

Abstract
With the high prevalence of obesity, diabetes, and other features of the metabolic syndrome in United States, nonalcoholic fatty liver disease (NAFLD) has inevitably become a very prevalent chronic liver disease and is now emerging as one of the leading indications for liver transplantation. Insulin resistance and derangement of lipid metabolism, accompanied by activation of the pro-inflammatory response and fibrogenesis, are essential pathways in the development of the more clinically significant form of NAFLD, known as nonalcoholic steatohepatitis (NASH). Recent advances in the functional characterization of bile acid receptors, such as farnesoid X receptor (FXR) and transmembrane G protein-coupled receptor (TGR) 5, have provided further insight in the pathophysiology of NASH and have led to the development of potential therapeutic targets for NAFLD and NASH. Beyond maintaining bile acid metabolism, FXR and TGR5 also regulate lipid metabolism, maintain glucose homeostasis, increase energy expenditure, and ameliorate hepatic inflammation. These intriguing features have been exploited to develop bile acid analogues to target pathways in NAFLD and NASH pathogenesis. This review provides a brief overview of the pathogenesis of NAFLD and NASH, and then delves into the biological functions of bile acid receptors, particularly with respect to NASH pathogenesis, with a description of the associated experimental data, and, finally, we discuss the prospects of bile acid analogues in the treatment of NAFLD and NASH.
Keywords: Bile acids, Bile acid receptors, Nonalcoholic steatohepatitis, Farnesoid X receptor, Transmembrane G protein-coupled receptor 5, Nonalcoholic fatty liver disease, Hepatic steatosis
Core tip: Bile acids and bile acid receptors play important roles in modulation of feature of the metabolic syndrome, hepatic steatosis, and hepatic inflammation. Development of bile acid analogues specifically targeting farnesoid X receptor and transmembrane G protein-coupled receptor 5 provide potential novel classes of drugs for the treatment of nonalcoholic steatohepatitis.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is now a very prevalent liver disease in the United States. It affects up to 20% of the United States population[1], with an estimated prevalence of 43%-60% in patients with diabetes[2], and 90% in patients with hyperlipidemia[3]. NAFLD, by definition, is macrovesicular fat accumulation in more than 5% of hepatocytes in patients who drink less than 20 g/d. NAFLD represent a spectrum of diseases ranging from simple hepatic steatosis to steatohepatitis. Simple steatosis rarely progresses to advanced fibrosis and thus does not carry an increased liver-related mortality. Nonalcoholic steatohepatitis (NASH) instead describes hepatic inflammation and hepatocyte damage within liver including lobular inflammation and hepatic ballooning in addition to macrovesicular fat. Fifteen percent to 30% of patients with NASH progresses to fibrosis, cirrhosis and cancer[4,5], leading to the need for a liver transplant. Based on the data from the United Network for Organ Sharing and Organ Procurement and Transplantation Network Registry, the percentage of patients who underwent a liver transplant for NASH has increased to 9.7% in 2009 compared to 1.2% in 2001[6]. The number of adults with NASH awaiting liver transplant has almost tripled in 2013, compared to the year 2004[7]. NASH is projected to become the leading etiology for liver transplant in the United States.
The “two hit hypothesis” and the “multiple hits hypothesis” have been proposed to explain the underlying pathogenesis of NAFLD[8,9] (Figure 1). Simple hepatic steatosis reflects the accumulation of triglyceride in the liver, as result of influx of lipids and de novo lipogenesis exceeding the export of lipids in the forms of lipoproteins. Insulin resistance has been considered a primary driving force for lipid influx by promoting the lipolysis of peripheral adipose tissue, and increasing the liver uptake of free fatty acids for de novo lipogenesis[10]. Hyperinsulinemia and hyperglycemia also inhibit fatty acid oxidation and accelerate lipogenesis[11]. Triglyceride is exported out of liver to peripheral tissues by incorporation into very-low-density lipoprotein (VLDL) carriers, and impairment of VLDL synthesis/export has been implicated in NAFLD pathogenesis[12]. Accumulation of lipids, including triglycerides and free fatty acids, as a first hit, primes the liver - making it susceptible to additional hepatotoxic insults (second or multiple hits), which then lead to hepatocyte injury, inflammation, and fibrosis. The second hit or multiple hits involve pro-inflammatory processes mediated by the gut-liver-axis with microbiota imbalance, mitochondrial dysfunction, oxidative stress pathway activation, and activation of intracellular signal such as nuclear factor κB and c-Jun N-terminal kinase (JNK) pathways[13-17].
