

Abstract
The sequence of events associated with the development of gastric cancer has been described as “the gastric precancerous cascade”. This cascade is a dynamic process that includes lesions, such as atrophic gastritis, intestinal metaplasia and dysplasia. According to this model, Helicobacter pylori (H. pylori) infection targets the normal gastric mucosa causing non-atrophic gastritis, an initiating lesion that can be cured by clearing H. pylori with antibiotics or that may then linger in the case of chronic infection and progress to atrophic gastritis. The presence of virulence factors in the infecting H. pylori drives the carcinogenesis process. Independent epidemiological and animal studies have confirmed the sequential progression of these precancerous lesions. Particularly long-term follow-up studies estimated a risk of 0.1% for atrophic gastritis/intestinal metaplasia and 6% in case of dysplasia for the long-term development of gastric cancer. With this in mind, a better understanding of the genetic and epigenetic changes associated with progression of the cascade is critical in determining the risk of gastric cancer associated with H. pylori infection. In this review, we will summarize some of the most relevant mechanisms and focus predominantly but not exclusively on the discussion of gene promoter methylation and miRNAs in this context.
Keywords: Helicobacter pylori, Methylation, Gastric cancer, Epigenetics, MicroRNA
Core tip: Helicobacter pylori infection increases the risk of developing gastric cancer. Intestinal type gastric cancer is characterized by a histological cascade in which aberrant methylation of CpG islands and deregulation of microRNAs are observed. An exacerbated host response and bacterial virulence factors contribute to these epigenetic changes by enhancing DNA methyl transferase activity via nitric oxide production and silencing of tumor suppressor genes and miRNAs. Interestingly, methylated Reprimo DNA is detectable in blood samples and is potentially useful as an early detection marker. Finally, also the role of gamma glutamyl transpeptidase related mechanisms in the loss of the anti-apoptotic protein Survivin and gastric carcinogenesis is discussed.
PRECANCEROUS CASCADE OF GASTRIC CANCER
Helicobacter pylori (H. pylori)-is a Gram negative bacteria that colonizes the gastric epithelium of more than 50% of the adult population worldwide and is responsible for 75 % of all gastric cancer cases[1]. Chronic infection with this pathogen is an ethiological agent responsible for gastric pathologies such as chronic gastritis, peptic ulcer, mucosa-associated lymphoid tissue (MALT) lymphoma and increased risk of developing gastric adenocarcinoma[2], the second leading cause of cancer-related deaths worldwide[3]. Histologically, gastric adenocarcinoma is divided into two sub-types following Lauren’s classification[4], termed Diffuse and Intestinal-type gastric cancer. For both types, a strong correlation with H. pylori-associated inflammation exists[5]. No identifiable precursor lesions are associated with the development of diffuse type gastric cancer; instead, this type of cancer appears to be closely linked to host genetic factors[6]. Alternatively, intestinal type gastric cancer follows a well-defined histological sequence of progression, initiating as chronic gastritis followed by atrophy, intestinal metaplasia, dysplasia and finally terminating in intestinal type gastric cancer[7,8].
One of the proposed mechanisms held responsible for initiation of the atrophic gastritis/intestinal metaplasia/dysplasia/carcinoma sequence, also referred to as the precancerous cascade of gastric cancer, is the loss of the specialized parietal cells, due to alterations in cellular cross-talk and cell differentiation that are crucial for gastric epithelial precursor cell maturation and the coordination of cell migration in the gastric pits[7]. In this local inflammatory environment, bacterial factors altering oncogenic signaling pathways are crucial to propagate the carcinogenic sequence of events. However, bacterial eradication with antibiotics can reverse metaplastic and dysplastic lesions, suggesting that the differentiation/transformation in the gastric mucosa depends on the local environment and cannot be ascribed exclusively to to genetic alterations[7]. Additionally, H. pylori alters the expression of tumor suppressor genes by epigenetic deregulation. Consistent with this notion, methylation of such promoter regions is frequently observable in vivo during H. pylori infection[9,10]. In the following sections, evidence is summarized describing how bacteria-host cell interactions trigger epigenetic changes crucial to understanding the process of H. pylori-induced gastric carcinogenesis.
H. PYLORI-INDUCED INFLAMMATION, EPIGENETICS AND GASTRIC CANCER
Persistent inflammation is a prevalent contributing factor to the development of many types of cancer[11-13]. For instance, infection with bacterial and viral agents is known to promote cancer in different epithelia[14]. Among the many potentially relevant intermediates produced during inflammation are molecules, such as cytokines, chemokines, free radicals (ROS and NOS), prostaglandins, growth factors and matrix metalloproteinases (Figure 1). These induce different epigenetic changes, including DNA methylation and histone modifications, which contribute to several steps important in tumorigenesis and progression involving leukocyte recruitment, neo-angiogenesis, proliferation, survival, invasion, and finally metastasis of tumor cells[13,15]. Specifically, H. pylori-induced gastric carcinogenesis has been associated with chronic inflammation, characterized by infiltration of neutrophils and macrophages to the gastric epithelium, which favor the accumulation of pro-inflammatory cytokines and reactive oxygen/nitrogen species (ROS/RNS). Accordingly, host factors exacerbating inflammatory responses are also responsible for the final outcome. For instance, polymorphisms present in IL-8, TNF-α and IL-1β promoter regions are associated with more severe signs of inflammation and an increased risk of developing gastric cancer[16-19]. H. pylori-associated chronic inflammation is linked to silencing of tumor suppressor genes via epigenetic modification[20]. Methylation of CpG islands in the promoter regions of tumor suppressor genes and deregulation of some onco-miRs have emerged as highly relevant players in the context of understanding H. pylori-linked gastric carcinogenesis.
Figure 1.
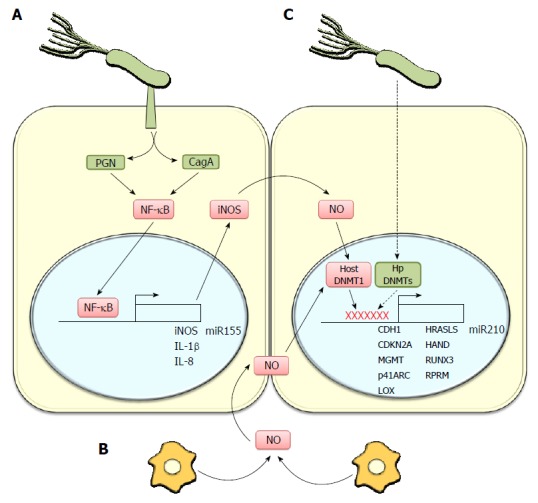
Helicobacter pylori-linked inflammation promotes methylation of genes associated with tumor suppression. A: Helicobacter pylori (H. pylori) induces, via injection of the virulence factors CagA and peptidoglycan (PGN), NF-κB activation, promoting a pro-inflammatory response that increases the expression of cytokines (IL-1β, IL-8) and inducible nitric oxide synthase (iNOS). The latter generates nitric oxide (NO) a mediator of inflammation. Furthermore, the H. pylori-linked activation of NF-κB promotes miR-155 expression, a microRNA associated with gastric pathologies; B: In addition to the direct effects of H. pylori on gastric epithelial cells, the infection activates macrophages, which increase NO production and NO levels in gastric epithelial cells. NO can activate DNA methyltransferase 1 (host DNMT1) to promote DNA methylation in several gene promoter regions; C: Also H. pylori DNA Methyltransferases (Hp DNMTs) may directly promote host DNA methylation in the promoter regions of several genes thought to function as tumor suppressors in gastric cancer. Moreover, infection is associated with promoter methylation and decreased expression of miR210, a miRNA which targets mRNAs implicated in stem cell survival, stalling of the DNA repair system, induction of angiogenesis and cellular differentiation, among others.
Importantly, studies in animal models of H. pylori-associated carcinogenesis further support this hypothesis. For instance, a study in the Mongolian gerbil model, demonstrated that H. pylori infection promotes DNA methylation as a consequence of chronic inflammation[21]. Interestingly, bacterial eradication did not decrease significantly overall DNA methylation in the gastric epithelium. However, upon suppression of inflammatory responses in gerbils treated with Cyclosporin A, the increased level of methylation was blocked without affecting bacterial colonization[21]. Following a similar rationale, the same authors described that prevention of DNA methylation using 5-aza-2-deoxycytidine decreased the incidence of gastric cancer induced by H. pylori infection[22]. However, in this study development of gastric cancer was not completely prevented, suggesting H. pylori could promote gastric cancer via additional mechanisms beyond aberrant DNA methylation, perhaps by favoring the development of genetic mutations. Likewise, a clinical study using human gastric samples showed that higher levels of DNA methylation in some genes typically hypermethylated in gastric cancer correlated with more severe gastric inflammation and more advanced precancerous lesions[23].
The effect of H. pylori induced-inflammation on epigenetic DNA modifications has also been observed in vitro. Katayama and colleagues[24] showed that H. pylori induces iNOS expression and NO production in macrophages, and the co-culture of macrophages and gastric cells promotes RUNX3 methylation in the gastric cells, without a requirement for direct interaction between gastric cells and the bacteria[24]. Alternatively, Huang et al[25] showed that E-cadherin gene methylation occurred in a manner independent of immune cells, because the direct interaction between H. pylori and gastric cells increased methylation of this gene. Furthermore, DNA methyl transferase (DNMT) activity was enhanced by increasing NO levels due to iNOS induction in host cells triggered by IL-1β secretion. Moreover, the participation of interleukin-1β in H. pylori-induced gastric inflammation and DNA methylation was confirmed using a receptor type 1 knockout (IL-1R1-/-) mouse model in which gastritis, NO production and methylation levels were reduced[26]. Despite some discrepancies between these studies, both point towards the importance of the pro-inflammatory molecule NO as a key mediator in the H. pylori-induced methylation of genes relevant to the genesis of gastric cancer. In agreement with this conclusion, H. pylori infection of iNOS-/- mice lead to a decreased incidence of gastric cancer compared with wild type controls[27].
DNA DAMAGE AND H. PYLORI INFECTION
During gastric cancer progression and similar to other types of cancer, chronic inflammation leads to epigenetic changes characterized by the hypermethylation of driver tumor-suppressor genes, as well as passenger genes, and the hypomethylation of repetitive DNA sequences generating genomic instability[20,28]. Consistent with these observations, dietary supplementation with folic acid (a methyl donor) prevented the decrease in global DNA methylation as well as gastric dysplasia and mucosal inflammation observed during H. pylori-associated carcinogenesis in mice[29]. However, for an important proportion of gastric cancers mutations in tumor suppressor genes are also detected, indicating that this type of cancer originates not only as an epigenetic disease. For instance, mutations, particularly in genes like TP53, APC, CTNNB1, CDH1 and KRAS are frequently observed in gastric cancer[30].
