

Abstract
AIM: To evaluate the inflammasome activation and the effect of peroxisome proliferator-activated receptors (PPAR)-δ agonist treatment in nonalcoholic fatty liver disease (NAFLD) models.
METHODS: Male C57BL/6J mice were classified according to control or high fat diet (HFD) with or without PPAR-δ agonist (GW) over period of 12 wk [control, HFD, HFD + lipopolysaccharide (LPS), HFD + LPS + GW group]. HepG2 cells were exposed to palmitic acid (PA) and/or LPS in the absence or presence of GW.
RESULTS: HFD caused glucose intolerance and hepatic steatosis. In mice fed an HFD with LPS, caspase-1 and interleukin (IL)-1β in the liver were significantly increased. Treatment with GW ameliorated the steatosis and inhibited overexpression of pro-inflammatory cytokines. In HepG2 cells, PA and LPS treatment markedly increased mRNA of several nucleotide-binding and oligomerization domain-like receptor family members (NLRP3, NLRP6, and NLRP10), caspase-1 and IL-1β. PA and LPS also exaggerated reactive oxygen species production. All of the above effects of PA and LPS were reduced by GW. GW also enhanced the phosphorylation of AMPK-α.
CONCLUSION: PPAR-δ agonist reduces fatty acid-induced inflammation and steatosis by suppressing inflammasome activation. Targeting the inflammasome by the PPAR-δ agonist may have therapeutic implication for NAFLD.
Keywords: Nonalcoholic fatty liver disease, Nonalcoholic steatohepatitis, Inflammasome, Nucleotide-binding and oligomerization domain-like receptor, Peroxisome proliferator-activated receptors delta
Core tip: Until now, the underlying mechanisms of disease progression and therapeutic targets were uncertain in nonalcoholic fatty liver disease (NAFLD). Our study were to evaluate the inflammasome activation and the effect of peroxisome proliferator-activated receptors (PPAR)-δ agonist treatment in NAFLD models. In our NAFLD models, mRNA of several NOD-like receptor family members, caspase-1 and interleukin-1β were markedly increased. All of those effects were reduced by PPAR-δ agonist treatment. It also ameliorated the steatosis and inhibited overexpression of pro-inflammatory cytokines. In conclusion, PPAR-δ agonist reduces fatty acid-induced inflammation and steatosis by suppressing inflammasome activation.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is one of the most common liver diseases, occurring in 10%-35% of the general population, and its prevalence has increased in parallel with the worldwide epidemic of obesity and its related insulin-resistant state[1,2]. The spectrum of NAFLD ranges from simple steatosis to nonalcoholic steatohepatitis (NASH) and fibrosis. Certain portion of NAFLD eventually progressed to liver cirrhosis and hepatocellular carcinoma[1,3]. Recently, the two-hit hypothesis was been reported in the literature[4], but the precise mechanism involved in the development and progression of NAFLD is not entirely understood.
Regarding the pathogenesis of NAFLD[5,6], steatosis sensitizes the liver and makes it susceptible to additional insults. Factors such as increased oxidative stress, pro-inflammatory cytokines and impaired adenosine triphosphate (ATP) production[7,8] could trigger necroinflammation and lead to the progression of steatohepatitis. Although many kind of drugs such as thiazolidinediones, vitamin E, losartan, and silybin have been evaluated in several studies, few pharmacological treatments can be recommended at present[9-11].
Inflammasome is a large, intracellular multi-protein complex that is a sensor of the exogenous and endogenous danger signals, such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), that promotes the cleavage and maturation of pro-inflammatory cytokines such as pro-interleukin (IL)-1β and pro-IL-18[12,13]. Most DAMPs induce the production of reactive oxygen species (ROS), which results in nucleotide-binding and oligomerization domain (NOD)-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation[14]. The NLRP3 inflammasome is known to be related to the pathogenesis of obesity, insulin resistance, and development of diabetes[15-18]. But, the role of inflammasome in the pathogenesis of NAFLD/NASH has not been elucidated.
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptor proteins, and three types have been identified in mammals: PPAR-α, PPAR-γ, and PPAR-δ. Pharmacological targets of PPAR-α and PPAR-γ are relatively well-known in the treatment of dyslipidemia and diabetes[19]. It has become increasingly apparent that PPAR-δ also play important roles in the regulation of metabolism, as its activation increases fatty acid (FA) oxidation, ameliorates glucose homeostasis, and attenuates macrophage inflammatory responses[19,20]. In the mouse model of NASH, PPAR-δ improves hepatic steatosis and inflammation by regulation of lipid metabolism and inhibition of inflammatory response[21].
In this study, we determined whether treatment with the PPAR-δ agonist could ameliorate high fat-induced inflammasome activation and the inflammatory response using in vivo and in vitro NAFLD models.
MATERIALS AND METHODS
Materials
PPAR-δ agonist, GW501516 (GW), was obtained from Enzo Life Sciences (Farmingdale, NY, United States). Lipopolysaccharide (LPS) (Escherichia coli 0111:B4) and palmitic acid (PA) were purchased from Sigma-Aldrich (St Louis, MO, United States). Solutions and reagents used for cell culture were obtained from Invitrogen (Carlsbad, CA, United States) unless otherwise noted. Antibodies were purchased from Cell Signaling Technology Inc. (Beverly, MA, United States).
Animals and experimental design
The animal protocol was designed to minimize pain or discomfort to the animals. Male 4-5-wk-old C57BL/6J mice were obtained from Japan SLC Inc. (Shizuoka, Japan). They were acclimatized to laboratory conditions (22-24 °C and 37%-64% humidity, with a 12-h dark-light cycle, free access to food and water) for one week prior to experimentation. Reflecting endotoxemia in NAFLD, we were decided to inject non-lethal very low dosage of LPS. Mice were randomly divided into four groups, which were treated for 12 wk as follows: standard diet (control, n = 5); HFD (HFD, 60% kcal from fat; D12492, Research Diets; New Brunswick, NJ, United States, n = 5); HFD plus one daily oral gavage of vehicle (0.5% carboxymethyl cellulose solution) with one weekly intraperitoneal (IP) injection of LPS (1 mg/kg per week) (HFD + LPS, n = 5); and HFD plus one daily oral dose of 3 mg/kg per day of GW501516, which was dissolved in the vehicle, with IP injection of LPS (HFD + LPS + GW, n = 6). Vehicle and GW501516 were administered for the last 3 wk. Body weight and food intake were recorded weekly. Body length (from nose to anus) was measured before sacrifice. Body mass index (BMI) was calculated by dividing body weight by the square of the body length (g/cm2)[22].