Figure 1.
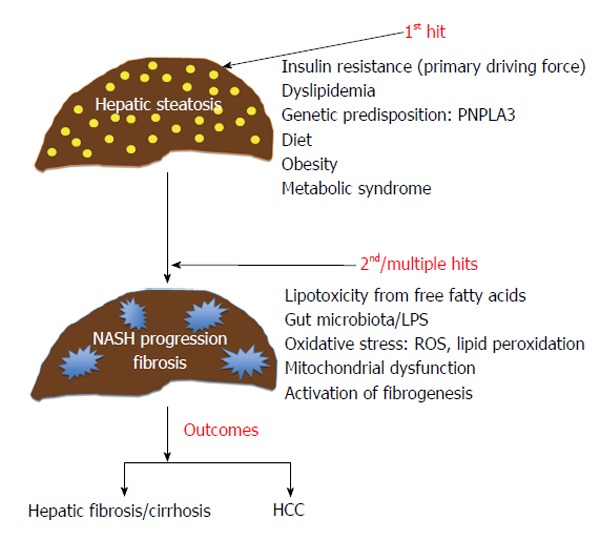
Pathogenesis of nonalcoholic steatohepatitis. Insulin resistance is considered a primary driving force for hepatic steatosis by promoting lipolysis of peripheral adipose tissue, and increasing hepatic uptake of free fatty acids for de novo lipogenesis. The second hit, or multiple hits, involve genetic predisposition such as PNPLA3, pro-inflammatory processes mediated by the gut-liver-axis with microbiota imbalance, mitochondrial dysfunction, activation of oxidative stress pathways, and induction of lipotoxicity from free fatty acids. HCC: Hepatocellular carcinoma; NASH: Nonalcoholic steatohepatitis; PNPLA3: Patatin-like phospholipase domain containing 3; LPS: Lipopolysaccharides; ROS: Reactive oxygen species.
Unraveling the pathogenesis of NAFLD has established several important drug targets in recent years. Among them are bile acid receptors including farnesoid X receptor (FXR) and transmembrane G protein-coupled receptor (TGR) 5, which play pivotal roles in regulation of metabolism, inflammation and cell proliferation. These receptors have emerged as attractive targets for drug development for the treatment of NAFLD, and are the focus of this review.
BILE ACID METABOLISM
Bile acid synthesis
Bile acids are generated from cholesterol oxidation in the liver through two major pathways: “classic pathway” also called the neutral pathway, and the “alternative pathway” also called the acidic pathway. Cholesterol 7α-hydroxylase (CYP7A1) is a rate-limiting enzyme in the classic pathway. Both of the primary bile acids, cholic acid (CA) and chenodeoxycholic acids (CDCA), are end products of the classic pathway. The alternative pathway of bile acid synthesis is initiated by sterol 27-hydroxylase (CYP27A1), an enzyme located on the inner membrane of mitochondria and widely expressed in various tissues. The alternative pathway produces oxysterols, notably 25-hydroxycholesterol and 27-hydroxycholesterol, which are important ligands in regulating inflammation, lipid metabolism, and cell proliferation.
