vary from one genomic location to another and that all induced DSBs will not behave equivalently toward clustering. In addition, DSB mobility in yeast is highly dependent on the resection machinery (Dion et al., 2012, Miné-Hattab and Rothstein, 2012). It is thus tempting to speculate that HR-prone DSBs might be more prone to cluster than others. This would be in agreement with recent data showing that clustering of damaged telomeres also depends on the HR machinery (Cho et al., 2014).
Furthermore, in agreement with previous studies (Aten et al., 2004, Krawczyk et al., 2006, Krawczyk et al., 2012) we showed that this event is driven by ATM, since ATM inhibition led to a complete dispersion of DSBs throughout the nucleus. In agreement, inhibition of Mre11 (a component of the MRN complex, whose retention at DSBs requires ATM activity) led to a clear decrease in DSB pairing (Roukos et al., 2013). Interestingly, other processes where distant DSBs are brought together, such as distal end rejoining, occurring during class switch and V(D)J recombination, or the fusion of deprotected telomeres, also depend on ATM (Difilippantonio et al., 2008, Dimitrova et al., 2008). Thus, DSB association is likely mediated by an ATM substrate. Of note, the 53BP1 repair protein, targeted by ATM, has been involved in both promoting telomere fusion and efficient V(D)J recombination (Difilippantonio et al., 2008, Dimitrova et al., 2008). Whether it also promotes AsiSI-induced DSB clustering remains to be investigated, even though a couple of studies indicated that depletion of 53BP1 did not impede DSB mobility (Krawczyk et al., 2012, Soutoglou et al., 2007). Furthermore, H2AX-deficient mice also display defects in both class switch and V(D)J recombination. Thus, a potential function of γH2AX domains in ATM-dependent DSB pairing is an exciting possibility that should be further tested.
An open and critical question resides in why would clustering be beneficial for repair, taking into account the high risk of translocation generated by the proximity of distal DNA ends. Given that ATM is both involved in this process and in accurate repair, but not required for end joining itself, an interesting hypothesis would be that these events might help to increase the repair fidelity. In this regard, it is notable that neither ATM nor H2AX are required for survival in mammals (H2AX knockout mice are viable, as are AT patients) although their lack of function severely increase tumor susceptibility, in agreement with a potential reduction of repair accuracy. Whether ATM-dependent H2AX phosphorylation mediates DSB clustering and whether this event promotes repair fidelity are exciting hypotheses to be tested by future investigations.
Experimental Procedures
Cell Culture
For AsiSI-dependent DSB induction, DIvA or AID-DIvA cells were treated with 300 nM 4OHT (Sigma; H7904) for 4 hr. When indicated, 4OHT-treated cells were incubated with 500 μg/mL auxin. For the treatment with inhibitors, the following final concentrations were used: KU55933(ATMi), 20 μM; Nu7441(DNAPKi), 2 μM; ETP-46464 (ATRi, a kind gift from Dr. O. Fernandez-Capetillo), 5 μM; rapamycin, 20 nM; and SP600125 (JNKi), 50 μM. Inhibitors were added to the medium 1 hr before the addition of 4OHT and during the 4-hr stage of break induction (4OHT) and/or during the repair step (auxin) as indicated.
ChIP
ChIP assays were carried out according to the protocol described in Iacovoni et al. (2010) and are detailed in the Supplemental Experimental Procedures. IP efficiencies were calculated as the percent of input DNA immunoprecipitated.
For ChIP-chip, 8 ng of inputs and samples were amplified as in Iacovoni et al. (2010), labeled, and hybridized on Affymetrix tiling arrays covering human chromosomes 1 and 6.
To plot data with respect to the 24 DSBs induced on chromosome 1 and 6 (Iacovoni et al., 2010), the ChIP-chip signal was averaged for 200-bp windows spanning 40 kb (P-ATM and P-DNA-PKcs) or 2 Mb (γH2AX) surrounding each annotated AsiSI site. For heatmap representations, the average ChIP-chip signal was determined in 500-bp bins for P-ATM and P-DNA-PKcs, and 50-kb bins for γH2AX, centered on each cleaved AsiSI site.
