

Abstract
Objective
Neointima formation is associated with stenosis and subsequent thrombosis in arteriovenous grafts (AVG). A role of integrin β3 in the neointima formation of AVGs remains poorly understood.
Approach and Results
In integrin β3−/− mice, we found significantly accelerated occlusion of AVGs compared to the wild type mice. This is caused by the development of neointima and lack of endothelial regeneration. The latter is a direct consequence of impaired functions of circulating angiogenic cells (CACs) and platelets in integrin β3−/− mice. Evidence suggests the involvement of platelet regulating CAC homing to and differentiation at graft sites via TGF-β1 and Notch signaling pathway. First, CACs deficient of integrin β3 impaired adhesion activity toward exposed subendothelium. Second, platelets from integrin β3−/− mice failed to sufficiently stimulate CACs to differentiate into mature endothelial cells. Finally, we found that TGF-β1 level was increased in platelets from integrin β3−/− mice and resulted in enhanced Notch1 activation in CACs in AVGs. These results demonstrate that integrin β3 is critical for endothelial cell homing and differentiation. The increased TGF-β1 and Notch1 signaling mediates integrin β3−/−-induced AVG occlusion. This accelerated occlusion of AVGs was reversed in integrin β3−/− mice transplanted with the bone marrow from WT mice.
Conclusion
Our results suggest that boosting integrin β3 function in the endothelial cells and platelets could prevent neointima and thrombosis in AVGs.
Keywords: integrin β3, arteriovenous graft, platelets, bone marrow, endothelial progenitor cell, Notch
Introduction
The creation of functional arteriovenous grafts (AVG) for patients who undergo coronary artery bypass or hemodialysis is a frequent and expensive surgical procedure. Even when initial surgery is successful, an AVG frequently fails. The primary patency rates of graft failure are 42% and 40–60% for coronary bypass surgery and hemodialysis, respectively,1, 2 primarily due to neointima formation and AVG thrombosis.
The integrin β3 family consists of αvβ3 and αIIbβ3. The former is expressed in endothelial cells and selective inflammatory cells, while the latter is primarily on platelets. Platelets are involved in the recruitment and differentiation of bone marrow-derived terminal progenitor cells,3, 4 mediated through the adhesive receptor integrin αIIbβ3.5 Recent reports suggest that integrin β3 expressed on endothelial cell and platelets influences the function of bone marrow-derived progenitor cells.6 Platelets from integrin β3−/− mice are also reported to secrete more chemokines and cytokines compared with the wild type mice, including TGF-β1.7
In a mouse AVG model, there was a massive loss of the endothelium 3 days after surgery. The subsequent endothelial regeneration plays a key role for vascular remodeling and maintaining a patent AVG.8 Platelets influence the homing and differentiation of circulating angiogenic cells (CAC) into endothelial cells at sites of vascular injury by serving as carriers9, 10 and by secreting SDF-1α.11 Here, we tested hypothesis that integrin β3 is critical for neointima formation and endothelial cell regeneration at sites of AVGs by studying the adhesion and differentiation of CACs in vitro and vascular repairs in a mouse AVG model. We demonstrate that CACs from integrin β3−/− mice homed poorly because of significant reduction in CAC adhesion and differentiation. The latter is caused by an altered TGFβ1-Notch1 signaling mediated by β3 deficient platelets.
Materials and Methods are available in the online-only Data Supplement.
Results
AVG occlusion accelerated in integrin β3−/− mice
Integrin β3 is expressed in artery, vein and heart tissues; there was no such expression in integrin β3−/− mice (Fig. 1A). In our model of AVGs in wild type mice, we used an enface analysis to study the endothelium of the vein of the AVG and a normal vena cava. Integrin β3 was expressed in endothelial cells in a clustered pattern (Fig. 1B). In a failed AVG from patients, integrin β3 expression was located in the endothelium and the neointima (Fig. 1C). At one month after surgery, similar results were found in AVGs created in wild type mice (Fig. 1D). Compared with results in a control vena cava, there was reduced integrin β3 expression in the endothelium of the AVG (Fig. 1D). Newly formed endothelial cells expressed integrin β3 on the basal side (Fig. 1D). In AVGs, there was marked thinning and attenuation of the endothelium compared to findings in normal vena cava.