Bile acid recycling
Once synthesized in the liver, bile acids are conjugated to glycine or taurine, excreted out of liver, and stored in the gallbladder. In response to a meal, the contraction of the gallbladder delivers bile salts to the small intestine, facilitating the digestion of dietary fat. In the gastrointestinal tract, CA and CDCA are further metabolized by intestinal microbiota to secondary bile acids: Lithocholic acid (LCA) and deoxycholic acid (DCA) by de-conjugation and dehydroxylation. DCA is unable to convert back to CA in the liver, and thus the proportion of DCA in the bile acid pool varies from 1% to 50%, depending on the level and activity of bile acid 7α-dehydroxylating gut bacteria and intestinal transient time. LCA is reabsorbed and reduced to CA in the liver. Overall, approximately 95% of bile acids are reabsorbed in the ileum and transported back to the liver via the enterohepatic circulation. Approximately 5% of bile acids are lost in the feces daily. But bile acids do not just aid in digestion and participate in the enterohepatic circulation, they also function as signaling molecules both within and outside of the liver.
FXR AND NAFLD
FXR belongs to the family of nuclear hormone receptors that regulate expression of genes involved in a wide array of biologic processes including, development, reproduction, and metabolism, and was first described in 1995[18-20]. Bile acids were subsequently identified as unique endogenous ligands for FXR at physiologic levels[21,22] in 1999. FXR is richly expressed at the ileum, and in liver parenchymal cells. It is also expressed in liver non-parenchymal cells such as endothelial cells, Kupffer cells and stellate cells at very low level. Various bile acids activate FXR in the following order of activity: CDCA > DCA > CA > LCA. The targets and effects of FXR are outlined in detail below and summarized in Figure 2.
Figure 2.
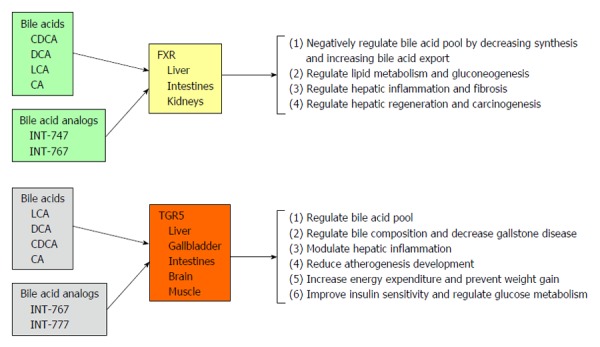
Characteristics of farnesoid X receptor and transmembrane G protein-coupled receptor 5, and their functions. CA: Cholic acid; LCA: Lithocholic acid; DCA: Deoxycholic acid; FXR: Farnesoid X receptor; TGR5: Transmembrane G protein-coupled receptor 5; CDCA: Chenodeoxycholic acids.
FXR biologic functions
Bile acid synthesis: FXR plays an essential role in the feedback regulation of bile acid biosynthesis by repression of CYP7A1, and CYP8B1, two key enzymes in bile acids synthesis. Repression of CYP7A1 is mediated by activation of the orphan nuclear receptor small heterodimer partner (SHP)[22-24], which in turn interact with liver receptor homolog (LRH-1). SHP protein blocks activities of LRH-1 that is known to positively regulate CYP7A1 expression. Mice lacking SHP (SHP-/-) failed to repress CYP7A1 in response to a specific agonist for FXR. Yet, Bile acid feeding can restore expression of CYP7A1 in SHP-null mice, indicating the existence of SHP-independent regulation pathways, as well. One of these pathways involves JNK mitogen-activated protein kinase activation[24,25] and fibroblast growth factor 19 (human FGF19, mouse FGF15)[26]. Additionally, FXR induces the expression of ATP-binding cassette transporters such as bile salt export pump, multidrug resistance protein 3 (MDR3) and multidrug resistance-associated protein 2. These transporters export bile acids from hepatocytes into bile canaliculi. Activation of FXR was also found to stimulate the expression of intestinal bile acid-binding protein at ileum, which facilitates enterohepatic recycling of bile acids[21,27].