The study was reviewed and approved by the Institutional Review Board of Korea University and it was approved by the institutional animal review board of Korea University, Seoul, Korea, KUIACUC-2013-66 and conducted in compliance with the Guide for the Care and Use of Laboratory Animals.
Glucose tolerance test
The glucose tolerance test was conducted in all animals at 11 wk after dietary manipulation. After overnight fasting, 2 g/kg glucose was injected intraperitoneally, and blood samples were taken from the tail vein at 0, 15, 30, 60, 90, and 120 min. Blood glucose was measured using an Accu-Check Compact kit (Roche Diagnostics GmbH; Mannheim, Germany).
Blood biochemistry
At 12 wk, the mice were intraperitoneally anesthetized with a mixture of tiletamine/zolazepam (30 mg/kg, Zoletil; Yuhan Corp.; Seoul, Korea) and xylazine (10 mg/kg, Rompun; Bayer, Inc.; Frankfurt, Germany), and sacrificed by exsanguination. Blood samples were extracted and the serum was isolated. The livers were rapidly excised and weighed. Serum alanine transaminase (ALT), aspartate transaminase (AST), triglyceride (TG), and total cholesterol (TC) levels were measured using common biochemical kits (Mindray Medical International Ltd.; Shenzhen, China).
Liver histological analysis
The right liver lobe was stored at -80 °C until analysis of mRNA and protein. The left liver lobe was immediately fixed in 10% neutral-buffered formalin, paraffin-embedded, sectioned, and sections were stained with hematoxylin and eosin (HE). To visualize the neutral lipids, some of the frozen sections of fresh liver were stained using Oil Red O reagent. The liver samples were examined histologically in a blind manner by an experienced pathologist using the histological scoring system for NAFLD[23].
Cell culture and treatment
The human hepatoma HepG2 cell line (ATCC; Manassas, VA, United States) cells were cultured as manufacture’s instruction. In all experiments, PA concentration of 0.2 mmol/L which had no influence on cell viability was selected. The cells were stimulated with PA-BSA (0.2 μmol/L), LPS (1 μg/mL), or both, with or without GW501516 (1 or 10 μmol/L).
RNA preparation and analysis
Total RNA was extracted from mouse liver tissues and HepG2 cells with TRIzol reagent according to the manufacturer’s instructions. RNA was reverse-transcribed into cDNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems; Foster City, CA, United States). Gene expressions of NLRP1, NLRP3, NLRP6, NLRP10, NLRC4, caspase-1, and IL-1β were quantified by real-time polymerase chain reaction (PCR) with a 7000 Real-time PCR system (Applied Biosystems). PCR reactions were prepared with the Power SYBR Green PCR Master mix (Applied Biosystems) in 20 μL. The cycling parameters were 10 min at 95 °C followed by 40 cycles of 95 °C (15 s) and 30 s at 60 °C, followed by melting curve analysis. Gene expression by real-time PCR was presented relative to glyceraldehyde 3-phosphate dehydrogenase. The PCR primers used are listed in the Table 1.
Table 1.
Sequence of primers employed for quantitative real-time polymerase chain reaction
| mRNA | Forward (5'-, -3') | Reverse (5'-, -3') |
| Mouse | ||
| Caspase-1 | CACAGCTCTGGAGATGGTGA | GGTCCCACATATTCCCTCC |
| IL-1β | GCTGCTTCCAAACCTTTGAC | AGCTTCTCCACAGCCACAAT |
| GAPDH | CAGCCTCAAGATCATCAGCA | GTCTTCTGGGTGGCAGTGAT |
| Human | ||
| NLRP1 | TCTCAAGGGGACCTGCATAC | AGCCAGCTACAGGGAAGTGA |
| NLRP3 | AAGGAAGTGGACTGCGAGA | AACGTTCGTCCTTCCTTCCT |
| NLRP6 | TTCATCACCAGCGTTCTGAG | GTTGCTCCAGTTCCTTCTCG |
| NLRP10 | GGATGAGAAGCAAGCTGACC | CTTTGCCTCTCTCCATCTGC |
| NLRC4 | GCAAGGCTCTGACCAAGTTC | TGTCTGCTTCCTGATTGTGC |
| Caspase-1 | CCACAATGGGCTCTGTTTTT | CATCTGGCTGCTCAAATGAA |
| IL-1β | GGACAAGCTGAGGAAGATGC | TCGTTATCCCATGTGTCGAA |
| GAPDH | CAGCCTCAAGATCATCAGCA | GTCTTCTGGGTGGCAGTGAT |
GAPDH: Glyceraldehyde 3-phosphate dehydrogenase.
Western blot analysis
Mouse liver tissues and HepG2 cell were lysed using M-PER Mammalian Protein Extraction Reagent (Pierce Chemical; Rockford, IL, United States). The protein concentration was determined using the bicinchoninic acid (BCA) method. Equal amounts (20 μg) of total proteins were boiled for 5 min and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by electrotransfer to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% BSA in Tris-buffered saline (TBS) for 1 h at room temperature, followed by incubation with antibodies: anti-capase-1 (D7F10), anti-IL-1β (3A6), anti-p-AMPK-α (Thr172;40H9), anti-AMPK-α (23A3), and anti-β-actin (AC-15). The bands were visualized with an enhanced chemiluminescent direct labeling (ECL) system. The Gel Pro Analyzer 4.5, 2000 software (Media Cybernetics; Silver Spring, MD, United States) was used to determine the band density.