Figure 1. Integrin β3 expression in arteriovenous graft (AVG).
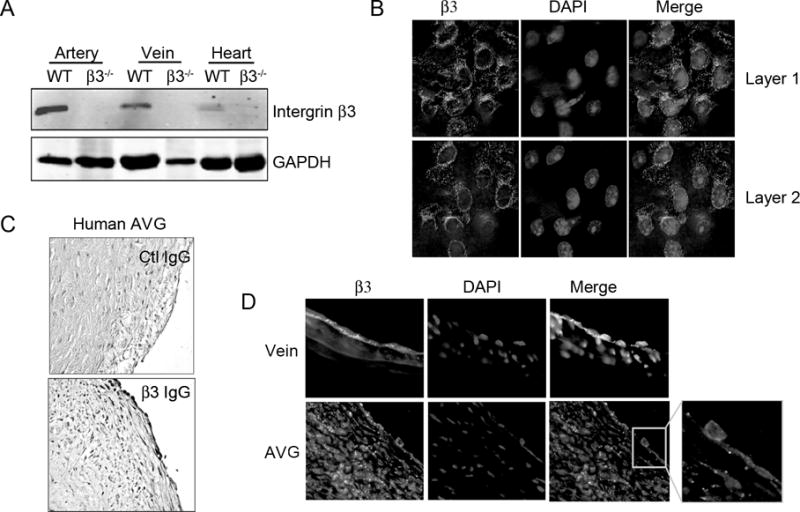
A. Integrin β3 expression in veins and arteries was detected by Western blot. The experiment was repeated for 3 times. B. Enface analysis of integrin β3 in vena cava. C. In AVGs from patient, immunostaining reveals integrin β3. D. In endothelial cells in a mouse AVG integrin β3 expression was decreased, representative data from 5 AVGs.
Integrin β3−/− and wild type mice were subjected to AVG and evaluated one month after surgery. The rate of graft failure due to occlusion was 86.7% for β3−/− mice as compared to 5% for wild type mice (Fig. 2A & B). An intact vascular lining of CD31+ endothelial cells was found in AVGs of WT mice, but not in integrin β3−/− mice (Fig. 2C). SMA-α was strongly expressed in neointima cells in WT AVGs but not in integrin β3−/− miced (Fig. 2C). When stained for markers for thrombosis (vWF, CD41, and CD42), we found much more expression of thrombotic positive markers in AVGs placed in integrin β3 KO mice vs. the expression in WT mice (Fig. 2D and E), suggesting that the integrin β3 deficiency prevents the formation of an intact endothelium and causes thrombosis, which could be the potential factor influencing the AVG patency.
Figure 2. Integrin β3 deficiency accelerates AVG failure.
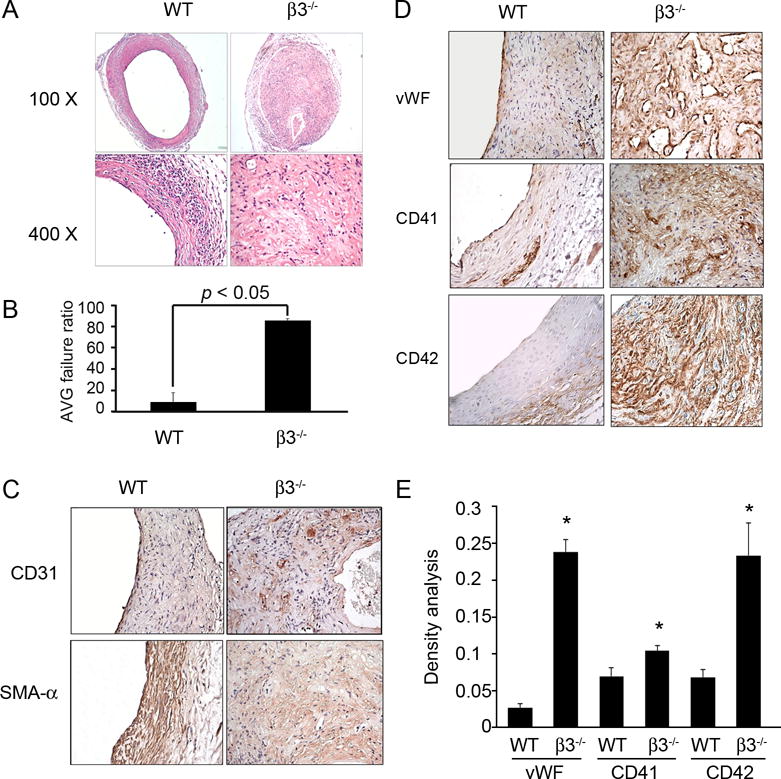
A. H & E staining of AVGs in wild type and integrin β3−/− mice show marked differences in patency. B. The ratio of failed to total AVGs was calculated. Total 15 AVGs were created in integrin β3−/− mice, and 9 in wild type mice. C and D. The difference in AVGs of wild type or integrin β3−/− mice is revealed by immunostaining with the endothelial marker, CD31 (C), smooth muscle marker SMA-α (C), and platelets markers (D). E. The density analysis of the expression of platelet markers in WT and integrin β3−/− mice (n=4).