Lipid and glucose metabolism: FXR also regulates lipid metabolism and gluconeogenesis[28,29]. FXR-null mice develop severe fatty liv er with elevated circulating plasma cholesterol, elevated triglycerides and free fatty acids. Several lipoproteins such as phospholipid transfer protein, apoC-II, apoC-III, and apoA-1 are FXR targets[30-32] and their decreased expression likely accounts for lipid derangements in FXR-null mice. FXR also regulates lipid synthesis by involving acetyl-CoA carboxylase 1 (Acc1), Acc2, Cd36, and sterol regulatory element-binding protein 1C, with the latter being a major regulator of lipogenesis via stimulation of de novo lipogenesis, and these FXR effects likely occur through activation of SHP[33] and FGF19[34]. Beyond regulation of lipid metabolism, FXR also plays an important role in glucose homeostasis. Loss of FXR in mice lead to development of impaired glucose tolerance and insulin resistance both in liver and skeletal muscles which is associated hepatic steatosis and elevated circulating free fatty acids[35]. Bile acids alter the expression of genes involved in gluconeogenesis, including phosphoenolpyruvate carboxykinase (PEPCK), glucose-6-phosphatase (G-6-Pase), and fructose-1,6-biphosphatase[36-38]. SHP activation may modulate gluconeogenesis through repression of PEPCK and G-6-Pase[38,39]. FGF15/19 appear also to be critical in glucose regulation. In the postprandial state, FGF15/19 are released from the small intestine and inhibit hepatic glucogenesis, like insulin, through dephosphorylation and inactivation of cAMP regulatory element-binding protein[40].
Hepatic inflammation and fibrosis: FXR also regulates hepatic inflammation and fibrosis[29]. FXR is expressed at very low levels on hepatic Kupffer, stellate, and endothelial cells. Porcine serum treatment or bile duct ligation (BDL) are commonly used experimental methods to induce cirrhosis in rats. Treatment of these rats with 6-ethyl chenodeoxycholic acid (6-ECDCA), an FXR ligand, prevents liver fibrosis in porcine serum-treated rats or BDL-treated rats, and decreases expression of matrix proteins including, α1-collagen, transforming growth factor β-1, αSMA, and tissue inhibitors of metalloproteinase 1 and 2[41]. Interestingly, Fickert et al[42] showed that FXR loss reduced fibrosis of the hepatic biliary tree. In the study, hepatic fibrosis was induced in wild type and FXR knock-out mice (FXR-/-) by a variety of methods, including carbon tetrachloride (CCl4) intoxication, 3,5-diethoxycarbonyl-1,4-dihydrocollidine feeding, BDL, or Schistosoma mansoni (S.m.)-infection. Only biliary-type hepatic fibrosis was reduced in FXR (-/-) mice with BDL and 3,5-diethoxycarbonyl-1,4-dihydrocollidine. FXR loss had no effect on the prevention of non-cholestatic liver fibrosis in the study.
Hepatic regeneration and carcinogenesis: FXR appears to regulate liver regeneration and carcinogenesis. CA feeding has been shown to induce liver growth and decrease mortality in mice that have undergone partial hepatectomy. This effect may involve activation of FGF15/19. Studies have shown that the protective effects with CA feeding after partial hepatectomy were significantly abolished in FGF15 (-/-) mice[43], and proliferation of hepatocytes and cholangiocytes was also noticeably reduced in CA-fed FGF15 (-/-) mice[43]. FXR (-/-) mice developed spontaneous hepatocellular carcinoma (HCC) at age > 12 mo[44,45]. And FXR had a direct effect in down-regulating a number of tumor suppressor genes such as N-myc downstream-regulated gene 2[46] and gankyrin, a proteasomal subunit that assists in degradation of a number of tumor suppressor proteins[46,47]. Interestingly, selective reactivation of intestinal FXR can restore bile acid enterohepatic circulation and protect FXR (-/-) mice from spontaneous HCC development[48].