Measurement of lipid peroxidation in the liver and intracellular ROS production
Liver was perfused with saline and homogenized in Tris-HCl buffer (20 mmol/L, pH 7.4). The homogenates were centrifuged at 2500 × g for 10 min at 4 °C. Two hundred microliters of homogenates was analyzed for malondialdehyde (MDA) levels using a kit (Cell Biolabs, Inc.; San Diego, CA, United States), and the value was read at 532 nm.
Intracellular ROS levels in HepG2 cells were determined using 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA). Cells were seeded on a 96-well bottom dishes. After the treatment described above in Cell culture and treatment, the cells were collected, and incubated with 10 μmol/L DCFDA (Molecular Probes; CA, United States) for 30 min at 37 °C. DCFDA fluorescence intensity was measured at 530 nm with an excitation wavelength of 488 nm using flow cytometry (Becton Dickinson; Heidelberg, Germany). The percentage of ROS-producing cells was determined by counting only those cells that produced high levels of ROS.
Statistical analysis
Data are presented as mean ± SD. Statistical significance was determined with one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons using the GraphPad Instant program (GraphPad Software version 5.00; GraphPad Software Inc.; San Diego, CA). P < 0.05 was considered statistically significant. The statistical methods of this study were reviewed by a biomedical statistician.
RESULTS
Effects of HFD and GW501516 on body weight, blood glucose, and hepatic function in mice fed the HFD
The body weight and BMI of the HFD group were significantly higher compared with those of control mice (all P < 0.05) (Table 2). The food intake was greater in the HFD group compared with the HFD + LPS and HFD + LPS + GW501516 groups. The proportion of liver weight to body weight or body length was similar among the four groups. Serum AST and ALT levels were significantly increased in the HFD group compared with the control group (AST, 123.8 ± 30.54 IU/L vs 52.6 ± 10.33 IU/L; ALT, 136.6 ± 69.43 IU/L vs 23.4 ± 4.04 IU/L, all P < 0.05). GW treatment in HFD+LPS group significantly reduced serum AST and ALT compared with HFD (AST, 45.67 ± 11.11 IU/L vs 123.8 ± 30.54 IU/L; ALT, 20.5 ± 6.12 IU/L vs 136.6 ± 69.43 IU/L, all P < 0.05). HFD and/or LPS injection had no significant effect on TG or TC levels.
Table 2.
Clinical and biochemical characteristics
| Control | HFD | HFD + LPS | HFD + LPS + GW | |
| (n = 5) | (n = 5) | (n = 5) | (n = 6) | |
| Food/wk (g) | 20.78 ± 1.41 | 17.33 ± 0.82a | 14.08 ± 1.28ac | 13.92 ± 1.23ac |
| BW (g) | 26.70 ± 0.69 | 41.04 ± 4.69a | 30.98 ± 3.86c | 29.87 ± 3.31c |
| BMI (g/cm2) | 0.33 ± 0.02 | 0.41 ± 0.04a | 0.36 ± 0.02c | 0.35 ± 0.02c |
| Liver/BW (%) | 3.50 ± 0.30 | 3.04 ± 0.46 | 2.77 ± 0.27 | 3.08 ± 0.94 |
| Liver/BL (g/cm) | 0.10 ± 0.01 | 0.13 ± 0.03 | 0.09 ± 0.01 | 0.10 ± 0.03 |
| AST (IU/L) | 52.6 ± 10.33 | 123.8 ± 30.54a | 62.0 ± 13.71c | 45.67 ± 11.11c |
| ALT (IU/L) | 23.4 ± 4.04 | 136.6 ± 69.43a | 49.6 ± 31.09c | 20.5 ± 6.12c |
| TG (mg/dL) | 97.8 ± 19.33 | 69.6 ± 21.55 | 65.2 ± 13.16 | 79.2 ± 23.82 |
| TC (mg/dL) | 86.6 ± 10.92 | 118.4 ± 37.16 | 116.0 ± 9.43 | 135.3 ± 34.09a |
Data are presented as mean ± SD. P values are presented as:
P < 0.05 vs control;
P < 0.05 vs HFD. BW: Body weight; BMI: Body mass index; BL: Body length; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; TG: Triglyceride; TC: Total cholesterol; HFD: High fat diet.
When subjected to a glucose tolerance test, glucose levels of control mice peaked at 30 min and returned to the basal level (Figure 1A). As expected, mice fed the HFD with or without LPS infection were more glucose-intolerant compared with controls, as demonstrated by the significant increase in the area under the curve (AUC) (53108 ± 9750 mg/dL and 52653 ± 9425 mg/dL vs 34545 ± 2776 mg/dL × 120 min, all P < 0.01) (Figure 1B). However, this glucose intolerance was not abolished by GW501516 treatment.
Figure 1.
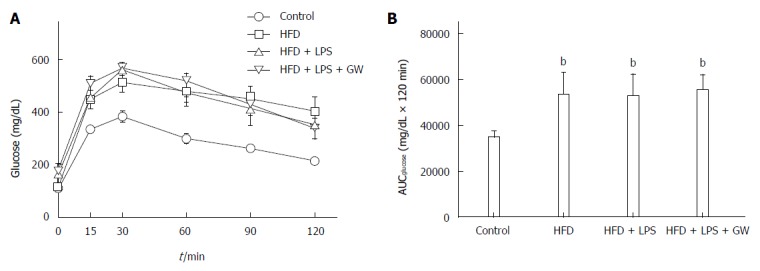
Effects of high-fat diet for 12 wk on glucose tolerance in male mice treated with GW501516 (GW, 3 mg/kg per day for 3 wk). Intraperitoneal glucose tolerance test (A) and area under the curve for glucose (AUCglucose) (B) showed glucose intolerance in mice fed the HFD with or without lipopolysaccharide (LPS) injection compared with the control group. GW treatment did not improve glucose intolerance. Data are expressed as the mean ± SD (5-6 mice per group). bP < 0.01 vs control group.