Integrin β3−/− delays endothelial regeneration in AVGs
Endothelial cells became disorganized 3 hrs after AVG surgery and most were lost by 24 hours in wild type and integrin β3−/− mice (Fig. 3A &C). These results could not be claimed on surgeries as a cross section of AVG just after surgery revealed that the endothelium was intact (Fig. 3B). However, endothelial cells regenerated to form the new endothelium around 7 to 14 days after surgery (Fig. 3A & C). In contrast, endothelial cells were scarcely present at an AVG site of integrin β3−/− mouse during the same period (Fig. 3D). This result was further verified by enface analysis, which detected significantly less CD31 signals in AVGs from integrin β3−/− mice as compared to that from WT mice (Fig. 3E).
Figure 3. Integrin β3 KO delays endothelial regeneration in AVGs.
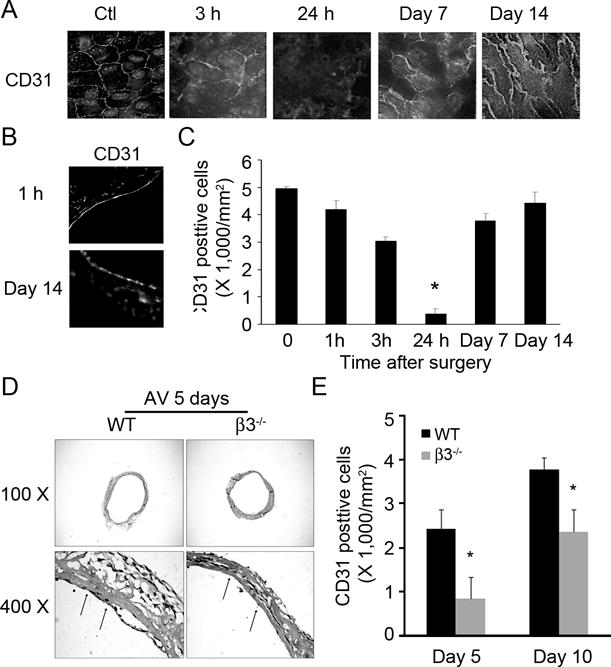
A. AVGs collected at different time points were examined by a deconvolution microscope after staining with the endothelial marker, CD31. B. Endothelial markers were present in cross sections of AVGs collected at different times from wild type mice. C. Statistical analysis of endothelial cell numbers shown in panel A. D. AVGs created in wild type and integrin β3−/− mice were collected after 5 days and examined by H & E staining. E. Statistical analysis of the endothelium in AVGs from wild type and integrin β3−/− mice after enface analysis, confirmed that the absence of integrin β3 delays regeneration. Representative data were from at least 3 mice.
Integrin β3−/− suppresses bone marrow-derived CAC adhesion in vitro
Alternatively, the reduced endothelial regeneration could be due to the impaired CAC homing capability. Reduced CAC homing could be caused by the lack of integrin β3-mediated interaction between CACs and the subendothelial matrix. Consistent with the notion, CACs from integrin β3−/− mice adhered poorly to vitronectin, the ligand for integrin β3. While the adherence of integrin β3-deficiency CACs to laminin and collagen I were not affected (Fig. 4A & B). These results are further confirmed with experiments testing lineage negative/c-Kit/Sca I positive CACs isolated from the bone marrow of wild type and β3−/− mice (Fig. 4C). The absence of integrin β3 suppressed CAC adhesion to extracellular matrix. Notably, the extracellular matrix tested included vitronectin and fibronectin which are interacting with Integrin β3 (Fig. 4C). Together, these results suggest that β3 deficiency CACs adhere poorly to the subendothelium exposed at sites of AVGs.