FXR agonists in the treatment of NAFLD
6-ECDCA, also known as INT-747 or obeticholic acid (OCA), is a lipophilic bile acid derivative and a potent selective FXR activator[49]. In animal studies, it improves hepatic steatosis[50], fibrosis[42], and portal hypertension[51]. The FLINT trial[52], a phase IIB randomized, placebo-controlled trial of OCA in human NASH, demonstrated that OCA significantly improved the NAFLD activity score in all components, including steatosis, lobular inflammation, and hepatocellular ballooning, compared to placebo, establishing a clear benefit of in alleviating liver injury and inflammation in NAFLD. There was also some improvement in fibrosis score in the OCA group in the FLINT trial, as well, but the trial was not powered to detect the statistical significance in fibrosis changes. It remains to be determined whether or not OCA will resolve NASH and ameliorate advanced fibrosis, but further trials are ongoing.
TGR5 and NAFLD
TGR5 is a classic G-protein coupled cell surface receptor[53,54] that is activated by bile acids in the order of LCA > DCA > CDCA > CA[53]. In the absence of bile acid binding, TGR5 is tightly associated with a G-protein complex consisting of α, β and γ subunits. Upon binding to bile acids, TGR5 allows the release of α subunit, which in turn activates adenylyl cyclase, leading to the accumulation of cAMP and activation of protein kinase A. TGR5 is widely expressed in various tissues including the liver, gallbladder, bile ducts, adipose tissue, spleen, intestines, and kidneys. Within the liver, TGR5 is abundantly expressed in Kupffer cells and endothelial cells, but not in hepatocytes. The targets and effects of TGR5 are outlined in detail below and summarized in Figure 2.
TGR5 functions and NAFLD
Regulation of the bile acid pool: TGR5 regulates the bile acid pool. The bile acid pool is significantly reduced in TGR (-/-) mice compared to wild-type mice. TGR5 also regulates bile composition as demonstrated by an experiment showing that, when fed with lithogenic diet, TGR (-/-) mice were protected from gallstone diseases. The expression of TGR5 was shown to be present in gallbladder epithelial cells and cholangiocytes, and TGR5 activation induced bicarbonate and chloride secretion from cholangiocytes, which may account for the alteration of bile composition.
Modulation of the immune response: TGR5 also modulates immune responses of immune cells via increasing intracellular cAMP[53], and this function appears relevant to the regulation of hepatic inflammation and atherosclerosis development. TGR5 is highly expressed in monocytes and macrophages. Activation of TGR5 increases cAMP in rat alveolar macrophages and, as a result, it reduces the phagocytic activity of the macrophage and inhibits lipopolysaccharide (LPS)-induced production of pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin-1 (IL-1), IL-6 and IL-8. This finding was also demonstrated in resident hepatic macrophage Kupffer cells. Activation of TGR5 in isolated Kupffer cells causes an increase in cAMP and reduced expression of pro-inflammatory cytokines, including: TNF-α, IL-1, IL-6 and IL-8, following LPS treatment. Notably, TGR5-deficient mice are more susceptible to LPS-induced liver injury[55]. TGR5 activation also attenuates the formation of atheromatous plaque in low-density lipoprotein receptor knockout mice (LDL-/-), a commonly used murine model for atherosclerosis studies that have functioning TGR5 (LDL-/-; TGR5+/+). This attenuation of plaque formation was achieved through decreases in intra-plaque inflammation and macrophage activation[56]. Recent study has also showed that a TGR5 agonist increased the production of nitric oxide (NO) in endothelial cells, a key anti-atherogenic molecule. This suggests that NO might be one of downstream effectors of TGR5 signaling.