Effects of GW501516 on IL-1β levels and inflammasome activation in mice and HepG2 cells
In addition to steatosis, chronic inflammation is an important contributing factor in NASH pathogenesis[5]. To investigate the inflammatory response in this model, we measured hepatic caspase-1 and IL-1β levels. The gene expression level of caspase-1, which is known to be activated by the NLRP3 inflammasome complex and to cleave pro-IL-1β to the active form, was increased in the HFD + LPS group relative to the control group (P < 0.01) (Figure 2A). The hepatic gene expression and protein levels of IL-1β were also increased in the HFD + LPS group relative to the control group (P < 0.05 for mRNA expression and P < 0.001 for protein-level expression) (Figure 2B and C). When compared with HFD+LPS, PPAR-delta agonist GW treatment reduced hepatic caspase-1 mRNA (2.8-fold vs 1.7-fold) and IL-1β mRNA (2.2-fold vs 1.6-fold).
Figure 2.
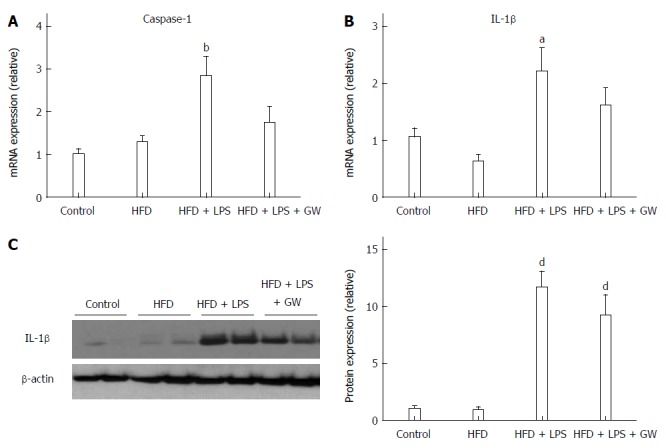
Effects of GW501516 on caspase-1 and IL-1β levels in mice fed a high fat diet. Hepatic mRNA levels of caspase-1 (A) and IL-1β (B) at 12 wk showed a significant increase in the HFD + lipopolysaccharide (LPS) group compared with the control group. GW501516 treatment tended to ameliorate this increase. Representative immunoblot and graphic presentation show hepatic protein levels of IL-1β (C) in different groups of mice as indicated. Mice fed an HFD with LPS injection with or without GW501516 treatment had increased protein levels of IL-1β. Data are expressed as the mean ± SD (5-6 mice per group). aP < 0.05, bP < 0.01, and dP < 0.001 vs control group.
We also investigated the effect of PPAR-δ activator on inflammasome induced by PA and LPS in HepG2 cells. First of all, we analyzed the PPAR-δ protein expression itself in various stimuli. In vitro exposure with- or without stimuli such as LPS only, PA only or GW only, or combined with it, PPAR-delta protein expressions were not different. After then, we analyzed the expression of several inflammasome-related mRNAs upon activation with PA and/or LPS. PA and LPS together elicited the mRNA expression of NLRP3, NRLP6, and NLRP10 (all P < 0.05) (Figure 3). We next tested whether GW501516 could prevent the overexpression of such inflammasome components and the downstream pro-inflammatory signals. As shown in Figure 4, treatment with 10 μmol/L GW501516 reduced the PA and LPS-induced mRNA expression of NLRP3, NRLP6, and NLRP10 (all P < 0.05). GW501516 also reduced the expression of caspase-1 and IL-1β mRNA in a dose-dependent manner (all P < 0.05) (Figure 4D and E). In concordance with mRNA expression, the presence of GW501516 reduced the PA and LPS-induced production of caspase-1 and IL-1β protein expression (Figure 4F).
Figure 3.

Expression of inflammasome components in HepG2 cells after stimulation with palmitic acid and lipopolysaccharide. Relative mRNA levels of NLRP1, NLRP3, NLRC4, NLRP6, and NLRP10 in HepG2 cells were analyzed by RT-PCR. Compared with the control group, significantly increased expression of NLPR3, NLRP6, and NLRP10 was observed in the PA and LPS stimulation group. Data are expressed as the mean ± SD. aP < 0.05 vs control cells.
Figure 4.
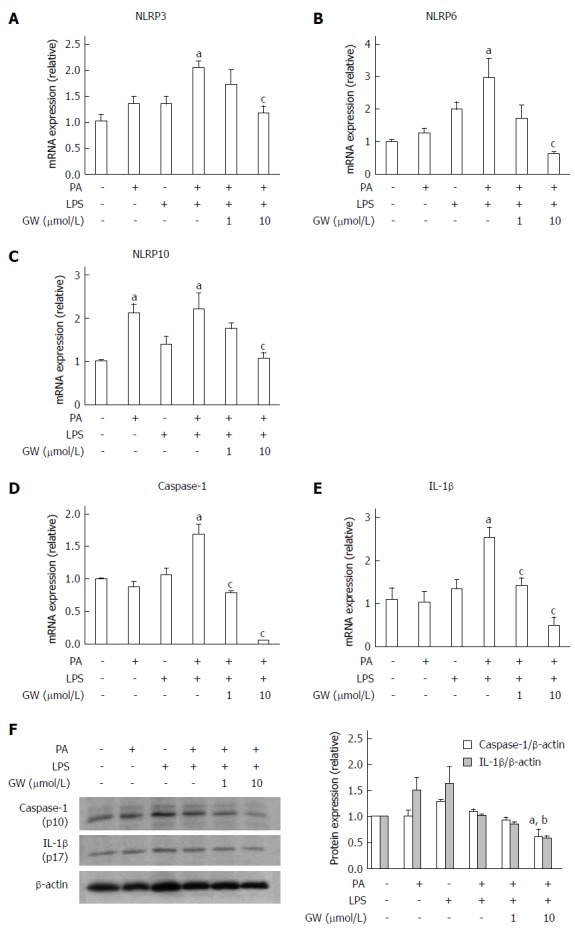
Effects of GW501516 on palmitic acid- and lipopolysaccharide-induced inflammasome and pro-inflammatory cytokine in HepG2 cells. Relative mRNA levels of NLRP3 (A), NLRP6 (B), NLRP10 (C), caspase-1 (D), and IL-1β (E) in HepG2 cells were analyzed by RT-PCR. GW501516 inhibited PA- and LPS-induced mRNA expression of several inflammasome components, caspase-1, and IL-1β. Representative immunoblot and graphic presentation show protein levels of caspase-1 and IL-1β (F) in HepG2 cells. PA and LPS tended to elicit caspase-1 and IL-1β release, and GW501516 reduced this processing significantly. Data are expressed as the mean ± SD. aP < 0.05 vs control cells, cP < 0.05 and bP < 0.01 vs PA + LPS-treated cells.