Inflammatory processes initiated in response to H. pylori infection are accompanied by the formation of reactive oxygen and nitrogen species (ROS/NOS), predominantly by activated neutrophils, macrophages and gastric cells in response to bacterial factors[31,32] (Figure 1). As a consequence, elevated levels of oxidized proteins and fragmented DNA are detected in the gastric mucosa[31]. Nitric oxide produced by inducible oxide synthase (iNOS) increases early in the infected mucosa and, as a consequence, DNA adducts, such as 8-nitroguanine and 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxo-dG or 8-OHdG) are readily detected in humans and animal models[33,34]. Another mechanism that contributes to mutagenesis of host DNA is the H. pylori-induced aberrant NF-κB-dependent expression of activation-induced cytidine deaminase (AID), an enzyme that functions as a DNA- and RNA-editing enzyme in response to H. pylori infection[35]. Upregulation of AID favored nucleotide alterations in the TP53 tumor suppressor gene in gastric cells in vitro, and similar alterations were also detected in the gastric mucosa of infected patients[35,36]. Studies in animal models indicate that oxidative stress is an important contributing factor to gastric carcinogenesis. For instance, nutritional supplementation with sulphoraphane, a natural compound and activator of the antioxidant transcriptional factor Nrf2, was shown to protect mice against gastric pathologies induced by H. pylori infection[37]. Furthermore, similar results were obtained upon supplementation with antioxidants like ascorbic acid in humans[38]. A possible explanation for these observations is that ROS/NOS are responsible for methylation of antioxidant enzyme genes[20]. Also, it is important to consider that ROS accumulation produces directly epigenetic changes. For instance, free radical adducts have been shown to alter DNA cytosine methylation[39] and NO can enhance DNMT activity[25].
H. PYLORI VIRULENCE FACTORS ASSOCIATED WITH INFLAMMATORY RESPONSES AND EPIGENETIC CHANGES IN THE GASTRIC MUCOSA
The experiments in Mongolian gerbils infected with H. pylori showing that the presence of the immunosuppressor drug Cyclosporine A reduces the ability of H. pylori to induce changes in the methylation status in gastric cells would suggest that H. pylori is not directly responsible for triggering aberrant methylation and that these alterations may be attributed to the inflammatory response. However, this point is still controversial because the H. pylori genome encodes a large number of cytosine and adenosine-specific type II DNA methyl transferases that would be expected to eventually methylate the host DNA because no additional factors are required for enzymatic activity. Relevant examples in this context is m6A or m4C modifications that normally are not considered in current studies[40,41]. Also, epidemiological and basic research data all point towards a number of bacterial factors as being responsible for the observed gastric inflammation, deregulation of cellular signaling pathways and cancer. Some 60% of Western H. pylori isolates bear a cag pathogenicity island (cagPAI)[11], which encodes among other components a type IV secretion system (T4SS) that is able to transfer macromolecules, peptidoglycans and DNA into the host cells[42] (Figure 1). The presence and functionality of the cagPAI correlates very strongly with the development of peptic ulcer and gastric cancer[43]. Interestingly, NF-κB activation is found to be dependent on an intact T4SS secretion system[44]. For instance, one study showed that peptidoglycans injected by the T4SS apparatus were responsible for NF-κB activation via the intracellular pathogen recognition receptor NOD1[45].
As mentioned previously, Mongolian gerbils infected with H. pylori undergo histological changes similar to those observed in human gastric carcinogenesis[46]. In a recent study, the inactivation of the peptidoglycan deacetylase (PgdA) enzyme in a particularly carcinogenic H. pylori strain (H. pylori 7.13, isolated following animal passage)[47], showed that this peptidoglycan modification enhanced the pro-inflammatory activity of this molecule[48]. However, it should be noted that this observation is controversial, because in another study PgdA inactivation contributed to bacterial survival by mitigating the immune response[49]. Together, these studies demonstrate once again the relevance of T4SS functionality in the promotion of inflammation and gastric cancer, although the precise contribution of peptidoglycan-dependent activation of the intracellular receptor NOD1 to aberrant methylation requires further investigation.
On the other hand, the T4SS is also implicated in the injection of another important carcinogenic factor, the immuno-dominant cytotoxin CagA (cytotoxin associated gene A), which is perhaps the best characterized factor in H. pylori-associated gastric cancer development[50] (Figure 1) Accordingly, patients infected with CagA+ strains develop more pronounced inflammation and higher levels of IL-8 and IL-1β[51]. CagA (125-145 KDa) is also encoded in the cagPAI and is present in 60% of H. pylori isolates. After bacterial adhesion, this factor is injected into host cells by the T4SS[52,53]. Once attached to the inner leaflet of the plasma membrane, multimeric CagA is phosphorylated by c-Src and c-Abl kinases on tyrosine residues present in the Glu-Pro-Ile-Tyr-Ala (EPIYA)- motifs of the C-terminal region of the protein[52,53]. Four different EPIYA motifs (A, B, C and D) exist, which determine the genetic variability of CagA due to differences in the number and the sequences of these motifs. In particular, the D motif is exclusively found in East-Asian CagA+ strains, where its presence is considered a major risk for developing gastric cancer[54]. The oncogenic activity of CagA has been demonstrated in a variety of settings where sustained expression of this factor promotes primary gastric cell immortalization[55], intestinal trans-differentiation[8], epithelial-mesenchymal transition[56], loss of cell-cell adhesion[57], extracellular matrix remodeling[58], metastasis[59] and gastrointestinal and hematopoietic neoplasms in mouse models[60]. Signaling pathways may be altered by CagA activity both in phosphorylation-dependent and -independent ways[61]. Phosphorylation-dependent downstream signaling processes include the recruitment of Src homology 2 (SH2)-domain containing molecules[62]. The ensuing events perturb the actin cytoskeleton, and in doing so trigger elongation and scattering of infected host cells in vitro, a morphological change referred to as the “hummingbird phenotype”[63]. Phosphorylation-independent signaling induces pro-inflammatory events, mitogenic responses and disruption of cell-cell junctions[62]. Interestingly, the Grb2 adapter can interact with CagA in a phosphorylation-dependent and -independent manner. Furthermore, transgenic expression of CagA mimics the eukaryotic Grb2-associated adaptor protein in Drosophila melanogaster[64]. This interaction promotes Ras activation and in turn stimulates the Raf-MEK-ERK signalling cascade. Particularly, this axis appears to be involved in the CagA phosphorylation-independent persistent activation of transcription factors, such as NF-κB (inflammation)[65-67]. Interestingly, in infected patients, the presence of CagA+ strains is associated with increased levels in the methylation of certain promoters when compared to CagA- infections[68]. Similar observations have also been reported in vitro. For instance, ectopic CagA expression can promote silencing of let-7 microRNA by enhancing histone and DNA methylation[69].
The cytotoxin VacA, another potentially relevant secreted bacterial factor, induces vacuole formation in cells treated with purified or recombinant forms of the protein[70,71]. Initially, its activity was associated with apoptosis of gastric cells[72], the inhibition of T and B cell proliferation[73] and induction of autophagy[74] in vitro. Despite the many functions attributed to VacA in vitro, their relevance in vivo is questionable[75]. Of note, vacuolization of cells has been observed in human biopsies and oral administration of partially purified toxin produces mucosal damage in mice[76]. However, infection of gnotobiotic piglets and Mongolian gerbils with isogenic H. pylori mutants lacking VacA causes indistinguishable degrees of gastritis in both cases[77]. Despite such results, epidemiology data indicate that the presence of more active variants of the toxin is associated with increased inflammation, peptic ulcers and even cancer[78,79]. Also, a group recently demonstrated that an H. pylori strain bearing a more active VacA induced metaplasia in mice[80]. Furthermore, VacA alleles with elevated activity are associated with enhanced secretion of pro-inflammatory cytokines such as TNF-α, MIP-1α, IL-1β, IL-8, IL-10 and IL-13 in vitro and expression of the s1m1 allele correlates with increased inflammation in the gastric mucosa[76,81,82]. In summary, it remains unclear whether VacA effects are relevant during early steps of carcinogenesis in promoting gastric epithelial damage or whether this factor promotes aberrant epigenetic changes during gastric progression and further research is required to resolve this issue.
THE EPIGENETIC BASIS OF THE GASTRIC PRECANCEROUS CASCADE
The control of gene expression via epigenetic mechanisms, including DNA methylation and microRNAs, are important regulatory events that modulate all pathways in the cellular network[83]. Moreover, these epigenetic modifications are considered excellent candidates to explain how environmental factors impact on the genome and cell function and increase the risk of cancer development[84].
DNA methylation in gastric cancer and the precancerous cascade
DNA methylation, a process whereby cytosine bases are modified with a methyl group in the 5’ position when they are followed by a guanine in the DNA sequence, is increasingly considered to be of great importance in gastric carcinogenesis[85]. Currently, a list of gastric cancer genes is emerging that are altered by DNA methylation and considered important in the progression of the cascade[86]. However, only few reports address the role of DNA hypermethylation in the gastric precancerous cascade. Importantly, higher methylation levels are detected in intestinal metaplasia than in atrophic gastritis samples[87,88]. However, no differences were observed in the methylation status between intestinal metaplasia and dysplasia. Further studies have shown methylation in multiple genes in H. pylori-induced chronic gastritis in comparison with healthy donors[89,90] suggesting that H. pylori infection potently induces aberrant DNA methylation.
As mentioned above, aberrant DNA methylation can be considered as a mechanism to explain how environmental factors alter the susceptibility of individuals to gastric cancer[84]. To support this idea, Chan et al[89] and Leung et al[91] evaluated changes in DNA methylation of the promoter region of the CDH-1 gene before and after the eradication of H. pylori, and in both studies complete regression of DNA methylation after H. pylori eradication was observed.
Genes modified by methylation upon H. pylori infection
Genes linked to intercellular junctions: As indicated above, extensive evidence is available linking gastric cancer to the silencing of the CDH1 gene by aberrant methylation. E-cadherin is a well-established tumor suppressor and represents an important component of adherent junctions[92]. A number of studies have pointed to the existence of a connection between the H. pylori infection status and CDH1 methylation in pre-cancer lesions. For instance, H. pylori infection and aberrant DNA methylation of CpG islands in the CDH1 promoter correlated in dyspeptic patients[89]. Perri et al[93] confirmed this observation, demonstrating that the CDH-1 promoter is also hypermethylated in gastritis patients infected with H. pylori, and that this was reduced following bacterial eradication. Interestingly, an in vitro study showed that H. pylori infection augmented E-cadherin promoter methylation via activation of the IL-1β receptor, iNOS induction and subsequently increased DNMT activity as a consequence of NO production[25] (Figure 1).