Repair Kinetics and Repair Fidelity at AsiSI Sites
Repair kinetics at specific AsiSI-induced DSBs were measured as described in Aymard et al. (2014) by a cleavage assay permitting the capture of unrepaired DSBs, at the indicated times after auxin addition. For fidelity assays, DIvA cells were treated with 4OHT to induce DSBs for 4 hr (300 nM) followed by an auxin treatment for 4 hr, in the presence or absence of ATMi (KU55933). The next day, cells were treated again with 4OHT for 4 hr. DNA was extracted and subjected to a cleavage assay as described above.
Immunofluorescence
Detailed methods for immunofluorescence against γH2AX (JBW301) in DIvA cells have already been described in Iacovoni et al. (2010). XRCC4 staining was performed according the protocol described in Britton et al. (2013). For high-resolution microscopy, a fluorescent widefield microscope was used to produce 3D image of the nucleus and subjected to deconvolution (Supplemental Experimental Procedures). Spot distribution was analyzed using the spatial analysis plug-in available in Icy. Live cell analysis was performed using an Andor Revolution Nipkow-disk confocal system. For the illustrations shown in Figure S5C, maximum projections using Image J were performed to generate 2D movies.
Author Contributions
J.C., P.C., B.B., and V.D. performed experiments. J.S.I., T.C., and M.A. performed bioinformatics analyses of the ChIP-chip data. T.M. performed high-resolution microscopy and γH2AX foci spatial distribution. A.A.-Q. and F.C.-L. performed induction of γH2AX foci in response to etoposide. G.L. conceived and analyzed experiments and wrote the manuscript. All authors commented and edited the manuscript.
Acknowledgments
We thank V. Benes at the EMBL Genomic Core Facility for microarrays hybridization. We thank Dr. C. Normand for time-lapse microscopy. We thank Dr. D. Jullien for the pEGFP-53BP1 and Dr. O. Fernandez Capetillo for the ATR inhibitor. T.C. and M.A. were supported by grants from the Fondation pour la Recherche Médicale (FRM). P.C. was supported by a grant from the Association Contre le Cancer (ARC). J.C. is supported by a grant from the Ligue Nationale contre le Cancer. Work in the F.C.-L. laboratory is funded by grants and fellowships from the Spanish Government (SAF2010-21017 and BES-2011-047351) and the regional Andalusian Government (CVI-7948). Funding in the G.L. laboratory was provided by grants from the Agence Nationale pour la Recherche (ANR-14-CE10-0002-01 and ANR-13-BSV8-0013), the Institut National contre le Cancer (INCA), the Ligue Nationale contre le Cancer (LNCC), and Research Innovation Therapeutic Cancerologie (RITC). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement 647344).
Published: November 12, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, five figures, and five movies and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.10.024.
Accession Numbers
The accession number for the ChIP-chip data reported in this paper is ArrayExpress: E-MTAB-2992. 4D movies for time-lapse microscopy are available upon request.
Supplemental Information
Time-lapse microscopy was performed on 4OHT-treated 53BP1-GFP DIvA using a spinning disk confocal microscope. Acquisition was started straight after 4OHT treatment, and all z stacks were acquired every 2 min. Maximum projection was computed with Image J. Full 4D movies are available upon request.
{kind=link}
Same as Movie S1.
{kind=link}
Same as Movie S1, except that acquisition was performed after 2 hr of 4OHT treatment and every 15 s. A magnification of 53BP1 foci ongoing clustering is presented. Related to Figure S5C bottom sequence.
{kind=link}
Same as Movie S1, except that acquisition was performed after 2 hr of 4OHT treatment and every 3 min. Related to Figure S5C top sequence.
{kind=link}
Same as Movie S1, except that acquisition was performed after 2 hr of 4OHT treatment and every 3 min.