Energy expenditure and metabolism: TGR5 enhances energy expenditure and mitigates obesity and insulin resistance in obese mice. TGR5 is expressed in human brown adipocytes and skeletal myocytes. In brown adipose and skeletal muscle, interactions between bile acids and TGR5 promote the expression of cAMP-dependent 2-iodothyronine de-iodinase (D2), which converts inactive thyroxine (T4) to active 3,5,3-tri-iodothyronine (T3), a major hormone in increasing basal metabolism and thus, inducing energy expenditure[57]. TGR5 signaling also regulates glucose homeostasis[58]. TGR5 is expressed on enteroendocrine L-cells, and activation of TGR5 on L-cells modulates mitochondrial oxidative phosphorylation and alters ATP/ADP ratio. This leads to the release of glucagon like peptide-1 (GLP-1) from L-cells. GLP-1 further stimulates insulin secretion from pancreas and maintains glucose homeostasis.
TGR5 agonists in the treatment of NALFD
Given the aforementioned biological effects of TGR5, TGR5 becomes an enticing potential target for NASH therapeutics. A specific CA derivative, 6α-ethyl-23(S)-methylcholic acid (INT-777) has been developed as a selective TGR5 agonist[59]. Treatment of high-fat fed mice with INT-777 increased energy expenditure and attenuated both weight gain and expansion of fat pad mass[58]. In addition, INT-777 treatment reduced hepatic steatosis and improved liver enzyme levels without evidence of hepatic fibrosis[58]. INT-777 treatment has also been shown to improve insulin sensitivity, likely through the release of GLP-1 in the intestines, in both diet-induced obese mice and in genetically obese mice that have a leptin receptor gene mutation (db/db), which is a well-established model of obesity and diabetes[58]. This effect was blunted in TGR5-/- mice, indicating the specificity of INT-777 treatment in targeting TGR5. With all these features, TGR5 agonists such as INT-777 are very attractive treatment candidates for NASH and other features of the metabolic syndrome[60].
TARGETING BOTH FXR AND TGR5 IN THE TREATMENT OF NAFLD
INT-767, the 23-sulphate derivative of OCA, is a dual FXR/TGR5 agonist[61]. INT-767 has been demonstrated to induce FXR-dependent lipid uptake by adipocytes, mobilizing lipid from the circulation and the liver to peripheral adipose tissue. INT-767 also promotes TGR5-dependant GLP-1 release. Treatment of obese mice with INT-767 significantly decreased total plasma cholesterol and triglyceride levels[61], and improved the histological features of NASH in these mice[62]. These effects have been postulated to be due to INT-767-mediated alterations in the phenotypes of intrahepatic macrophage populations and modulation of cytokine production[62]. Uniquely, INT-767, but not INT-777 or INT-747, ameliorates hepatic injury in MDR2 (-/-) mice, a model for chronic cholangiopathy. This hepatoprotective effect is manifested by a reduction in bile acid synthesis and an increase in bile flow and biliary HCO3- output[63].
CONCLUSION
Bile acids and bile acid receptors have pluripotent functions in energy expenditure, regulation of lipids and glucose metabolism, modulation of hepatic inflammation, fibrosis, regeneration, and carcinogenesis. These effects translate into attractive therapeutic targets for NASH and to improve metabolic profiles, ameliorate hepatic injury, and halt hepatic fibrosis. One currently very promising drug is OCA, but additional new drugs are expected in the not-too-distant future that will target the pathways in NASH pathogenesis.