Effects of GW501516 on hepatic steatosis in mice fed the HFD
Histological images of liver pathology were obtained. Liver sections were stained with HE and Oil Red O. In mice fed the standard diet, there was no detectable fatty change in the microscopic image. By contrast, relative to the control group, mice on a 12-wk HFD with or without LPS injection developed extensive macrovesicular steatosis (0.0 vs 17.0% ± 5.7% and 16.4% ± 14.7%, all P < 0.05) and inflammation around the perisinusoidal area. Ballooned hepatocytes and fibrosis were not observed in any study group. In mice that were administrated GW501516, the intensity of fat accumulation (2.7% ± 2.6%) and NAS (0.5) were significantly decreased (P < 0.05) compared to those of the HFD + LPS group (16.4% ± 14.7% and 1.8%, respectively) (Figure 5A, B and C).
Figure 5.

Histopathological features of livers in mice fed a high fat diet with or without lipopolysaccharide injection and GW501516 treatment. A: Hematoxylin and eosin (HE) staining and Oil Red O staining of hepatic lipid accumulation (magnification × 100). In mice fed an HFD, moderate macrovesicular steatosis (17.0%) and inflammatory cell infiltration were observed compared with the control group. The macrovesicular steatosis was improved to 2.7% following GW501516 treatment; B: Histogram of the percentage of hepatocytes showing macrovesicular fatty change; C: Nonalcoholic fatty liver disease (NAFLD) activity score. The NAFLD activity score in the GW501516-treated group was significantly lower than that in the HFD + LPS group (1.8 vs 0.5, P < 0.05). Data are expressed as the mean ± standard deviation (SD) (5-6 mice per group). aP < 0.05, bP < 0.01, and dP < 0.001 vs control group. cP < 0.05 vs the HFD + LPS group.
Effects of GW501516 on phosphorylation of AMPKα and ROS generation in mice and HepG2 cells
Oxidative stress is thought to play a role in the pathogenesis of NASH, and ROS are essential for inflammasome activation[7,14]. A large body of evidence indicates that AMP-activated protein kinase (AMPK) is an essential regulator of fatty acid metabolism[21], and suppresses ROS generation by regulating intracellular nicotinamide adenine dinucleotide phosphate (NADPH) production[18,24]. In the in vivo study, levels of MDA, a product of lipid peroxidation, in liver homogenates, there was no statistically significant difference in four groups (Figure 6A). We also examined the effect of GW501516 on AMPK activation. As the activity of AMPK correlates with phosphorylation at Thr-172, the activation of AMPK was assessed by determining phosphorylation of AMPKα. As shown in Figure 6B, treatment with GW501516 increased the levels of AMPKα phosphorylation in mice fed the HFD with LPS injection by 1.6-fold compared to the control group.
Figure 6.
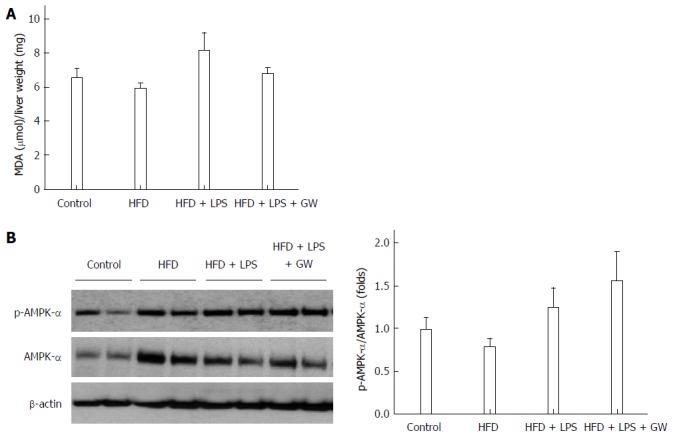
Effects of GW501516 on lipid peroxidation and associated molecular pattern-activated protein kinase-α phosphorylation in mice fed a high fat diet. Liver malondialdehyde (MDA) levels (A) showed a slight increase in the HFD + lipopolysaccharide (LPS) group without statistical significance; B: Representative immunoblot and graphic presentation of p-AMPK-α and total AMPK-α protein levels in the livers of mice are shown. Treatment of GW501516 enhanced phosphorylation of AMPK-α in mice fed an HFD with LPS injection. Data are expressed as the mean ± SD (5-6 mice per group).
In the in vitro study, the effect of GW501516 on PA and LPS-induced ROS production was investigated using DCFDA to detect cellular ROS levels. As illustrated in Figure 7A, incubating HepG2 cells with LPS and LPS + PA induced a significant increase in the cellular ROS level at 16 h (all P < 0.05 vs control group). Treatment with GW501516 (1 μmol/L and 10 μmol/L) attenuated the ROS generation in the PA- and LPS-stimulated HepG2 cells (all P < 0.05 vs PA + LPS group). In addition, we assessed AMPK activation with PA and LPS. PA or LPS alone had little effect on phosphorylation of the AMPKα subunit. However, adding GW501516 (1 μmol/L and 10 μmol/L) substantially increased phosphorylation of the AMPKα subunit in HepG2 cells (2.3- and 2.2-fold vs control group) (Figure 7B). These in vivo and in vitro results demonstrated that GW501516 could suppress hepatic oxidative stress and enhance phosphorylation of the AMPKα under high fat conditions.
Figure 7.