Vezatin (VEZT), another Adherent junction protein, is also silenced by gene promoter hypermethylation and is proposed to represent a novel tumor suppressor in gastric cancer[94]. As for CDH-1, the VEZT promoter is found to be hypermethylated in biopsies from H. pylori-positive chronic gastritis patients when compared with the non-infected control group. Additionally, an in vitro study demonstrated that H. pylori infection of the non-transformed gastric epithelial GES-1 cell line was associated with VEZT promoter methylation and a reduction in Vezatin mRNA levels was also observed[94]. Aberrant methylation of the VEZT promoter is also observed in other types of cancer, and re-expression of the protein reduces malignancy of these cells[94], indicating that Vezatin might be an important target during H. pylori-associated aberrant methylation and gastric carcinogenesis.
Connexin 32 (Cx32) and Connexin 43 (Cx43) are structural components of gap junctions between epithelial cells[95]. Wang and colleagues demonstrated that expression of these connexins was decreased in gastric pre-neoplastic lesions associated with H. pylori infection when compared to non-infected individuals and that these changes were closely associated with methylation in the respective promoter regions. Furthermore, hypermethylation of Cx32 and Cx43 promoters increased during the progression from pre-neoplasic to neoplastic lesions in infected patients[96].
Genes linked to cell cycle regulation: CDKN2A, another important tumor suppressor gene that is normally methylated in gastric cancer, encodes the p16 (INK4A) protein, which is involved in cell cycle arrest in the G1 phase[92]. Maekita et al[90] showed that methylation of this promoter was significantly elevated in the mucosa of H. pylori-positive compared with H. pylori-negative healthy volunteers[90]. Additionally, another study demonstrated that methylation levels of CDKN2A in the gastric mucosa did not differ from those observed in intestinal metaplasia patients, indicating that CDKN2A promoter methylation is an early event during H. pylori-associated gastric carcinogenesis. Furthermore, eradication of H. pylori infection reverted the aberrant methylation status of this gene[93].
Genes linked to DNA repair: The gene hMHL-1 (the human homolog of E. coli MutL), encoding a DNA repair protein in humans, is also hypermethylated in the promoter region following H. pylori gastric infection. However, this event appears to occur in later stages of cancer progression, as revealed in a study of intestinal metaplasia lesions[93]. In a similar study, higher levels of CpG methylation were observed for the promoter of a gene encoding another DNA repair protein, O6-methylguanine DNA methyltransferase (MGMT), in the gastric mucosa of gastritis patients compared to control patients. Following H. pylori eradication, methylation was reduced from 70% to 48% of the cases, coincident with significantly reduced CpG methylation and increased MGMT expression. Furthermore, in this same study, the authors demonstrated that MGMT methylation was remarkably higher in those patients infected with CagA bearing strains[68]. A recent study showed that hMHL-1 and MGMT promoters are hypermethylated in biopsies from H. pylori-associated chronic gastritis adult patients, but not in biopsy samples from infected children, and that hypermethylation was associated with H. pylori infection in the case of the MGMT promoter[97].
Genes linked to inflammation: TFF-2 gene encodes the Trefoil factor 2 protein, involved in wound healing and modulating the inflammatory response in the stomach[98,99]. Chronic H. pylori infection was shown to enhance promoter methylation of TFF-2 gene in the gastric mucosa. Furthermore, methylation of the TFF2 promoter is an early event that increases throughout gastric tumor progression[100]. On the other hand, the pro-inflammatory enzyme cycloxygenase-2 (COX-2) has been described to be hypermethylated in gastritis associated with H. pylori infection and bacterial eradication reverts completely the methylation[93]. COX enzymes are important to maintain tissue homeostasis and wound healing in the gastric mucosa. Therefore, methylation-mediated silencing of these genes might tend to favor the appearance of pre-neoplasic lesions.
Genes encoding transcriptional factors: RUNX3 is a transcription factor that is thought to function as a tumor suppressor by regulating the expression of several cancer-related genes, including p53, p27 and caspase-3, among others[101,102]. The RUNX3 promoter is hypermethylated in pre-neoplasic gastric lesions, increasing progressively from chronic atrophic gastritis to gastric cancer[103]. Additionally, H. pylori infection contributed to inactivation of the RUNX3 gene in gastric epithelial cells by augmenting promoter hypermethylation[103]. Moreover, an in vitro study confirmed that H. pylori promotes RUNX3 promoter methylation via NO produced during co-incubation with macrophages[24].
FOXD3 is a member of the forkhead box (Fox) transcription factor family, whose promoter is hypermethylated in gastric mucosa infected with H. pylori. The evidence provided indicates that silencing of FOXD3 could be important during gastric carcinogenesis given that ectopic expression of FOXD3 in cancer cell lines reduces cell proliferation and their invasive capacity[104].
The upstream stimulatory factors, USF1 and USF2, are pleiotropic transcriptional factors that regulate the expression of genes linked to immune responses, cell cycle control and cell proliferation[105]. Bussière and colleagues demonstrated in vitro that H. pylori decreases the expression of USF1 and USF2 genes by hypermethylation of their promoters regions. These observations were confirmed in gastric tissue samples from infected mice, where they observed decreased expression of USF1 and USF2 and hypermethylation of USF1 and USF2 promoters in H. pylori-associated gastritis[106]. These factors control cell growth and have been shown to block cMyc/Ras-mediated transformation of primary rat cells[105].
For the transcription factors GATA-4 and GATA-5, both tumor suppressors, a higher methylation status was detected in gastric samples from H. pylori infected patients than in those from non-infected patients. Changes in methylation were particularly notable for the GATA-4 promoter. In addition, among gastric tissues with or without chronic gastritis, those with GATA-4 methylation were from H. pylori infected patients and no GATA-4 methylation was observed in biopsies from individuals without H. pylori infection[107].
Other tumor suppressor genes: The LOX and HRASLS genes encode a lysyl oxidase and HRAS-like suppressor proteins, respectively. The promoters of these genes are highly methylated in H. pylori-infected patients as compared to those without infection. In the same study, higher levels of methylation were observed for the CpG isIands present in the promoter regions of the genes THBD, HAND1 and FLN in H. pylori-positive than in H. pylori-negative individuals[90]. On the other hand, Perri et al[93] determined that APC is hypermethylated in gastritis associated with H. pylori infection and that bacterial eradication was sufficient to reduce methylation of the promoter. p41ARC is a protein that regulates Arp2/3 complex formation and is necessary for cell migration[108]. For p41ARC, a higher degree of methylation was observed in an exonic CpG island in H. pylori-positive individuals than in non-infected individuals[90]. The tumor suppressor gene WWOX encodes the WW-domain containing oxidoreductase, a protein frequently down-regulated in several cancers. An in vitro study demonstrated that in gastric cancer cell lines infected with H. pylori, methylation of WWOX is elevated and, furthermore, in this model methylation was associated with H. pylori-enhanced expression of DNMT1 and DNMT3[109].
MICRORNAS IN GASTRIC CANCER, THE PRECANCEROUS CASCADE AND H. PYLORI INFECTION
MicroRNA biology
MiRNAs are 20-24 nucleotides single-strand RNAs that alter the stability and translation of target mRNAs and, in doing so, modulate gene expression at the post-transcriptional level[83,110]. A considerable number of reports in the literature suggest that miRNAs control several processes during carcinogenesis that favor tumor development and progression[83]. Reduced translation and stability of messenger RNAs (mRNAs) can be triggered by miRNA binding to the 3’ untranslated region (3’UTR) of messenger RNAs (mRNAs). Most miRNAs are found in introns and intergenic regions and possess their own promoter and regulatory elements[111]. miRNAs have many physiological functions and control a variety of biological processes, including cellular differentiation, development, proliferation, metabolism, apoptosis and immune responses[112]. Not surprisingly, deregulation of miRNA levels is observed in many pathological conditions beyond cancer, including neurodegenerative and cardiovascular diseases[113,114]. Notably, miRNA genes are frequently found in regions of chromosomal instability (amplification, translocation or deletion) or near chromosomal breakpoints[115]. In tumorigenesis, the target mRNAs affected by miRNAs include those involved in inflammation, cell cycle regulation, stress responses (autophagy), differentiation (epithelial-mesenchymal transition, EMT), apoptosis and invasion[116-118]. Several of the relevant target genes include oncogenes or tumor suppressors that are up-regulated or downregulated, respectively, in tumor progression and metastasis[112]. In addition to their function in physiological and pathological processes, miRNAs play an important role during microbial infection caused by viruses, bacteria, parasites and fungi[119].
MiRNA signatures in gastric cancer
Tong et al[120] reported on frequent aberrant expression of miRNAs in gastric cancer. An association between miRNA expression and alterations in cell proliferation (miR-139, by targeting the CXCR4 gene; and miR-1, -34a and -504 by targeting the FOXP1 gene), cell cycle progression (miR-221, -222, -160b, -93 and -25, by targeting the p57, p21 and p27 genes) and invasion/metastasis (miR-21, by targeting the RECK gene) were detected. Ueda et al[121] analyzed 160 paired tumor and non-tumor mucosa samples and detected 22 miRNAs that were upregulated and 13 that were downregulated in gastric cancer. Interestingly, for intestinal and diffuse types of gastric cancer two different miRNA signatures were identified. Importantly, the miRNAs (miR-105, miR-100, miR-125b, miR-199a, miR-99a, miR-143, miR-145 and miR-133a) and (miR-373*, miR-498, miR-202*, and miR-494) were upregulated in diffuse-type and intestinal-type gastric cancer, respectively. Differential analysis of miRNA expression also revealed that progression in TNM staging correlated with higher expression levels of miR-125b, miR-199a, and miR-100. Also, reduced expression of let-7g and miR-433 and elevated expression of miR-214 were identified as independent predictors of poor survivial that were not related to depth of invasion, lymph-node metastasis or TNM stage. These findings show that miRNAs are differentially expressed in histological gastric cancer subtypes and that these are characterized by specific miRNA signatures.
Moreover, the expression of particular miRNAs is also linked to gastric cancer progression and prognosis. Ueda et al[121] identified miRNA expression profiles for gastric cancer by RT-PCR in 100 patients and identified a group of seven miRNAs as independent predictors of overall survival and relapse-free survival. In this study, hazard ratios (HRs) assessed by Cox regression analysis, demonstrated that miRNA expression levels correlated directly or indirectly with death probability: 3 miRNAs (let-7a, miR-126, miR-30a-5p) were protective (HR < 1) and 4 miRNAs (miR-10b, miR-21, miR-223, miR-338) were associated with higher risk of relapse and poorer survival (HR > 1).