Footnotes
P- Reviewer: Garcia-Ruiz I, Kayadibi H, Tomizawa M S- Editor: Kong JX L- Editor: A E- Editor: Liu SQ
Conflict-of-interest statement: The authors have no conflicts of interest regarding this manuscript submission to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 17, 2015
First decision: September 2, 2015
Article in press: November 25, 2015
References
- 1.Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology. 1990;12:1106–1110. doi: 10.1002/hep.1840120505. [DOI] [PubMed] [Google Scholar]
- 2.Williamson RM, Price JF, Glancy S, Perry E, Nee LD, Hayes PC, Frier BM, Van Look LA, Johnston GI, Reynolds RM, et al. Prevalence of and risk factors for hepatic steatosis and nonalcoholic Fatty liver disease in people with type 2 diabetes: the Edinburgh Type 2 Diabetes Study. Diabetes Care. 2011;34:1139–1144. doi: 10.2337/dc10-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5:1544–1560. doi: 10.3390/nu5051544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angulo P. Long-term mortality in nonalcoholic fatty liver disease: is liver histology of any prognostic significance? Hepatology. 2010;51:373–375. doi: 10.1002/hep.23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 6.Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249–1253. doi: 10.1053/j.gastro.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 7.Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, Ahmed A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology. 2015;148:547–555. doi: 10.1053/j.gastro.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 8.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 9.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 10.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–152. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azzout-Marniche D, Bécard D, Guichard C, Foretz M, Ferré P, Foufelle F. Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem J. 2000;350 Pt 2:389–393. [PMC free article] [PubMed] [Google Scholar]
- 12.Fon Tacer K, Rozman D. Nonalcoholic Fatty liver disease: focus on lipoprotein and lipid deregulation. J Lipids. 2011;2011:783976. doi: 10.1155/2011/783976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarzian A. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28:1026–1033. doi: 10.1111/j.1478-3231.2008.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giguère V, Yang N, Segui P, Evans RM. Identification of a new class of steroid hormone receptors. Nature. 1988;331:91–94. doi: 10.1038/331091a0. [DOI] [PubMed] [Google Scholar]
- 20.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, Noonan DJ, Burka LT, McMorris T, Lamph WW, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 21.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 23.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, Wei P, Heyman RA, Karin M, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 25.Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, Shan B, Russell DW, Schwarz M. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell. 2002;2:713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang ST, Urizar NL, Moore DD, Henning SJ. Bile acids regulate the ontogenic expression of ileal bile acid binding protein in the rat via the farnesoid X receptor. Gastroenterology. 2002;122:1483–1492. doi: 10.1053/gast.2002.32982. [DOI] [PubMed] [Google Scholar]
- 28.Claudel T, Staels B, Kuipers F. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol. 2005;25:2020–2030. doi: 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- 29.Schreuder TC, Marsman HA, Lenicek M, van Werven JR, Nederveen AJ, Jansen PL, Schaap FG. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298:G440–G445. doi: 10.1152/ajpgi.00322.2009. [DOI] [PubMed] [Google Scholar]
- 30.Urizar NL, Dowhan DH, Moore DD. The farnesoid X-activated receptor mediates bile acid activation of phospholipid transfer protein gene expression. J Biol Chem. 2000;275:39313–39317. doi: 10.1074/jbc.M007998200. [DOI] [PubMed] [Google Scholar]
- 31.Claudel T, Inoue Y, Barbier O, Duran-Sandoval D, Kosykh V, Fruchart J, Fruchart JC, Gonzalez FJ, Staels B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology. 2003;125:544–555. doi: 10.1016/s0016-5085(03)00896-5. [DOI] [PubMed] [Google Scholar]
- 32.Claudel T, Sturm E, Duez H, Torra IP, Sirvent A, Kosykh V, Fruchart JC, Dallongeville J, Hum DW, Kuipers F, et al. Bile acid-activated nuclear receptor FXR suppresses apolipoprotein A-I transcription via a negative FXR response element. J Clin Invest. 2002;109:961–971. doi: 10.1172/JCI14505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyata M, Sakaida Y, Matsuzawa H, Yoshinari K, Yamazoe Y. Fibroblast growth factor 19 treatment ameliorates disruption of hepatic lipid metabolism in farnesoid X receptor (Fxr)-null mice. Biol Pharm Bull. 2011;34:1885–1889. doi: 10.1248/bpb.34.1885. [DOI] [PubMed] [Google Scholar]
- 35.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Fabiani E, Mitro N, Gilardi F, Caruso D, Galli G, Crestani M. Coordinated control of cholesterol catabolism to bile acids and of gluconeogenesis via a novel mechanism of transcription regulation linked to the fasted-to-fed cycle. J Biol Chem. 2003;278:39124–39132. doi: 10.1074/jbc.M305079200. [DOI] [PubMed] [Google Scholar]