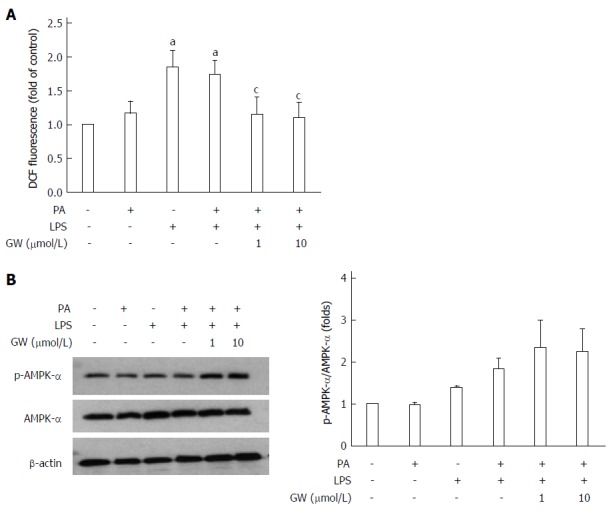
Effects of GW501516 on reactive oxygen species production and associated molecular pattern-activated protein kinase-α phosphorylation in HepG2 cells. Intracellular ROS production was quantified using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) (A). GW501516 inhibited lipopolysaccharide (LPS)- and palmitic acid (PA)-induced ROS generation. Representative immunoblot and graphic presentation of p-AMPK-α and total AMPK-α protein levels in HepG2 cells are shown (B). GW501516 tended to increase the phosphorylation of AMPK-α in PA- and LPS-treated cells. Data are expressed as the mean ± SD (5-6 mice per group). aP < 0.05 vs control cells, cP < 0.05 vs PA + LPS-treated cells.
DISCUSSION
Although inflammasome is known to have an important role in metabolic syndrome such as obesity and diabetes, there is few study to assess its role in the pathogenesis of NAFLD/NASH and the therapeutic implication of the PPAR-δ activator on inflammasome activation in NAFLD model.
The results of our study suggested that IL-1β, caspase, NLRP6 and NLRP10, as well as NLRP3, were activated in NAFLD models and PPAR-δ activator GW501516 has the ability to reduce hepatic steatosis, inflammation, and oxidative stress in mice fed with HFD. Also, these results were confirmed in the in vitro models.
A recent study demonstrated that NLRP6 is important in the self-renewal and integrity of the intestinal epithelium[25]. NLRP6 seemed to negatively regulate the progression from NAFLD to NASH by preventing the increase in colitogenic bacteria, indicating a role for NLRP6 in the pathogenesis of NAFLD[17,26]. On the other hand, the function of NLRP10 remains largely uncharacterized. With the exception of the present study, few studies have described the association between NLRP10 inflammasome and NAFLD. Further research is required into the exact mechanisms of how NLRP6 or NLRP10 functions in NAFLD.
To date, NLRP3 is the most widely studied inflammasome linked to obesity, insulin resistance, and atherosclerosis[15-18]. Excessive formation of ROS, which results in oxidative stress, is the central and common element for activating the NLRP3 inflammasome[14]. In addition, IL-1β, which is stimulated by islet amyloid polypeptide, promotes β-cell dysfunction and cell death, linking NLPR3 activation to insulin resistance[27]. Therefore, the role of inflammasome in the pathogenesis of NAFLD, the hepatic manifestation of metabolic syndrome, has been receiving increased attention.
In the present study, we showed that several inflammasomes, including NLRP3, NRLP6, and NLRP10, were activated in parallel with the production of ROS and the overexpression of caspase-1 and IL-1β in HepG2 cells after stimulation with PA and LPS. Overproduction of these pro-inflammatory cytokines was also confirmed in mice fed the HFD with LPS injection. These results are consistent with those from previous diet-induced NASH models including the methionine-choline-deficient (MCD) diet[28], the HFD [18], and the choline-deficient amino acid-defined (CDAA) diet[29].
PPAR-δ is the only subtype in the PPAR subfamily of nuclear receptors that is not a target for currently used drugs. Several studies have revealed that PPAR-δ activation exerts many metabolic effects, including reducing hepatic glucose production, increasing fatty acid catabolism in adipose tissue and muscle, and lowering the inflammatory status[19-21,30,31]. Until now, studies on the relationship between PPAR-δ activation and inflammasome have been scarce. In a recent study, the PPAR-δ agonist GW0742 was shown to attenuate the renal dysfunction and inflammation caused by chronic high-fructose corn syrup (HFCS-55) exposure by preventing activation of the NLRP3 inflammasome in the kidney[32]. In the present study, the PPAR-δ agonist GW501516 was found to reduce the activation of inflammasome as well as the overproduction of pro-inflammatory cytokines in HepG2 cells and the livers of mice. Moreover, GW501516 alleviated hepatic steatosis in vivo.
AMPK is a key metabolic regulator in cellular and organismal survival owing to its ability to maintain metabolic homeostasis; it is an essential mediator for fatty acid metabolism[18,21,33]. Several studies have demonstrated that PPAR-δ prevents the downregulation of AMPK[21,30]. In this study, although GW501516 treatment did not have an influence on the lipid profile in mice, it did increase phosphorylation of the AMPK-α subunit, both in vivo and in vitro. AMPK also has an anti-inflammatory effect and is associated with oxidative stress via suppression of ROS production[18,24]. The current study also demonstrated, for the first time, that the PPAR-δ agonist suppressed the ROS production induced by PA and LPS in the hepatocyte cell line. In immune cells such as macrophages, the AMPK-ROS signaling pathway is known to be associated with the activation of the NLRP3 inflammasome[18]. The current study may provide a clue into the complex role of PPAR-δ in metabolism, and suggests that the negative effect of PPAR-δ agonist on inflammasome activation in hepatocytes may be associated with the AMPK-ROS signaling pathway.