The role of miRNAs in the precancerous cascade of gastric cancer was evaluated by Wang et al[122]. Using an in silico approach with Gene Expression Omnibus (GEO) datasets, these authors identified 20 differentially expressed miRNAs between H. pylori-related atrophic gastritis and intestinal metaplasia samples. These miRNAs modulated a variety of cell functions, including signal transduction, cell proliferation and death, as well as metabolite transport and catabolism. Among the target genes, RAB22A, SOX4, IKZF2, PLAG1 and BTBD7 were found to be simultaneously regulated by several differentially expressed miRNAs. On the other hand, miR-204 was decreased in H. pylori-associated atrophic gastritis[123]. Knockdown of this miRNA enhanced gastric cancer cell proliferation and invasion in vitro. Alternatively, miR-204 down-regulation and over-expression of SOX4 promoted the EMT process[123]. Additionally, the potential of microRNAs as biomarkers for early gastric detection was recently assessed[124-126]. In a population-based study in Linqu, a high-risk area of gastric cancer in China, Song et al[124] investigated the relevance of serum miRNAs as biomarkers. Serum pools from GC control and validated gastric cancer and dysplasia subjects were correctly identified by several differentially expressed miRNAs. A signature of 16 miRNAs that were upregulated in gastric cancer patients was identified. miR-221, miR-744, and miR-376c were subsequently validated as non-invasive biomarkers for gastric cancer detection with 82.4% sensitivity and 58.8% specificity. Fu et al[127] identified an additional miRNA, miR-222, for which circulating plasma levels increased in atrophic gastritis patients compared to healthy controls (P < 0.001) with a diagnostic accuracy of 0.850 and 66.1% sensitivity as well as 88.3% specificity. Also, the expression patterns of serum let-7 microRNA (miRNA) and its target gene pepsinogen C (PGC) were correlated with different stages of the multistage cascade of gastric cancer[126]. Finally, the feasibility of using gastric juice as test material was examined by Yu et al[125]. Significantly lower levels of gastric juice miR-129-1-3p and miR-129-2-3p with AUC values of 0.639 and 0.65 to distinguish gastric cancer from healthy controls.
Deregulation of miRNAs expression during H. pylori infection
H. pylori infection in vivo and in vitro alters the expression of many miRNAs[128]. Several of these miRNAs were also found to be deregulated in gastric cancer. On the one hand, down-regulation of let7a, mir-31, mir-101, miR-141, miR-203, miR-210, miR-218, miR-375, miR-449 is observed, while miR-17, miR-20a, miR-21, miR-146a, miR-155 and miR-223 are up-regulated[129]. Interestingly, it has been suggested that these miRNAs may also be relevant to understanding H. pylori induced inflammation and carcinogenesis[129]. These miRNAs target mRNAs involved in biological processes, such as invasion and metastasis, proliferation, cell cycle progression, apoptosis, epithelial mesenchymal transition and the immune response[129].
MiR-155-5p, generically referred to as miR-155[130] (Figure 1). It is a typical, multifunctional miRNA involved in several biological processes, such as hematopoiesis, inflammation, immunity and carcinogenesis[130]. This miRNA is considered an oncogenic microRNA (oncomiR), given that it is up-regulated in different types of cancer, such as lymphoma, breast, cervical, lung, colon, pancreatic and thyroid cancer[130]. The miR155HG/miR-155 gene is induced upon exposure to lipopolysaccharide exposure via Toll-like receptor activation[130]. This characteristic was ascribed to the presence of two relevant NF-κB binding sequences present in the BIC/miR-155 promoter at -178 and -1150[131]. Similar to other pathogens, H. pylori infection elevates miR-155 levels in vitro and in vivo[132]. Several studies have focused on hematological and inflammation-associated malignances linked to this miRNA. For instance, gastric MALT lymphoma can be reverted by 60%-80% following antibiotic eradication of bacteria; however, resistant MALT lymphomas maintain miR-155 overexpression even after bacterial eradication[133]. Other studies show that miR-155 is a negative regulator of pro-inflammatory responses by targeting the receptor-associated adapter MyD88, thereby reducing NF-κB activation and IL-8 secretion[134]. In another study, H. pylori-dependent miR-155 induction reduced DNA-damage and induced apoptosis in macrophages, in a manner dependent on the cagPAI secretory system and TLRs, but independent of CagA[135]. In summary, miR-155 represents a multifunctional miRNA, whose potential role in H. pylori-associated pathologies is apparent, but still poorly understood.
As mentioned previously, several miRNAs are known to be down-regulated during gastric cancer progression and, similar to tumor suppressor genes, they can be also silenced by promoter hypermethylation. For instance, the expression of several microRNAs, such as miR-10a, -10b, -127, -148a, -181c, -195, -219-2-3p, -224, -338-3p, -340, -378, -433 and -452 among others was recently reported to be silenced by aberrant methylation in gastric cancer[136-143]. Interestingly, Kiga et al[144] demonstrated that H. pylori-associated inflammation promoted DNA methylation of the miR-210 locus and enhanced proliferation of gastric cells as a consequence of augmented STMN1 and DIMT1 expression. MIR-210 (Figure 1) is an hypoxia responsive multifunctional microRNA whose promoter contains Hypoxia Inducible factor (HIF1α and 2α) response elements (HRE) and the target mRNAs are implicated in the regulation of cell growth arrest, repression of mitochondrial metabolism, stem cell survival under hypoxic conditions, stalling the DNA repair system (promoting genetic instability), angiogenesis induction and cellular differentiation[145]. Again, as for miR-155, silencing of miR-210 following H. pylori infection is likely to contribute to disease, but further studies are required.
Taken together, the data presented here suggest that miRNAs play essential roles in gastric cancer by modulating cell proliferation, cell cycle and invasion/metastasis. In the gastric precancerous condition, miRNAs are linked to H. pylori-related gastritis and intestinal metaplasia. From a biomarker point of view, miRNAs have a strong potential as novel molecular tools for non-invasive risk assessment of gastric cancer and precancerous lesions.
REPRIMO AS A MODEL FOR EPIGENETIC CHANGES IN TUMOR SUPPRESSOR GENES IN GASTRIC CANCER
The Reprimo gene (official symbol RPRM, MIM 612171, GeneID 56475) is located in chromosome 2q23.3[146]. Sequence analysis indicates that RPRM is 303 bp long and does not have introns. The protein sequence includes 109 aa and has two glycosylation sites (AA 7-10 and 18-21), one phosphorylation site (AA 95-98) and one myristylation site (AA 13-18) (http://hits.isb-sib.ch/cgi-bin/motif_scan). Computational analysis shows a possible transmembrane domain between aa 56-78 (www.ensembl.org). According to Ohki and coworkers, RPRM is an intracellular cytoplasmic protein[147] that is induced after X-ray-irradiation in a p53-dependent manner. Ectopic p53 expression also results in the induction of the expression of RPRM. In both scenarios, RPRM causes G2 arrest of the cell cycle in association with the inhibition of nuclear translocation of cyclin B1 and Cdc2 activity[147]. An inverse association between RPRM gene expression and promoter methylation (Spearman rank R = -1; P = 0.042) was recently identified. Additionally, RPRM overexpression robustly inhibited colony formation and anchorage-independent growth. Furthermore, in primary gastric cancer cases, RPRM protein was not detected in tumor tissues but was found to be present in non-tumor adjacent mucosa (P = 0.001). Furthermore, the loss of expression is associated with invasive stages GC (stage I to II-IV, P = 0.006). Interestingly, these findings correlated with p73 (P < 0.0001 and kappa value = 0,363) but not p53 expression, suggesting a potential link to other members of the p53 family[148]. In a quantitative analysis of RPRM DNA promoter methylation, the H. pylori virulence factors cagA (including segments of the 3 end, encoding EPIYA polymorphisms) and vacA s1 and m1 regions were associated with increased RPRM promoter methylation[149]. These results favor the notion that H. pylori regulates RPRM gene expression[149]. Bernal et al[150] reported that methylated RPRM promoter sequences are not only a common finding in primary tissues of gastric cancer patients, but also that they can be detected in the plasma of these patients and rarely in healthy donors. A translational potentiality of this observation is the possibility that methylated circulating cell-free DNA may be employed as a non-invasive biomarker for detection of gastric cancer and precancerous lesions.
THE H. PYLORI γ-GLUTAMYL TRANSPEPTIDASE CONNECTION TO ADDITIONAL MODES OF REGULATION IN GASTRIC CANCER: SURVIVIN AS A MODEL
GGT is an enzyme that catalyzes the transpeptidation and hydrolysis of the γ-glutamyl moiety from glutathione and glutathione-conjugated compounds, to amino acids[151]. H. pylori γ-glutamyl transpeptidase (HpGGT) is constitutively expressed and detectable in all H. pylori strains[152], suggesting it is relevant to bacterial physiology. Among the many effects observed in gastric cells, GGT induces apoptosis[153], cell cycle arrest[154], and generates ROS, in particular H2O2, that potentially damages DNA[155]. Importantly, significantly higher GGT activity has been observed in H. pylori isolates obtained from patients with peptic ulcer disease than in those from patients with non-ulcer dyspepsia[155]. Gastric ulcers are associated with an elevated risk of developing gastric cancer[156] and these observations implicate HpGGT as being clinically relevant to the progression of precancerous gastric lesions.
Beyond the indicated alterations in gastric cells induced by GGT, recent evidence suggests that this virulence factor may also enhance proteasome-mediated degradation of proteins required for cell viability, as is the case for survivin, a member of the inhibitor of apoptosis (IAP) family of proteins. Survivin expression levels are controlled at the transcriptional level by several transcription factors and at the post-transcriptional level by phosphorylation and proteasome-mediated degradation[157]. Survivin is generally not expressed in normal differentiated tissues, except in proliferating stem cell populations, but is strongly upregulated in most human cancer cells where it enhances proliferation, viability, metastasis and angiogenesis[157-159]. Interestingly, the gastric mucosa apparently represents an exception to this general rule, because normal mucosa cells express high levels of survivin and it has been suggested that presence of the protein may favor mucosa cell survival in the adverse gastric environment. Importantly in biopsies from gastritis patients with H. pylori infection, survivin protein levels are decreased as compared to control samples from gastritis patients without infection[160]. In the same study, infection in vitro of cells of the gastrointestinal lineage with H. pylori was shown to trigger the loss of survivin protein and as a consequence reduces cell viability. Subsequently, this ability of H. pylori was linked to the secretion of HpGGT and enhanced turnover of the protein via the proteasome pathway, possibly via a mechanism involving the generation of ROS[161]. It is interesting to note here that this mechanism specifically affected survivin but not other anti-apoptotic proteins, like Bcl2 or Bcl-xL, and one may speculate that related events might control turnover of a number of yet to be identified components in infected cells. Finally, this GGT-proteasome connection may explain how early in disease development H. pylori damages the gastric tissue in zones with high levels of bacterial infection, favoring the genesis of a pro-inflammatory environment and potentially subsequent invasion by bone marrow-derived cells and intestinal metaplasia[162]. Further experimentation is required to substantiate this intriguing hypothesis that could also open up new avenues for the treatment of H. pylori positive gastritis patients.