Figure 4. Integrin β3−/− inhibits CAC adhesion.
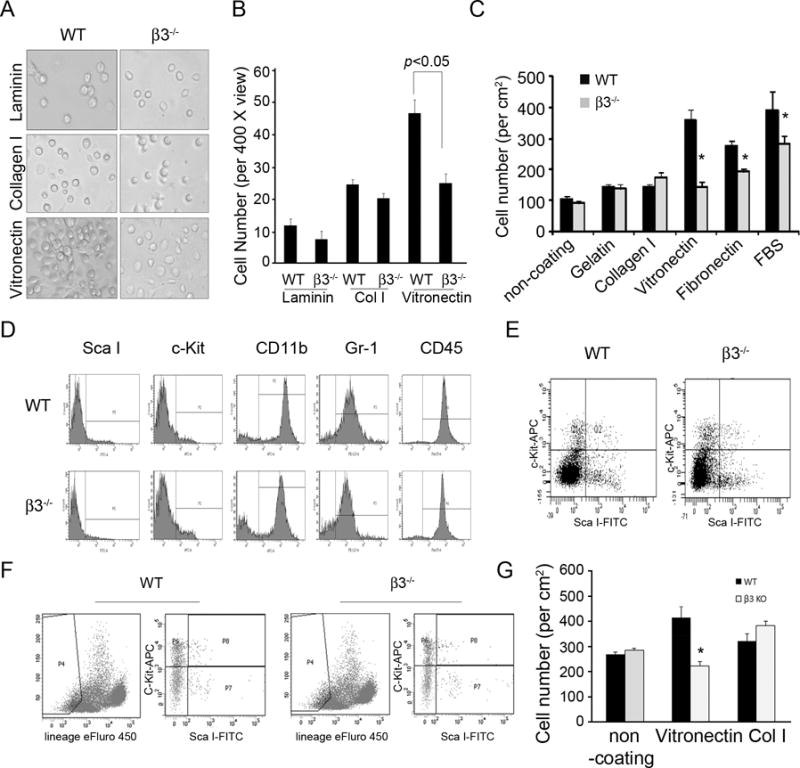
A and B. After 7 days, bone marrow cells from integrin β3−/− have limited attachment to extracellular matrices. C. ScaI and c-Kit positive CACs were isolated from bone marrow of wild type mice and the purity was confirmed. The purified cells from bone marrow and integrin β3−/− mice were seeded (~1 ×104) on 48-well plates coated with varied extracellular cellular matrices. After 7 days culture, cell numbers were counted. D and E. Bone marrow cells from wild type and integrin β3−/− mice were labeled with different antibodies and subjected to flow cytometry. Monocyte (CD45), macrophage (CD11b), neutrophil (Gr-1) populations and Sca I/c-Kit double positive CACs were not different between the two types of mice. F and G. The CACs from lineage negative population in bone marrow cells were isolated from wild type and integrin β3−/− mice (F). The lineage negative population was gated for isolation of Sca I+/c-Kit+ CACs and the adhesion analysis was performed (G). The data shown are representative of 2 experiments. *, p < 0.05 compared with WT.
This reduced CAC homing is not caused by a reduced number of CACs because integrin β3−/− mice had a comparable number of CACs in the bone marrow of WT mice (Fig. 4D and E). Moreover, the lineage negative CACs (lineage−/c-kit+/Sca I+ cells) were also purified and analyzed. The results indicate that that β3 deficiency did not affect CAC numbers but impair their adhesion capability to vitronectin (Fig. 4F & G). Similar results were found from circulating Sca I+/c-Kit+ cells (data not shown).