In the present study, we could not demonstrate statistical significant positive effect of PPAR-δ activator on insulin resistance and hyperlipidemia. Consistent with these findings, PPAR-δ protein expression were not different in controls and PA treatment with or without GW501516. The lipid-lowering and anti-diabetic effects of PPAR-δ, as well as its liver-protective effect, are already well documented[21,34-36]. In recent human clinical trials, 8 wk of therapy with MBX-8025 and GFT505, which are a novel PPAR-δ agonist and a dual PPAR-α/PPAR-δ agonist, respectively, significantly improved the plasma lipid profile, insulin resistance, and, interestingly, also decreased liver enzyme levels[34,37]. In our study, the treatment duration of GW501516 in mice was relatively short (3 wk), which may explain the insufficient effect observed on hyperlipidemia and glucose intolerance. Also, increasing oxidative capacity of muscle cells without provoking insulin sensitivity might supports our findings because PPAR-δ is highly expressed in the skeletal muscle, heart, and pancreatic β-cells, as well as in the liver[38]. Future challenges in determining the complex mechanism and cross-talk of PPAR-δ in different tissues and cell types are expected.
Recently, accumulating evidence has shown that bacterial endotoxins play a key role in the pathogenesis of NASH[28,39,40]. LPS, as an exogenous ligand for Toll-like receptor 4 (TLR4), may be capable of stimulating inflammasome expression, cytokine production, and the accumulation of inflammatory cells[28,39]. Therefore, we expected that the chronic exposure of low-dose LPS would lead to liver inflammation and fibrosis in mice fed an HFD. As for our study, we were decided to inject non-lethal dose of LPS which can reflect low level of endotoxemia of NAFLD. In our animal model, mice fed the HFD with LPS injection developed hepatic steatosis, although it failed to produce the severe form of NASH characterized by balloon degeneration and severe inflammation. However, unlike the MCD diet model[41], HFD with LPS injection resulted in weight gain and glucose intolerance, and therefore might more closely reflect the characteristics of human NAFLD. Future studies using endotoxins as HFD-enhancing factors in the murine NAFLD model are required to support the results of this study.
In conclusion, the results from our in vivo and in vitro studies demonstrate that the PPAR-δ agonist GW501516 suppresses the activation of inflammasome and reduces IL-1β levels, possibly by modulation of AMPK phosphorylation and decreased production of ROS. This anti-inflammatory effect might be associated with improvement of hepatic steatosis in mice. The targeting of inflammasome by the PPAR-δ agonist may have therapeutic implications for the treatment of NAFLD.
COMMENTS
Background
Nonalcoholic fatty liver disease (NAFLD) is one of the most common disease of the liver. Although thiazolidinediones and antioxidants such as vitamin E have been evaluated in several clinical trials, few pharmacological treatments can be recommended at present. Inflammasome is a large, intracellular multi-protein complex that is a sensor of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) that govern the cleavage of pro-inflammatory cytokines such as pro-interleukin (IL)-1β and pro-IL-18. Previous studies have shown that NLRP3 inflammasome is involved in the pathogenesis of obesity-induced inflammation, insulin resistance, and development of type 2 diabetes. However, the role of inflammasome in the pathogenesis of NAFLD/NASH has not been elucidated.
Research frontiers
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear receptor superfamily, and three isotypes exist in mammals. It has become increasingly evident that PPAR-δ is also an important metabolic regulator, as its activation enhances fatty acid oxidation, improves glucose homeostasis, and attenuates macrophage inflammatory responses. Current research hot spot is to whether the treatment with the PPAR-δ agonist could ameliorate high fat-induced inflammasome activation and the inflammatory response using in vivo and in vitro NAFLD models.
Innovative and breakthroughs
Until now, studies on the relationship between PPAR-δ activation and inflammasome have been scarce. In a recent study, the PPAR-δ agonist GW0742 was shown to attenuate the renal dysfunction and inflammation caused by chronic high-fructose corn syrup-55 exposure by preventing activation of the NLRP3 inflammasome in the kidney. To investigate whether the treatment with the PPAR-δ agonist could ameliorate high fat-induced inflammasome activation and the inflammatory response in the liver, the authors were using in vivo and in vitro NAFLD models. This study showed that PPAR-δ agonist GW501516 was found to reduce the activation of inflammasome as well as the overproduction of pro-inflammatory cytokines in HepG2 cells and the livers of mice. Moreover, GW501516 alleviated hepatic steatosis in vivo.
Applications
The study results suggested that the PPAR-δ agonist GW501516 suppresses the activation of inflammasome and reduces IL-1β levels in the liver, possibly by modulation of AMPK phosphorylation and decreased production of ROS. This anti-inflammatory effect might be associated with improvement of hepatic steatosis in mice. The targeting of inflammasome by the PPAR-δ agonist may have therapeutic implications for the treatment of NAFLD.
Terminology
NAFLD is a spectrum of disease entity including simple steatosis, steatosis with inflammation, fibrosis and cirrhosis. Inflammasome is a large, intracellular multi-protein complex that is a sensor of PAMPs or DAMPs that govern the cleavage of pro-inflammatory cytokines such as pro-IL-1β and pro-IL-18.
Peer-review
The authors studied effects of PPAR-δ activator; GW501516 on inflamasome pathway in mouse model of NASH (high fat diet, LPS/PA) and HepG2 cells culture. They show that the beneficial role in mouse liver was through increased NLRP3-10 in HepG2 cells.
Footnotes
Institutional review board statement: This study was approved by the institutional review board Korea University and conducted in compliance with the Guide for the care and Use of Laboratory Animals.
Institutional animal care and use committee statement: All procedure involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Korea University, Seoul, Korea, KUIACUC-2013-66 and carried out according to the Guide for the Care and Use of Laboratory Animals.