CONCLUSION
In summary, this review highlights how in addition to H. pylori-associated virulence factors, epigenetic changes in infected host cells, such as DNA methylation and miRNAs, are likely to play a significant role in gastric cancer development and the progression of the precancerous cascade. Moreover, recent results revealing how HpGGT modulates the turnover of specific protective host proteins underscores the tremendous variability and selectivity of changes that may be attributed to H. pylori infection. Importantly, from the clinical perspective, some of the epigenetic factors discussed can be determined by analyzing blood and other body fluid samples, thus opening up a new avenue for non-invasive risk assessment of gastric cancer. In doing so, we may anticipate that earlier diagnosis and, as a consequence, more effective treatment of this still very deadly cancer will become possible.
Footnotes
Supported by CONICYT-FONDAP project, No. 15130011 (to Corvalánand AH and Quest AFG); FONDECYT project, No. 1151411 (to Corvalán AH); and a CONICYT PhD fellowship award (to Canales J).
Conflict-of-interest statement: Authors declare no conflict of interests for this article
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 29, 2015
First decision: July 14, 2015
Article in press: October 13, 2015
P- Reviewer: El-Zahaby SA, Smith SM S- Editor: Yu J L- Editor: A E- Editor: Wang CH
References
- 1.Peleteiro B, Bastos A, Ferro A, Lunet N. Prevalence of Helicobacter pylori infection worldwide: a systematic review of studies with national coverage. Dig Dis Sci. 2014;59:1698–1709. doi: 10.1007/s10620-014-3063-0. [DOI] [PubMed] [Google Scholar]
- 2.Watari J, Chen N, Amenta PS, Fukui H, Oshima T, Tomita T, Miwa H, Lim KJ, Das KM. Helicobacter pylori associated chronic gastritis, clinical syndromes, precancerous lesions, and pathogenesis of gastric cancer development. World J Gastroenterol. 2014;20:5461–5473. doi: 10.3748/wjg.v20.i18.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu JB, Wu XM, Cai J, Zhang JY, Zhang JL, Zhou SH, Shi MX, Qiang FL. CpG island methylator phenotype and Helicobacter pylori infection associated with gastric cancer. World J Gastroenterol. 2012;18:5129–5134. doi: 10.3748/wjg.v18.i36.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 5.Handa Y, Saitoh T, Kawaguchi M, Misaka R, Ohno H, Tsai CR, Tani Y, Tsurui M, Yoshida H, Morita S, et al. Association of Helicobacter pylori and diffuse type gastric cancer. J Gastroenterol. 1996;31 Suppl 9:29–32. [PubMed] [Google Scholar]
- 6.Nardone G, Rocco A, Malfertheiner P. Review article: helicobacter pylori and molecular events in precancerous gastric lesions. Aliment Pharmacol Ther. 2004;20:261–270. doi: 10.1111/j.1365-2036.2004.02075.x. [DOI] [PubMed] [Google Scholar]
- 7.Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology. 2007;133:659–672. doi: 10.1053/j.gastro.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 8.Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, Aburatani H, Akiyama T, Peek RM, Azuma T, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26:4617–4626. doi: 10.1038/sj.onc.1210251. [DOI] [PubMed] [Google Scholar]
- 9.Kaise M, Yamasaki T, Yonezawa J, Miwa J, Ohta Y, Tajiri H. CpG island hypermethylation of tumor-suppressor genes in H. pylori-infected non-neoplastic gastric mucosa is linked with gastric cancer risk. Helicobacter. 2008;13:35–41. doi: 10.1111/j.1523-5378.2008.00572.x. [DOI] [PubMed] [Google Scholar]
- 10.Nardone G, Compare D, De Colibus P, de Nucci G, Rocco A. Helicobacter pylori and epigenetic mechanisms underlying gastric carcinogenesis. Dig Dis. 2007;25:225–229. doi: 10.1159/000103890. [DOI] [PubMed] [Google Scholar]
- 11.Peek RM, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–248. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 12.Hirahara K, Poholek A, Vahedi G, Laurence A, Kanno Y, Milner JD, O’Shea JJ. Mechanisms underlying helper T-cell plasticity: implications for immune-mediated disease. J Allergy Clin Immunol. 2013;131:1276–1287. doi: 10.1016/j.jaci.2013.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yasmin R, Siraj S, Hassan A, Khan AR, Abbasi R, Ahmad N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediators Inflamm. 2015;2015:201703. doi: 10.1155/2015/201703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawanishi S, Hiraku Y, Pinlaor S, Ma N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol Chem. 2006;387:365–372. doi: 10.1515/BC.2006.049. [DOI] [PubMed] [Google Scholar]
- 15.Fernandes JV, Cobucci RN, Jatobá CA, Fernandes TA, de Azevedo JW, de Araújo JM. The role of the mediators of inflammation in cancer development. Pathol Oncol Res. 2015;21:527–534. doi: 10.1007/s12253-015-9913-z. [DOI] [PubMed] [Google Scholar]
- 16.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 17.Sugimoto M, Furuta T, Shirai N, Nakamura A, Xiao F, Kajimura M, Sugimura H, Hishida A. Different effects of polymorphisms of tumor necrosis factor-alpha and interleukin-1 beta on development of peptic ulcer and gastric cancer. J Gastroenterol Hepatol. 2007;22:51–59. doi: 10.1111/j.1440-1746.2006.04442.x. [DOI] [PubMed] [Google Scholar]
- 18.Taguchi A, Ohmiya N, Shirai K, Mabuchi N, Itoh A, Hirooka Y, Niwa Y, Goto H. Interleukin-8 promoter polymorphism increases the risk of atrophic gastritis and gastric cancer in Japan. Cancer Epidemiol Biomarkers Prev. 2005;14:2487–2493. doi: 10.1158/1055-9965.EPI-05-0326. [DOI] [PubMed] [Google Scholar]
- 19.Zabaleta J, Camargo MC, Piazuelo MB, Fontham E, Schneider BG, Sicinschi LA, Ferrante W, Balart L, Correa P, Ochoa AC. Association of interleukin-1beta gene polymorphisms with precancerous gastric lesions in African Americans and Caucasians. Am J Gastroenterol. 2006;101:163–171. doi: 10.1111/j.1572-0241.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 20.Na HK, Woo JH. Helicobacter pylori Induces Hypermethylation of CpG Islands Through Upregulation of DNA Methyltransferase: Possible Involvement of Reactive Oxygen/Nitrogen Species. J Cancer Prev. 2014;19:259–264. doi: 10.15430/JCP.2014.19.4.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- 22.Niwa T, Toyoda T, Tsukamoto T, Mori A, Tatematsu M, Ushijima T. Prevention of Helicobacter pylori-induced gastric cancers in gerbils by a DNA demethylating agent. Cancer Prev Res (Phila) 2013;6:263–270. doi: 10.1158/1940-6207.CAPR-12-0369. [DOI] [PubMed] [Google Scholar]
- 23.Schneider BG, Piazuelo MB, Sicinschi LA, Mera R, Peng DF, Roa JC, Romero-Gallo J, Delgado AG, de Sablet T, Bravo LE, et al. Virulence of infecting Helicobacter pylori strains and intensity of mononuclear cell infiltration are associated with levels of DNA hypermethylation in gastric mucosae. Epigenetics. 2013;8:1153–1161. doi: 10.4161/epi.26072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katayama Y, Takahashi M, Kuwayama H. Helicobacter pylori causes runx3 gene methylation and its loss of expression in gastric epithelial cells, which is mediated by nitric oxide produced by macrophages. Biochem Biophys Res Commun. 2009;388:496–500. doi: 10.1016/j.bbrc.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Huang FY, Chan AO, Rashid A, Wong DK, Cho CH, Yuen MF. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer. 2012;118:4969–4980. doi: 10.1002/cncr.27519. [DOI] [PubMed] [Google Scholar]
- 26.Huang FY, Chan AO, Lo RC, Rashid A, Wong DK, Cho CH, Lai CL, Yuen MF. Characterization of interleukin-1β in Helicobacter pylori-induced gastric inflammation and DNA methylation in interleukin-1 receptor type 1 knockout (IL-1R1(-/-)) mice. Eur J Cancer. 2013;49:2760–2770. doi: 10.1016/j.ejca.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 27.Nam KT, Oh SY, Ahn B, Kim YB, Jang DD, Yang KH, Hahm KB, Kim DY. Decreased Helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut. 2004;53:1250–1255. doi: 10.1136/gut.2003.030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ushijima T, Hattori N. Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Cancer Res. 2012;18:923–929. doi: 10.1158/1078-0432.CCR-11-2011. [DOI] [PubMed] [Google Scholar]
- 29.Gonda TA, Kim YI, Salas MC, Gamble MV, Shibata W, Muthupalani S, Sohn KJ, Abrams JA, Fox JG, Wang TC, et al. Folic acid increases global DNA methylation and reduces inflammation to prevent Helicobacter-associated gastric cancer in mice. Gastroenterology. 2012;142:824–833.e7. doi: 10.1053/j.gastro.2011.12.058. [DOI] [PubMed] [Google Scholar]
- 30.Akhavan-Niaki H, Samadani AA. Molecular insight in gastric cancer induction: an overview of cancer stemness genes. Cell Biochem Biophys. 2014;68:463–473. doi: 10.1007/s12013-013-9749-7. [DOI] [PubMed] [Google Scholar]
- 31.Handa O, Naito Y, Yoshikawa T. Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflamm Res. 2010;59:997–1003. doi: 10.1007/s00011-010-0245-x. [DOI] [PubMed] [Google Scholar]
- 32.Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, Blake DR, Rampton DS. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut. 1994;35:179–185. doi: 10.1136/gut.35.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Touati E, Michel V, Thiberge JM, Wuscher N, Huerre M, Labigne A. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology. 2003;124:1408–1419. doi: 10.1016/s0016-5085(03)00266-x. [DOI] [PubMed] [Google Scholar]
- 34.Ma N, Adachi Y, Hiraku Y, Horiki N, Horiike S, Imoto I, Pinlaor S, Murata M, Semba R, Kawanishi S. Accumulation of 8-nitroguanine in human gastric epithelium induced by Helicobacter pylori infection. Biochem Biophys Res Commun. 2004;319:506–510. doi: 10.1016/j.bbrc.2004.04.193. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, Azuma T, Okazaki IM, Honjo T, Chiba T. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13:470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu T, Marusawa H, Matsumoto Y, Inuzuka T, Ikeda A, Fujii Y, Minamiguchi S, Miyamoto S, Kou T, Sakai Y, et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology. 2014;147:407–417.e3. doi: 10.1053/j.gastro.2014.04.036. [DOI] [PubMed] [Google Scholar]