{kind=link}
References
- Álvarez-Quilón A., Serrano-Benítez A., Lieberman J.A., Quintero C., Sánchez-Gutiérrez D., Escudero L.M., Cortés-Ledesma F. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 2014;5:3347. doi: 10.1038/ncomms4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aten J.A., Stap J., Krawczyk P.M., van Oven C.H., Hoebe R.A., Essers J., Kanaar R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004;303:92–95. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P., Legube G. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beamish H.J., Jessberger R., Riballo E., Priestley A., Blunt T., Kysela B., Jeggo P.A. The C-terminal conserved domain of DNA-PKcs, missing in the SCID mouse, is required for kinase activity. Nucleic Acids Res. 2000;28:1506–1513. doi: 10.1093/nar/28.7.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A., Durante M., Taucher-Scholz G., Jakob B. ATM alters the otherwise robust chromatin mobility at sites of DNA double-strand breaks (DSBs) in human cells. PLoS ONE. 2014;9:e92640. doi: 10.1371/journal.pone.0092640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo N., Stark J.M. ATM limits incorrect end utilization during non-homologous end joining of multiple chromosome breaks. PLoS Genet. 2010;6:e1001194. doi: 10.1371/journal.pgen.1001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beucher A., Birraux J., Tchouandong L., Barton O., Shibata A., Conrad S., Goodarzi A.A., Krempler A., Jeggo P.A., Löbrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009;28:3413–3427. doi: 10.1038/emboj.2009.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S., Coates J., Jackson S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013;202:579–595. doi: 10.1083/jcb.201303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S., Chen B.P., Murphy M., Kurimasa A., Chen D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Callén E., Jankovic M., Wong N., Zha S., Chen H.T., Difilippantonio S., Di Virgilio M., Heidkamp G., Alt F.W., Nussenzweig A., Nussenzweig M. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol. Cell. 2009;34:285–297. doi: 10.1016/j.molcel.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calsou P., Delteil C., Frit P., Drouet J., Salles B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. J. Mol. Biol. 2003;326:93–103. doi: 10.1016/s0022-2836(02)01328-1. [DOI] [PubMed] [Google Scholar]
- Caron P., Aymard F., Iacovoni J.S., Briois S., Canitrot Y., Bugler B., Massip L., Losada A., Legube G. Cohesin protects genes against γH2AX induced by DNA double-strand breaks. PLoS Genet. 2012;8:e1002460. doi: 10.1371/journal.pgen.1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chailleux C., Aymard F., Caron P., Daburon V., Courilleau C., Canitrot Y., Legube G., Trouche D. Quantifying DNA double-strand breaks induced by site-specific endonucleases in living cells by ligation-mediated purification. Nat. Protoc. 2014;9:517–528. doi: 10.1038/nprot.2014.031. [DOI] [PubMed] [Google Scholar]
- Chan D.W., Chen B.P., Prithivirajsingh S., Kurimasa A., Story M.D., Qin J., Chen D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–2338. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler H., Patel H., Palermo R., Brookes S., Matthews N., Peters G. Role of polycomb group proteins in the DNA damage response—a reassessment. PLoS ONE. 2014;9:e102968. doi: 10.1371/journal.pone.0102968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N.W., Dilley R.L., Lampson M.A., Greenberg R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014;159:108–121. doi: 10.1016/j.cell.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T., Legube G. DNA double strand break repair pathway choice: a chromatin based decision? Nucleus. 2015;6:107–113. doi: 10.1080/19491034.2015.1010946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X., Yu Y., Gupta S., Cho Y.M., Lees-Miller S.P., Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell. Biol. 2005;25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio L.G., Stansel R.M., Griffith J.D., Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002;21:3192–3200. doi: 10.1093/emboj/cdf299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deriano L., Roth D.B. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- Difilippantonio S., Gapud E., Wong N., Huang C.Y., Mahowald G., Chen H.T., Kruhlak M.J., Callen E., Livak F., Nussenzweig M.C. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N., Chen Y.C., Spector D.L., de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V., Kalck V., Horigome C., Towbin B.D., Gasser S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat. Cell Biol. 2012;14:502–509. doi: 10.1038/ncb2465. [DOI] [PubMed] [Google Scholar]