Conflict-of-interest statement: The authors declare no conflicts of interest.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 11, 2015
First decision: June 2, 2015
Article in press: October 13, 2015
P- Reviewer: Abenavoli L, Balaban YH, Laguna JC, Pirola CJ S- Editor: Qi Y L- Editor: A E- Editor: Liu XM
References
- 1.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 2.Vuppalanchi R, Chalasani N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology. 2009;49:306–317. doi: 10.1002/hep.22603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–649. doi: 10.3109/07853890.2010.518623. [DOI] [PubMed] [Google Scholar]
- 4.Abenavoli L, Milic N, Peta V, Alfieri F, De Lorenzo A, Bellentani S. Alimentary regimen in non-alcoholic fatty liver disease: Mediterranean diet. World J Gastroenterol. 2014;20:16831–16840. doi: 10.3748/wjg.v20.i45.16831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 7.Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. doi: 10.1016/j.freeradbiomed.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Carabelli J, Burgueño AL, Rosselli MS, Gianotti TF, Lago NR, Pirola CJ, Sookoian S. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J Cell Mol Med. 2011;15:1329–1338. doi: 10.1111/j.1582-4934.2010.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Musso G, Gambino R, Cassader M, Pagano G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology. 2010;52:79–104. doi: 10.1002/hep.23623. [DOI] [PubMed] [Google Scholar]
- 10.Abenavoli L, Greco M, Nazionale I, Peta V, Milic N, Accattato F, Foti D, Gulletta E, Luzza F. Effects of Mediterranean diet supplemented with silybin-vitamin E-phospholipid complex in overweight patients with non-alcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol. 2015;9:519–527. doi: 10.1586/17474124.2015.1004312. [DOI] [PubMed] [Google Scholar]
- 11.Rosselli MS, Burgueño AL, Carabelli J, Schuman M, Pirola CJ, Sookoian S. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis. 2009;206:119–126. doi: 10.1016/j.atherosclerosis.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 12.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 13.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 15.Stienstra R, Tack CJ, Kanneganti TD, Joosten LA, Netea MG. The inflammasome puts obesity in the danger zone. Cell Metab. 2012;15:10–18. doi: 10.1016/j.cmet.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 16.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulsen Ll, Siersbæk M, Mandrup S. PPARs: fatty acid sensors controlling metabolism. Semin Cell Dev Biol. 2012;23:631–639. doi: 10.1016/j.semcdb.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–818. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Barroso E, Rodríguez-Calvo R, Serrano-Marco L, Astudillo AM, Balsinde J, Palomer X, Vázquez-Carrera M. The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1α-Lipin 1-PPARα pathway leading to increased fatty acid oxidation. Endocrinology. 2011;152:1848–1859. doi: 10.1210/en.2010-1468. [DOI] [PubMed] [Google Scholar]
- 22.Buchner DA, Burrage LC, Hill AE, Yazbek SN, O’Brien WE, Croniger CM, Nadeau JH. Resistance to diet-induced obesity in mice with a single substituted chromosome. Physiol Genomics. 2008;35:116–122. doi: 10.1152/physiolgenomics.00033.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 24.Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu C, Viollet B, Yan D, Zou MH. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010;106:1117–1128. doi: 10.1161/CIRCRESAHA.109.212530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA. 2011;108:9601–9606. doi: 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kempster SL, Belteki G, Forhead AJ, Fowden AL, Catalano RD, Lam BY, McFarlane I, Charnock-Jones DS, Smith GC. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300:G253–G263. doi: 10.1152/ajpgi.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–144. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology. 2013;57:577–589. doi: 10.1002/hep.26081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coll T, Alvarez-Guardia D, Barroso E, Gómez-Foix AM, Palomer X, Laguna JC, Vázquez-Carrera M. Activation of peroxisome proliferator-activated receptor-{delta} by GW501516 prevents fatty acid-induced nuclear factor-{kappa}B activation and insulin resistance in skeletal muscle cells. Endocrinology. 2010;151:1560–1569. doi: 10.1210/en.2009-1211. [DOI] [PubMed] [Google Scholar]
- 31.Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci USA. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collino M, Benetti E, Rogazzo M, Mastrocola R, Yaqoob MM, Aragno M, Thiemermann C, Fantozzi R. Reversal of the deleterious effects of chronic dietary HFCS-55 intake by PPAR-δ agonism correlates with impaired NLRP3 inflammasome activation. Biochem Pharmacol. 2013;85:257–264. doi: 10.1016/j.bcp.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 33.Sid B, Verrax J, Calderon PB. Role of AMPK activation in oxidative cell damage: Implications for alcohol-induced liver disease. Biochem Pharmacol. 2013;86:200–209. doi: 10.1016/j.bcp.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Bays HE, Schwartz S, Littlejohn T, Kerzner B, Krauss RM, Karpf DB, Choi YJ, Wang X, Naim S, Roberts BK. MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab. 2011;96:2889–2897. doi: 10.1210/jc.2011-1061. [DOI] [PubMed] [Google Scholar]
- 35.Cariou B, Zaïr Y, Staels B, Bruckert E. Effects of the new dual PPAR α/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care. 2011;34:2008–2014. doi: 10.2337/dc11-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, Yamazaki Y, Kuroda J, Shibata N. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 37.Cariou B, Hanf R, Lambert-Porcheron S, Zaïr Y, Sauvinet V, Noël B, Flet L, Vidal H, Staels B, Laville M. Dual peroxisome proliferator-activated receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care. 2013;36:2923–2930. doi: 10.2337/dc12-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Lange P, Lombardi A, Silvestri E, Goglia F, Lanni A, Moreno M. Peroxisome Proliferator-Activated Receptor Delta: A Conserved Director of Lipid Homeostasis through Regulation of the Oxidative Capacity of Muscle. PPAR Res. 2008;2008:172676. doi: 10.1155/2008/172676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kudo H, Takahara T, Yata Y, Kawai K, Zhang W, Sugiyama T. Lipopolysaccharide triggered TNF-alpha-induced hepatocyte apoptosis in a murine non-alcoholic steatohepatitis model. J Hepatol. 2009;51:168–175. doi: 10.1016/j.jhep.2009.02.032. [DOI] [PubMed] [Google Scholar]
- 40.Shanab AA, Scully P, Crosbie O, Buckley M, O’Mahony L, Shanahan F, Gazareen S, Murphy E, Quigley EM. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig Dis Sci. 2011;56:1524–1534. doi: 10.1007/s10620-010-1447-3. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]