- 37.Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, Kuipers F, Staels B. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–898. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 38.Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, Fukamizu A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. 2004;279:23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 39.Kim JY, Kim HJ, Kim KT, Park YY, Seong HA, Park KC, Lee IK, Ha H, Shong M, Park SC, et al. Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/Foxa transactivation via inhibition of its DNA binding. Mol Endocrinol. 2004;18:2880–2894. doi: 10.1210/me.2004-0211. [DOI] [PubMed] [Google Scholar]
- 40.Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, Suino-Powell K, Xu HE, Gerard RD, Finck BN, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1α pathway. Cell Metab. 2011;13:729–738. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, Orlandi S, Pellicciari R, Morelli A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology. 2004;127:1497–1512. doi: 10.1053/j.gastro.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 42.Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, Halilbasic E, Stöger U, Arrese M, Pizarro M, Solís N, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol. 2009;175:2392–2405. doi: 10.2353/ajpath.2009.090114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, Vespasiani-Gentilucci U, Morini S, Vicente E, Concepcion AR, et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut. 2013;62:899–910. doi: 10.1136/gutjnl-2012-302945. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Ge X, Heemstra LA, Chen WD, Xu J, Smith JL, Ma H, Kasim N, Edwards PA, Novak CM. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol Endocrinol. 2012;26:272–280. doi: 10.1210/me.2011-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28:940–946. doi: 10.1093/carcin/bgl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deuschle U, Schüler J, Schulz A, Schlüter T, Kinzel O, Abel U, Kremoser C. FXR controls the tumor suppressor NDRG2 and FXR agonists reduce liver tumor growth and metastasis in an orthotopic mouse xenograft model. PLoS One. 2012;7:e43044. doi: 10.1371/journal.pone.0043044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang Y, Iakova P, Jin J, Sullivan E, Sharin V, Hong IH, Anakk S, Mayor A, Darlington G, Finegold M, et al. Farnesoid X receptor inhibits gankyrin in mouse livers and prevents development of liver cancer. Hepatology. 2013;57:1098–1106. doi: 10.1002/hep.26146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Degirolamo C, Modica S, Vacca M, Di Tullio G, Morgano A, D’Orazio A, Kannisto K, Parini P, Moschetta A. Prevention of spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice by intestinal-specific farnesoid X receptor reactivation. Hepatology. 2015;61:161–170. doi: 10.1002/hep.27274. [DOI] [PubMed] [Google Scholar]
- 49.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 50.Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–784. doi: 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verbeke L, Farre R, Trebicka J, Komuta M, Roskams T, Klein S, Elst IV, Windmolders P, Vanuytsel T, Nevens F, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59:2286–2298. doi: 10.1002/hep.26939. [DOI] [PubMed] [Google Scholar]
- 52.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 54.Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 2002;520:97–101. doi: 10.1016/s0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- 55.Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor κ light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology. 2011;54:1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, Rizzo G, Gioiello A, Adorini L, Pellicciari R, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–757. doi: 10.1016/j.cmet.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 58.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, et al. Discovery of 6alpha-ethyl-23(S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem. 2009;52:7958–7961. doi: 10.1021/jm901390p. [DOI] [PubMed] [Google Scholar]
- 60.Tiwari A, Maiti P. TGR5: an emerging bile acid G-protein-coupled receptor target for the potential treatment of metabolic disorders. Drug Discov Today. 2009;14:523–530. doi: 10.1016/j.drudis.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 61.Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, Orlandi S, Sadeghpour B, Wang XX, Jiang T, et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol. 2010;78:617–630. doi: 10.1124/mol.110.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, Pruzanski M, Adorini L, Golden-Mason L, Levi M, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288:11761–11770. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Baghdasaryan A, Claudel T, Gumhold J, Silbert D, Adorini L, Roda A, Vecchiotti S, Gonzalez FJ, Schoonjans K, Strazzabosco M, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO-3 output. Hepatology. 2011;54:1303–1312. doi: 10.1002/hep.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]