- 37.Yanaka A. Sulforaphane enhances protection and repair of gastric mucosa against oxidative stress in vitro, and demonstrates anti-inflammatory effects on Helicobacter pylori-infected gastric mucosae in mice and human subjects. Curr Pharm Des. 2011;17:1532–1540. doi: 10.2174/138161211796196945. [DOI] [PubMed] [Google Scholar]
- 38.Drake IM, Davies MJ, Mapstone NP, Dixon MF, Schorah CJ, White KL, Chalmers DM, Axon AT. Ascorbic acid may protect against human gastric cancer by scavenging mucosal oxygen radicals. Carcinogenesis. 1996;17:559–562. doi: 10.1093/carcin/17.3.559. [DOI] [PubMed] [Google Scholar]
- 39.Weitzman SA, Turk PW, Milkowski DH, Kozlowski K. Free radical adducts induce alterations in DNA cytosine methylation. Proc Natl Acad Sci USA. 1994;91:1261–1264. doi: 10.1073/pnas.91.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sitaraman R. Helicobacter pylori DNA methyltransferases and the epigenetic field effect in cancerization. Front Microbiol. 2014;5:115. doi: 10.3389/fmicb.2014.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krebes J, Morgan RD, Bunk B, Spröer C, Luong K, Parusel R, Anton BP, König C, Josenhans C, Overmann J, et al. The complex methylome of the human gastric pathogen Helicobacter pylori. Nucleic Acids Res. 2014;42:2415–2432. doi: 10.1093/nar/gkt1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terradot L, Waksman G. Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J. 2011;278:1213–1222. doi: 10.1111/j.1742-4658.2011.08037.x. [DOI] [PubMed] [Google Scholar]
- 43.Backert S, Schwarz T, Miehlke S, Kirsch C, Sommer C, Kwok T, Gerhard M, Goebel UB, Lehn N, Koenig W, et al. Functional analysis of the cag pathogenicity island in Helicobacter pylori isolates from patients with gastritis, peptic ulcer, and gastric cancer. Infect Immun. 2004;72:1043–1056. doi: 10.1128/IAI.72.2.1043-1056.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glocker E, Lange C, Covacci A, Bereswill S, Kist M, Pahl HL. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-kappaB activation. Infect Immun. 1998;66:2346–2348. doi: 10.1128/iai.66.5.2346-2348.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 47.Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, Neish AS, Collier-Hyams L, Perez-Perez GI, Hatakeyama M, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suarez G, Romero-Gallo J, Piazuelo MB, Wang G, Maier RJ, Forsberg LS, Azadi P, Gomez MA, Correa P, Peek RM. Modification of Helicobacter pylori Peptidoglycan Enhances NOD1 Activation and Promotes Cancer of the Stomach. Cancer Res. 2015;75:1749–1759. doi: 10.1158/0008-5472.CAN-14-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang G, Maier SE, Lo LF, Maier G, Dosi S, Maier RJ. Peptidoglycan deacetylation in Helicobacter pylori contributes to bacterial survival by mitigating host immune responses. Infect Immun. 2010;78:4660–4666. doi: 10.1128/IAI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Perez GI, Peek RM, Legath AJ, Heine PR, Graff LB. The role of CagA status in gastric and extragastric complications of Helicobacter pylori. J Physiol Pharmacol. 1999;50:833–845. [PubMed] [Google Scholar]
- 51.Yamaoka Y, Kita M, Kodama T, Sawai N, Kashima K, Imanishi J. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut. 1997;41:442–451. doi: 10.1136/gut.41.4.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu J, Xu S, Zhu Y. Helicobacter pylori CagA: a critical destroyer of the gastric epithelial barrier. Dig Dis Sci. 2013;58:1830–1837. doi: 10.1007/s10620-013-2589-x. [DOI] [PubMed] [Google Scholar]
- 53.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 54.Azuma T, Yamazaki S, Yamakawa A, Ohtani M, Muramatsu A, Suto H, Ito Y, Dojo M, Yamazaki Y, Kuriyama M, et al. Association between diversity in the Src homology 2 domain--containing tyrosine phosphatase binding site of Helicobacter pylori CagA protein and gastric atrophy and cancer. J Infect Dis. 2004;189:820–827. doi: 10.1086/381782. [DOI] [PubMed] [Google Scholar]
- 55.Zhu Y, Zhong X, Zheng S, Du Q, Xu W. Transformed immortalized gastric epithelial cells by virulence factor CagA of Helicobacter pylori through Erk mitogen-activated protein kinase pathway. Oncogene. 2005;24:3886–3895. doi: 10.1038/sj.onc.1208551. [DOI] [PubMed] [Google Scholar]
- 56.Bessède E, Staedel C, Acuña Amador LA, Nguyen PH, Chambonnier L, Hatakeyama M, Belleannée G, Mégraud F, Varon C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-mesenchymal transition-like changes. Oncogene. 2014;33:4123–4131. doi: 10.1038/onc.2013.380. [DOI] [PubMed] [Google Scholar]
- 57.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwamoto J, Mizokami Y, Takahashi K, Nakajima K, Ohtsubo T, Miura S, Narasaka T, Takeyama H, Omata T, Shimokobe K, et al. Expressions of urokinase-type plasminogen activator, its receptor and plasminogen activator inhibitor-1 in gastric cancer cells and effects of Helicobacter pylori. Scand J Gastroenterol. 2005;40:783–793. doi: 10.1080/00365520510015665. [DOI] [PubMed] [Google Scholar]
- 59.Wandler AM, Guillemin K. Transgenic expression of the Helicobacter pylori virulence factor CagA promotes apoptosis or tumorigenesis through JNK activation in Drosophila. PLoS Pathog. 2012;8:e1002939. doi: 10.1371/journal.ppat.1002939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003–1008. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hatakeyama M, Higashi H. Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis. Cancer Sci. 2005;96:835–843. doi: 10.1111/j.1349-7006.2005.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Backert S, Tegtmeyer N, Selbach M. The versatility of Helicobacter pylori CagA effector protein functions: The master key hypothesis. Helicobacter. 2010;15:163–176. doi: 10.1111/j.1523-5378.2010.00759.x. [DOI] [PubMed] [Google Scholar]
- 63.Backert S, Moese S, Selbach M, Brinkmann V, Meyer TF. Phosphorylation of tyrosine 972 of the Helicobacter pylori CagA protein is essential for induction of a scattering phenotype in gastric epithelial cells. Mol Microbiol. 2001;42:631–644. doi: 10.1046/j.1365-2958.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 64.Botham CM, Wandler AM, Guillemin K. A transgenic Drosophila model demonstrates that the Helicobacter pylori CagA protein functions as a eukaryotic Gab adaptor. PLoS Pathog. 2008;4:e1000064. doi: 10.1371/journal.ppat.1000064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatakeyama M. Helicobacter pylori and gastric carcinogenesis. J Gastroenterol. 2009;44:239–248. doi: 10.1007/s00535-009-0014-1. [DOI] [PubMed] [Google Scholar]
- 66.Brandt S, Kwok T, Hartig R, König W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lamb A, Yang XD, Tsang YH, Li JD, Higashi H, Hatakeyama M, Peek RM, Blanke SR, Chen LF. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009;10:1242–1249. doi: 10.1038/embor.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sepulveda AR, Yao Y, Yan W, Park DI, Kim JJ, Gooding W, Abudayyeh S, Graham DY. CpG methylation and reduced expression of O6-methylguanine DNA methyltransferase is associated with Helicobacter pylori infection. Gastroenterology. 2010;138:1836–1844. doi: 10.1053/j.gastro.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 69.Hayashi Y, Tsujii M, Wang J, Kondo J, Akasaka T, Jin Y, Li W, Nakamura T, Nishida T, Iijima H, et al. CagA mediates epigenetic regulation to attenuate let-7 expression in Helicobacter pylori-related carcinogenesis. Gut. 2013;62:1536–1546. doi: 10.1136/gutjnl-2011-301625. [DOI] [PubMed] [Google Scholar]
- 70.de Bernard M, Moschioni M, Papini E, Telford J, Rappuoli R, Montecucco C. Cell vacuolization induced by Helicobacter pylori VacA toxin: cell line sensitivity and quantitative estimation. Toxicol Lett. 1998;99:109–115. doi: 10.1016/s0378-4274(98)00140-4. [DOI] [PubMed] [Google Scholar]
- 71.de Bernard M, Arico B, Papini E, Rizzuto R, Grandi G, Rappuoli R, Montecucco C. Helicobacter pylori toxin VacA induces vacuole formation by acting in the cell cytosol. Mol Microbiol. 1997;26:665–674. doi: 10.1046/j.1365-2958.1997.5881952.x. [DOI] [PubMed] [Google Scholar]
- 72.Galmiche A, Rassow J, Doye A, Cagnol S, Chambard JC, Contamin S, de Thillot V, Just I, Ricci V, Solcia E, et al. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000;19:6361–6370. doi: 10.1093/emboj/19.23.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol. 2007;179:5433–5440. doi: 10.4049/jimmunol.179.8.5433. [DOI] [PubMed] [Google Scholar]
- 74.Terebiznik MR, Raju D, Vázquez CL, Torbricki K, Kulkarni R, Blanke SR, Yoshimori T, Colombo MI, Jones NL. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5:370–379. doi: 10.4161/auto.5.3.7663. [DOI] [PubMed] [Google Scholar]
- 75.Lu H, Yamaoka Y, Graham DY. Helicobacter pylori virulence factors: facts and fantasies. Curr Opin Gastroenterol. 2005;21:653–659. doi: 10.1097/01.mog.0000181711.04529.d5. [DOI] [PubMed] [Google Scholar]
- 76.Supajatura V, Ushio H, Wada A, Yahiro K, Okumura K, Ogawa H, Hirayama T, Ra C. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J Immunol. 2002;168:2603–2607. doi: 10.4049/jimmunol.168.6.2603. [DOI] [PubMed] [Google Scholar]