- Falck J., Coates J., Jackson S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Goodarzi A.A., Noon A.T., Deckbar D., Ziv Y., Shiloh Y., Löbrich M., Jeggo P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell. 2008;31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Iacovoni J.S., Caron P., Lassadi I., Nicolas E., Massip L., Trouche D., Legube G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29:1446–1457. doi: 10.1038/emboj.2010.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob B., Splinter J., Durante M., Taucher-Scholz G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl. Acad. Sci. USA. 2009;106:3172–3177. doi: 10.1073/pnas.0810987106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob B., Splinter J., Taucher-Scholz G. Positional stability of damaged chromatin domains along radiation tracks in mammalian cells. Radiat. Res. 2009;171:405–418. doi: 10.1667/RR1520.1. [DOI] [PubMed] [Google Scholar]
- Jasin M., Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013;5:a012740. doi: 10.1101/cshperspect.a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S., Kruhlak M.J., Kim J., Tran A.D., Liu J., Nyswaner K., Shi L., Jailwala P., Sung M.H., Hakim O., Oberdoerffer P. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014;8:1049–1062. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk P.M., Stap J., van Oven C., Hoebe R., Aten J.A. Clustering of double strand break-containing chromosome domains is not inhibited by inactivation of major repair proteins. Radiat. Prot. Dosimetry. 2006;122:150–153. doi: 10.1093/rpd/ncl479. [DOI] [PubMed] [Google Scholar]
- Krawczyk P.M., Borovski T., Stap J., Cijsouw T., ten Cate R., Medema J.P., Kanaar R., Franken N.A., Aten J.A. Chromatin mobility is increased at sites of DNA double-strand breaks. J. Cell Sci. 2012;125:2127–2133. doi: 10.1242/jcs.089847. [DOI] [PubMed] [Google Scholar]
- Kruhlak M.J., Celeste A., Dellaire G., Fernandez-Capetillo O., Müller W.G., McNally J.G., Bazett-Jones D.P., Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagache T., Lang G., Sauvonnet N., Olivo-Marin J.C. Analysis of the spatial organization of molecules with robust statistics. PLoS ONE. 2013;8:e80914. doi: 10.1371/journal.pone.0080914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.S., Lee K., Legube G., Haber J.E. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat. Struct. Mol. Biol. 2014;21:103–109. doi: 10.1038/nsmb.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M., Rothstein R., Mortensen U.H. Rad52 forms DNA repair and recombination centers during S phase. Proc. Natl. Acad. Sci. USA. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Zhu F., Cho Y.Y., Tang F., Zykova T., Ma W.Y., Bode A.M., Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell. 2006;23:121–132. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkle D., Douglas P., Moorhead G.B., Leonenko Z., Yu Y., Cramb D., Bazett-Jones D.P., Lees-Miller S.P. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry. 2002;41:12706–12714. doi: 10.1021/bi0263558. [DOI] [PubMed] [Google Scholar]
- Miné-Hattab J., Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 2012;14:510–517. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- Noon A.T., Shibata A., Rief N., Löbrich M., Stewart G.S., Jeggo P.A., Goodarzi A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010;12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- Renkawitz J., Lademann C.A., Jentsch S. γH2AX spreading linked to homology search. Cell Cycle. 2013;12:2526–2527. doi: 10.4161/cc.25836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riballo E., Kühne M., Rief N., Doherty A., Smith G.C., Recio M.J., Reis C., Dahm K., Fricke A., Krempler A. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Roukos V., Voss T.C., Schmidt C.K., Lee S., Wangsa D., Misteli T. Spatial dynamics of chromosome translocations in living cells. Science. 2013;341:660–664. doi: 10.1126/science.1237150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic V., Yin B., Maas N.L., Bredemeyer A.L., Carpenter A.C., Helmink B.A., Yang-Iott K.S., Sleckman B.P., Bassing C.H. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell. 2009;34:298–310. doi: 10.1016/j.molcel.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R., Xie A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013;750:5–14. doi: 10.1016/j.mrfmmm.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J., Kim S.C., Lee H.S., Kim J.K., Shon H.J., Salleh N.L., Desai K.V., Lee J.H., Kang E.S., Kim J.S., Choi J.K. Genome-wide profiles of H2AX and γ-H2AX differentiate endogenous and exogenous DNA damage hotspots in human cells. Nucleic Acids Res. 2012;40:5965–5974. doi: 10.1093/nar/gks287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C., Oswald D., Phelps D., Cam H., Pelloski C.E., Pang Q., Houghton P.J. Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res. 2013;73:3393–3401. doi: 10.1158/0008-5472.CAN-12-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A., Barton O., Noon A.T., Dahm K., Deckbar D., Goodarzi A.A., Löbrich M., Jeggo P.A. Role of ATM and the damage response mediator proteins 53BP1 and MDC1 in the maintenance of G(2)/M checkpoint arrest. Mol. Cell. Biol. 2010;30:3371–3383. doi: 10.1128/MCB.01644-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A., Conrad S., Birraux J., Geuting V., Barton O., Ismail A., Kakarougkas A., Meek K., Taucher-Scholz G., Löbrich M., Jeggo P.A. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–1092. doi: 10.1038/emboj.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu B.M., Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb. Perspect. Biol. 2013;5:a012724. doi: 10.1101/cshperspect.a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E., Zhao G.Y., Kohzaki M., Dhar P.K., Kikuchi K., Redon C., Pilch D.R., Bonner W.M., Nakano A., Watanabe M. Collaborative roles of gammaH2AX and the Rad51 paralog Xrcc3 in homologous recombinational repair. DNA Repair (Amst.) 2007;6:280–292. doi: 10.1016/j.dnarep.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Soutoglou E., Dorn J.F., Sengupta K., Jasin M., Nussenzweig A., Ried T., Danuser G., Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T., O’Driscoll M., Rief N., Iwabuchi K., Löbrich M., Jeggo P.A. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Toledo L.I., Murga M., Zur R., Soria R., Rodriguez A., Martinez S., Oyarzabal J., Pastor J., Bischoff J.R., Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Wang M., Wang H., Böcker W., Iliakis G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J. Cell. Physiol. 2005;202:492–502. doi: 10.1002/jcp.20141. [DOI] [PubMed] [Google Scholar]
- Ward I.M., Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 2001;276:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- Xie A., Puget N., Shim I., Odate S., Jarzyna I., Bassing C.H., Alt F.W., Scully R. Control of sister chromatid recombination by histone H2AX. Mol. Cell. 2004;16:1017–1025. doi: 10.1016/j.molcel.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., McCord R.P., Ho Y.J., Lajoie B.R., Hildebrand D.G., Simon A.C., Becker M.S., Alt F.W., Dekker J. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Thomas H.D., Batey M.A., Cowell I.G., Richardson C.J., Griffin R.J., Calvert A.H., Newell D.R., Smith G.C., Curtin N.J. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66:5354–5362. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Paull T.T. DNA-dependent protein kinase regulates DNA end resection in concert with Mre11-Rad50-Nbs1 (MRN) and ataxia telangiectasia-mutated (ATM) J. Biol. Chem. 2013;288:37112–37125. doi: 10.1074/jbc.M113.514398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time-lapse microscopy was performed on 4OHT-treated 53BP1-GFP DIvA using a spinning disk confocal microscope. Acquisition was started straight after 4OHT treatment, and all z stacks were acquired every 2 min. Maximum projection was computed with Image J. Full 4D movies are available upon request.
Same as Movie S1.
Same as Movie S1, except that acquisition was performed after 2 hr of 4OHT treatment and every 15 s. A magnification of 53BP1 foci ongoing clustering is presented. Related to Figure S5C bottom sequence.
Same as Movie S1, except that acquisition was performed after 2 hr of 4OHT treatment and every 3 min. Related to Figure S5C top sequence.
Same as Movie S1, except that acquisition was performed after 2 hr of 4OHT treatment and every 3 min.