- 77.Salama NR, Otto G, Tompkins L, Falkow S. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect Immun. 2001;69:730–736. doi: 10.1128/IAI.69.2.730-736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Atherton JC, Cao P, Peek RM, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- 79.Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, Atherton JC. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133:926–936. doi: 10.1053/j.gastro.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 80.Winter JA, Letley DP, Cook KW, Rhead JL, Zaitoun AA, Ingram RJ, Amilon KR, Croxall NJ, Kaye PV, Robinson K, et al. A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori-induced metaplasia in the stomach. J Infect Dis. 2014;210:954–963. doi: 10.1093/infdis/jiu154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sun J, Aoki K, Zheng JX, Su BZ, Ouyang XH, Misumi J. Effect of NaCl and Helicobacter pylori vacuolating cytotoxin on cytokine expression and viability. World J Gastroenterol. 2006;12:2174–2180. doi: 10.3748/wjg.v12.i14.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sugimoto M, Furuta T, Yamaoka Y. Influence of inflammatory cytokine polymorphisms on eradication rates of Helicobacter pylori. J Gastroenterol Hepatol. 2009;24:1725–1732. doi: 10.1111/j.1440-1746.2009.06047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 84.Mikeska T, Craig JM. DNA methylation biomarkers: cancer and beyond. Genes (Basel) 2014;5:821–864. doi: 10.3390/genes5030821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qu Y, Dang S, Hou P. Gene methylation in gastric cancer. Clin Chim Acta. 2013;424:53–65. doi: 10.1016/j.cca.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 86.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang GH, Shim YH, Jung HY, Kim WH, Ro JY, Rhyu MG. CpG island methylation in premalignant stages of gastric carcinoma. Cancer Res. 2001;61:2847–2851. [PubMed] [Google Scholar]
- 88.Park SY, Yoo EJ, Cho NY, Kim N, Kang GH. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for Helicobacter pylori infection. J Pathol. 2009;219:410–416. doi: 10.1002/path.2596. [DOI] [PubMed] [Google Scholar]
- 89.Chan AO, Lam SK, Wong BC, Wong WM, Yuen MF, Yeung YH, Hui WM, Rashid A, Kwong YL. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–506. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 91.Leung WK, Man EP, Yu J, Go MY, To KF, Yamaoka Y, Cheng VY, Ng EK, Sung JJ. Effects of Helicobacter pylori eradication on methylation status of E-cadherin gene in noncancerous stomach. Clin Cancer Res. 2006;12:3216–3221. doi: 10.1158/1078-0432.CCR-05-2442. [DOI] [PubMed] [Google Scholar]
- 92.Ushijima T, Nakajima T, Maekita T. DNA methylation as a marker for the past and future. J Gastroenterol. 2006;41:401–407. doi: 10.1007/s00535-006-1846-6. [DOI] [PubMed] [Google Scholar]
- 93.Perri F, Cotugno R, Piepoli A, Merla A, Quitadamo M, Gentile A, Pilotto A, Annese V, Andriulli A. Aberrant DNA methylation in non-neoplastic gastric mucosa of H. Pylori infected patients and effect of eradication. Am J Gastroenterol. 2007;102:1361–1371. doi: 10.1111/j.1572-0241.2007.01284.x. [DOI] [PubMed] [Google Scholar]
- 94.Miao R, Guo X, Zhi Q, Shi Y, Li L, Mao X, Zhang L, Li C. VEZT, a novel putative tumor suppressor, suppresses the growth and tumorigenicity of gastric cancer. PLoS One. 2013;8:e74409. doi: 10.1371/journal.pone.0074409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meşe G, Richard G, White TW. Gap junctions: basic structure and function. J Invest Dermatol. 2007;127:2516–2524. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- 96.Wang Y, Huang LH, Xu CX, Xiao J, Zhou L, Cao D, Liu XM, Qi Y. Connexin 32 and 43 promoter methylation in Helicobacter pylori-associated gastric tumorigenesis. World J Gastroenterol. 2014;20:11770–11779. doi: 10.3748/wjg.v20.i33.11770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alvarez MC, Santos JC, Maniezzo N, Ladeira MS, da Silva AL, Scaletsky IC, Pedrazzoli J, Ribeiro ML. MGMT and MLH1 methylation in Helicobacter pylori-infected children and adults. World J Gastroenterol. 2013;19:3043–3051. doi: 10.3748/wjg.v19.i20.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Farrell JJ, Taupin D, Koh TJ, Chen D, Zhao CM, Podolsky DK, Wang TC. TFF2/SP-deficient mice show decreased gastric proliferation, increased acid secretion, and increased susceptibility to NSAID injury. J Clin Invest. 2002;109:193–204. doi: 10.1172/JCI12529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kurt-Jones EA, Cao L, Sandor F, Rogers AB, Whary MT, Nambiar PR, Cerny A, Bowen G, Yan J, Takaishi S, et al. Trefoil family factor 2 is expressed in murine gastric and immune cells and controls both gastrointestinal inflammation and systemic immune responses. Infect Immun. 2007;75:471–480. doi: 10.1128/IAI.02039-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peterson AJ, Menheniott TR, O’Connor L, Walduck AK, Fox JG, Kawakami K, Minamoto T, Ong EK, Wang TC, Judd LM, et al. Helicobacter pylori infection promotes methylation and silencing of trefoil factor 2, leading to gastric tumor development in mice and humans. Gastroenterology. 2010;139:2005–2017. doi: 10.1053/j.gastro.2010.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yamada C, Ozaki T, Ando K, Suenaga Y, Inoue K, Ito Y, Okoshi R, Kageyama H, Kimura H, Miyazaki M, et al. RUNX3 modulates DNA damage-mediated phosphorylation of tumor suppressor p53 at Ser-15 and acts as a co-activator for p53. J Biol Chem. 2010;285:16693–16703. doi: 10.1074/jbc.M109.055525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen W, Gao N, Shen Y, Cen JN. Hypermethylation downregulates Runx3 gene expression and its restoration suppresses gastric epithelial cell growth by inducing p27 and caspase3 in human gastric cancer. J Gastroenterol Hepatol. 2010;25:823–831. doi: 10.1111/j.1440-1746.2009.06191.x. [DOI] [PubMed] [Google Scholar]
- 103.Lu XX, Yu JL, Ying LS, Han J, Wang S, Yu QM, Wang XB, Fang XH, Ling ZQ. Stepwise cumulation of RUNX3 methylation mediated by Helicobacter pylori infection contributes to gastric carcinoma progression. Cancer. 2012;118:5507–5517. doi: 10.1002/cncr.27604. [DOI] [PubMed] [Google Scholar]
- 104.Cheng AS, Li MS, Kang W, Cheng VY, Chou JL, Lau SS, Go MY, Lee CC, Ling TK, Ng EK, et al. Helicobacter pylori causes epigenetic dysregulation of FOXD3 to promote gastric carcinogenesis. Gastroenterology. 2013;144:122–133.e9. doi: 10.1053/j.gastro.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 105.Corre S, Galibert MD. Upstream stimulating factors: highly versatile stress-responsive transcription factors. Pigment Cell Res. 2005;18:337–348. doi: 10.1111/j.1600-0749.2005.00262.x. [DOI] [PubMed] [Google Scholar]
- 106.Bussière FI, Michel V, Mémet S, Avé P, Vivas JR, Huerre M, Touati E. H. pylori-induced promoter hypermethylation downregulates USF1 and USF2 transcription factor gene expression. Cell Microbiol. 2010;12:1124–1133. doi: 10.1111/j.1462-5822.2010.01457.x. [DOI] [PubMed] [Google Scholar]
- 107.Wen XZ, Akiyama Y, Pan KF, Liu ZJ, Lu ZM, Zhou J, Gu LK, Dong CX, Zhu BD, Ji JF, et al. Methylation of GATA-4 and GATA-5 and development of sporadic gastric carcinomas. World J Gastroenterol. 2010;16:1201–1208. doi: 10.3748/wjg.v16.i10.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R, Kumar R. p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep. 2004;5:154–160. doi: 10.1038/sj.embor.7400079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yan J, Zhang M, Zhang J, Chen X, Zhang X. Helicobacter pylori infection promotes methylation of WWOX gene in human gastric cancer. Biochem Biophys Res Commun. 2011;408:99–102. doi: 10.1016/j.bbrc.2011.03.127. [DOI] [PubMed] [Google Scholar]
- 110.Kang C, Song JJ, Lee J, Kim MY. Epigenetics: an emerging player in gastric cancer. World J Gastroenterol. 2014;20:6433–6447. doi: 10.3748/wjg.v20.i21.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 112.Stahlhut Espinosa CE, Slack FJ. The role of microRNAs in cancer. Yale J Biol Med. 2006;79:131–140. [PMC free article] [PubMed] [Google Scholar]
- 113.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 114.Vickers KC, Rye KA, Tabet F. MicroRNAs in the onset and development of cardiovascular disease. Clin Sci (Lond) 2014;126:183–194. doi: 10.1042/CS20130203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Raisch J, Darfeuille-Michaud A, Nguyen HT. Role of microRNAs in the immune system, inflammation and cancer. World J Gastroenterol. 2013;19:2985–2996. doi: 10.3748/wjg.v19.i20.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhai H, Fesler A, Ju J. MicroRNA: a third dimension in autophagy. Cell Cycle. 2013;12:246–250. doi: 10.4161/cc.23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang J, Ma L. MicroRNA control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012;31:653–662. doi: 10.1007/s10555-012-9368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eulalio A, Schulte L, Vogel J. The mammalian microRNA response to bacterial infections. RNA Biol. 2012;9:742–750. doi: 10.4161/rna.20018. [DOI] [PubMed] [Google Scholar]
- 120.Tong F, Cao P, Yin Y, Xia S, Lai R, Liu S. MicroRNAs in gastric cancer: from benchtop to bedside. Dig Dis Sci. 2014;59:24–30. doi: 10.1007/s10620-013-2887-3. [DOI] [PubMed] [Google Scholar]
- 121.Ueda T, Volinia S, Okumura H, Shimizu M, Taccioli C, Rossi S, Alder H, Liu CG, Oue N, Yasui W, et al. Relation between microRNA expression and progression and prognosis of gastric cancer: a microRNA expression analysis. Lancet Oncol. 2010;11:136–146. doi: 10.1016/S1470-2045(09)70343-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang XW, Wu Y, Wang D, Qin ZF. MicroRNA network analysis identifies key microRNAs and genes associated with precancerous lesions of gastric cancer. Genet Mol Res. 2014;13:8695–8703. doi: 10.4238/2014.October.27.10. [DOI] [PubMed] [Google Scholar]
- 123.Zhou X, Li L, Su J, Zhang G. Decreased miR-204 in H. pylori-associated gastric cancer promotes cancer cell proliferation and invasion by targeting SOX4. PLoS One. 2014;9:e101457. doi: 10.1371/journal.pone.0101457. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Song MY, Pan KF, Su HJ, Zhang L, Ma JL, Li JY, Yuasa Y, Kang D, Kim YS, You WC. Identification of serum microRNAs as novel non-invasive biomarkers for early detection of gastric cancer. PLoS One. 2012;7:e33608. doi: 10.1371/journal.pone.0033608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yu X, Luo L, Wu Y, Yu X, Liu Y, Yu X, Zhao X, Zhang X, Cui L, Ye G, et al. Gastric juice miR-129 as a potential biomarker for screening gastric cancer. Med Oncol. 2013;30:365. doi: 10.1007/s12032-012-0365-y. [DOI] [PubMed] [Google Scholar]
- 126.Liu WJ, Xu Q, Sun LP, Dong QG, He CY, Yuan Y. Expression of serum let-7c, let-7i, and let-7f microRNA with its target gene, pepsinogen C, in gastric cancer and precancerous disease. Tumour Biol. 2015;36:3337–3343. doi: 10.1007/s13277-014-2967-9. [DOI] [PubMed] [Google Scholar]
- 127.Fu Z, Qian F, Yang X, Jiang H, Chen Y, Liu S. Circulating miR-222 in plasma and its potential diagnostic and prognostic value in gastric cancer. Med Oncol. 2014;31:164. doi: 10.1007/s12032-014-0164-8. [DOI] [PubMed] [Google Scholar]
- 128.Matsushima K, Isomoto H, Inoue N, Nakayama T, Hayashi T, Nakayama M, Nakao K, Hirayama T, Kohno S. MicroRNA signatures in Helicobacter pylori-infected gastric mucosa. Int J Cancer. 2011;128:361–370. doi: 10.1002/ijc.25348. [DOI] [PubMed] [Google Scholar]
- 129.Noto JM, Peek RM. The role of microRNAs in Helicobacter pylori pathogenesis and gastric carcinogenesis. Front Cell Infect Microbiol. 2011;1:21. doi: 10.3389/fcimb.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene. 2013;532:1–12. doi: 10.1016/j.gene.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 131.Thompson RC, Vardinogiannis I, Gilmore TD. Identification of an NF-κB p50/p65-responsive site in the human MIR155HG promoter. BMC Mol Biol. 2013;14:24. doi: 10.1186/1471-2199-14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xiao B, Liu Z, Li BS, Tang B, Li W, Guo G, Shi Y, Wang F, Wu Y, Tong WD, et al. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J Infect Dis. 2009;200:916–925. doi: 10.1086/605443. [DOI] [PubMed] [Google Scholar]
- 133.Saito Y, Suzuki H, Tsugawa H, Imaeda H, Matsuzaki J, Hirata K, Hosoe N, Nakamura M, Mukai M, Saito H, et al. Overexpression of miR-142-5p and miR-155 in gastric mucosa-associated lymphoid tissue (MALT) lymphoma resistant to Helicobacter pylori eradication. PLoS One. 2012;7:e47396. doi: 10.1371/journal.pone.0047396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tang B, Xiao B, Liu Z, Li N, Zhu ED, Li BS, Xie QH, Zhuang Y, Zou QM, Mao XH. Identification of MyD88 as a novel target of miR-155, involved in negative regulation of Helicobacter pylori-induced inflammation. FEBS Lett. 2010;584:1481–1486. doi: 10.1016/j.febslet.2010.02.063. [DOI] [PubMed] [Google Scholar]
- 135.Koch M, Mollenkopf HJ, Klemm U, Meyer TF. Induction of microRNA-155 is TLR- and type IV secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc Natl Acad Sci USA. 2012;109:E1153–E1162. doi: 10.1073/pnas.1116125109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Deng H, Guo Y, Song H, Xiao B, Sun W, Liu Z, Yu X, Xia T, Cui L, Guo J. MicroRNA-195 and microRNA-378 mediate tumor growth suppression by epigenetical regulation in gastric cancer. Gene. 2013;518:351–359. doi: 10.1016/j.gene.2012.12.103. [DOI] [PubMed] [Google Scholar]
- 137.Jia H, Zhang Z, Zou D, Wang B, Yan Y, Luo M, Dong L, Yin H, Gong B, Li Z, et al. MicroRNA-10a is down-regulated by DNA methylation and functions as a tumor suppressor in gastric cancer cells. PLoS One. 2014;9:e88057. doi: 10.1371/journal.pone.0088057. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 138.Lei H, Zou D, Li Z, Luo M, Dong L, Wang B, Yin H, Ma Y, Liu C, Wang F, et al. MicroRNA-219-2-3p functions as a tumor suppressor in gastric cancer and is regulated by DNA methylation. PLoS One. 2013;8:e60369. doi: 10.1371/journal.pone.0060369. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 139.Guo LH, Li H, Wang F, Yu J, He JS. The Tumor Suppressor Roles of miR-433 and miR-127 in Gastric Cancer. Int J Mol Sci. 2013;14:14171–14184. doi: 10.3390/ijms140714171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Li P, Chen X, Su L, Li C, Zhi Q, Yu B, Sheng H, Wang J, Feng R, Cai Q, et al. Epigenetic silencing of miR-338-3p contributes to tumorigenicity in gastric cancer by targeting SSX2IP. PLoS One. 2013;8:e66782. doi: 10.1371/journal.pone.0066782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sakamoto N, Naito Y, Oue N, Sentani K, Uraoka N, Zarni Oo H, Yanagihara K, Aoyagi K, Sasaki H, Yasui W. MicroRNA-148a is downregulated in gastric cancer, targets MMP7, and indicates tumor invasiveness and poor prognosis. Cancer Sci. 2014;105:236–243. doi: 10.1111/cas.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hashimoto Y, Akiyama Y, Yuasa Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS One. 2013;8:e62589. doi: 10.1371/journal.pone.0062589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li Z, Lei H, Luo M, Wang Y, Dong L, Ma Y, Liu C, Song W, Wang F, Zhang J, et al. DNA methylation downregulated mir-10b acts as a tumor suppressor in gastric cancer. Gastric Cancer. 2015;18:43–54. doi: 10.1007/s10120-014-0340-8. [DOI] [PubMed] [Google Scholar]
- 144.Kiga K, Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, Kim M, Ogawa M, Iwasaki YW, Kayo H, et al. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic Helicobacter pylori infection. Nat Commun. 2014;5:4497. doi: 10.1038/ncomms5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chan YC, Banerjee J, Choi SY, Sen CK. miR-210: the master hypoxamir. Microcirculation. 2012;19:215–223. doi: 10.1111/j.1549-8719.2011.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Corvalán AH, Torres VA. RPRM (reprimo, TP53 dependent G2 arrest mediator candidate) Atlas Genet Cytogenet Oncol Haematol. 2012;16:223–224. [Google Scholar]
- 147.Ohki R, Nemoto J, Murasawa H, Oda E, Inazawa J, Tanaka N, Taniguchi T. Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J Biol Chem. 2000;275:22627–22630. doi: 10.1074/jbc.C000235200. [DOI] [PubMed] [Google Scholar]
- 148.Saavedra K, Valbuena J, Olivares W, Marchant MJ, Rodríguez A, Torres-Estay V, Carrasco-Avino G, Guzmán L, Aguayo F, Roa JC, et al. Loss of Expression of Reprimo, a p53-induced Cell Cycle Arrest Gene, Correlates with Invasive Stage of Tumor Progression and p73 Expression in Gastric Cancer. PLoS One. 2015;10:e0125834. doi: 10.1371/journal.pone.0125834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schneider BG, Peng DF, Camargo MC, Piazuelo MB, Sicinschi LA, Mera R, Romero-Gallo J, Delgado AG, Bravo LE, Wilson KT, et al. Promoter DNA hypermethylation in gastric biopsies from subjects at high and low risk for gastric cancer. Int J Cancer. 2010;127:2588–2597. doi: 10.1002/ijc.25274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bernal C, Aguayo F, Villarroel C, Vargas M, Díaz I, Ossandon FJ, Santibáñez E, Palma M, Aravena E, Barrientos C, et al. Reprimo as a potential biomarker for early detection in gastric cancer. Clin Cancer Res. 2008;14:6264–6269. doi: 10.1158/1078-0432.CCR-07-4522. [DOI] [PubMed] [Google Scholar]
- 151.Hanigan MH. gamma-Glutamyl transpeptidase, a glutathionase: its expression and function in carcinogenesis. Chem Biol Interact. 1998;111-112:333–342. doi: 10.1016/s0009-2797(97)00170-1. [DOI] [PubMed] [Google Scholar]
- 152.Chevalier C, Thiberge JM, Ferrero RL, Labigne A. Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol Microbiol. 1999;31:1359–1372. doi: 10.1046/j.1365-2958.1999.01271.x. [DOI] [PubMed] [Google Scholar]
- 153.Kim KM, Lee SG, Park MG, Song JY, Kang HL, Lee WK, Cho MJ, Rhee KH, Youn HS, Baik SC. Gamma-glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem Biophys Res Commun. 2007;355:562–567. doi: 10.1016/j.bbrc.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 154.Kim KM, Lee SG, Kim JM, Kim DS, Song JY, Kang HL, Lee WK, Cho MJ, Rhee KH, Youn HS, et al. Helicobacter pylori gamma-glutamyltranspeptidase induces cell cycle arrest at the G1-S phase transition. J Microbiol. 2010;48:372–377. doi: 10.1007/s12275-010-9293-8. [DOI] [PubMed] [Google Scholar]
- 155.Gong M, Ling SS, Lui SY, Yeoh KG, Ho B. Helicobacter pylori gamma-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology. 2010;139:564–573. doi: 10.1053/j.gastro.2010.03.050. [DOI] [PubMed] [Google Scholar]
- 156.Ricci V, Giannouli M, Romano M, Zarrilli R. Helicobacter pylori gamma-glutamyl transpeptidase and its pathogenic role. World J Gastroenterol. 2014;20:630–638. doi: 10.3748/wjg.v20.i3.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lladser A, Sanhueza C, Kiessling R, Quest AF. Is survivin the potential Achilles’ heel of cancer? Adv Cancer Res. 2011;111:1–37. doi: 10.1016/B978-0-12-385524-4.00001-5. [DOI] [PubMed] [Google Scholar]
- 158.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fernández JG, Rodríguez DA, Valenzuela M, Calderon C, Urzúa U, Munroe D, Rosas C, Lemus D, Díaz N, Wright MC, et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced β-catenin/Tcf-Lef dependent transcription. Mol Cancer. 2014;13:209. doi: 10.1186/1476-4598-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Valenzuela M, Pérez-Pérez G, Corvalán AH, Carrasco G, Urra H, Bravo D, Toledo H, Quest AF. Helicobacter pylori-induced loss of the inhibitor-of-apoptosis protein survivin is linked to gastritis and death of human gastric cells. J Infect Dis. 2010;202:1021–1030. doi: 10.1086/656143. [DOI] [PubMed] [Google Scholar]
- 161.Valenzuela M, Bravo D, Canales J, Sanhueza C, Díaz N, Almarza O, Toledo H, Quest AF. Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial gamma-glutamyl transpeptidase activity. J Infect Dis. 2013;208:1131–1141. doi: 10.1093/infdis/jit286. [DOI] [PubMed] [Google Scholar]
- 162.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]