Platelets from integrin β3−/− mice delay homing and differentiation of bone marrow-derived CACs
Platelets are known to play a critical role in CAC homing 10. We next examine whether integrin β3 deficiency in platelets contributes to the poor regeneration of endothelial cells at an AVG site. We found that there was significantly less ScaI+-CACs adhere to AVGs at day 5 in integrin β3−/− mice compared to controls (Fig. 5A and B). Moreover, CACs has less potential to differentiate into mature endothelial cells when co-cultured with platelets from integrin β3−/− mice compared with those on WT platelets (Fig. 5C). Furthermore, these β3 deficient platelets expressed a 2-fold higher level of TGF-β1 (Fig. 5D). Using ELISA assay, we found that thrombin (0.125 U/ml) or collagen I (5 µg/ml) treatments stimulated more TGF-β1 release from platelets isolated from integrin β3 KO mice compared with that of WT mice (Fig. 5E). In AVGs, we found that TGF-β1 colocalized with platelets and more TGF-β1 level was detected in integrin β3 KO mice vs that in wild type mice (Fig. 5F).
Figure 5. Integrin β3−/− affects CAC homing and differentiation.
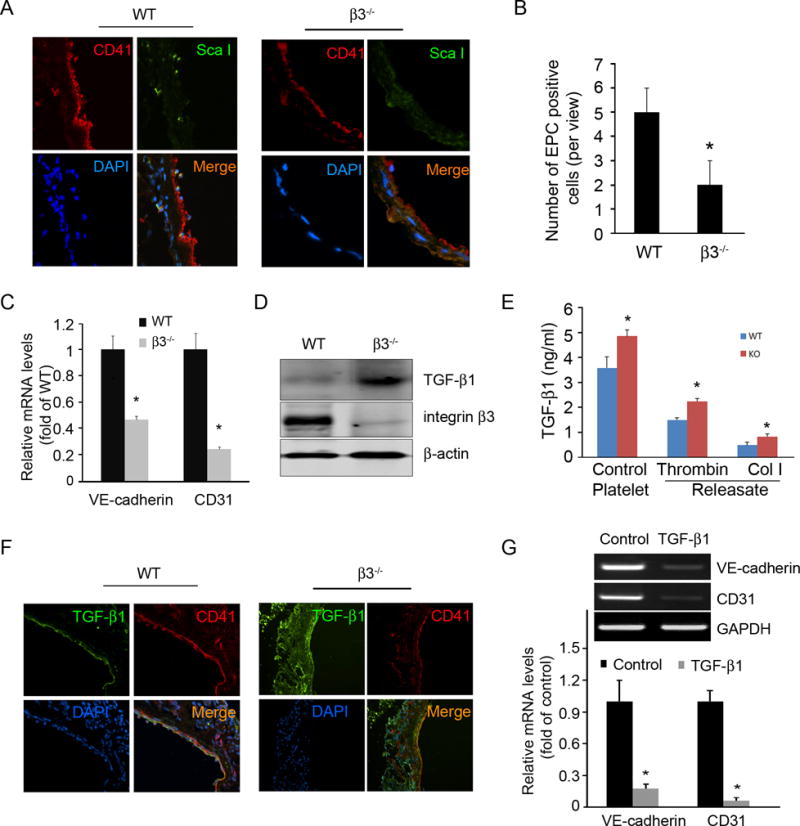
A. Detection of CD41+ platelets and Sca I+ CACs in AVGs in wild type and integrin β3 KO mice. B. Statistical analysis of ScaI+ cell accumulation in AVGs. n = 4 mice from both wild type and integrin β3 KO mice. C. Platelets isolated from wild type and integrin β3 KO mice were cocultured with CACs for 7 days. Endothelial markers (real time RT-PCR) revealed that platelets promote CAC differentiation. This was more prominent with wild type platelets. D. Integrin β3 null platelets express excess TGF-β1 detected by western blot. E. Washed platelets isolated from wild type and integrin β3 KO mice were stimulated with 0.125 U/ml thrombin or 5 μg/ml collagen I at 37°C for 5 minutes and centrifuged at 14 000 g for 20 minutes. The TGF-β1 level in the platelets and releasates was measured by ELISA after acidification (n=3). F. In vivo, prominent TGF-β1 expression in AVGs from integrin β3 KO mice colocalized with platelets. G. CACs isolated from bone marrow and cultured in EGM2 with or without incubation with TGF-β1 (3 ng/ml) for 7 days. Excess TGF-β1 inhibits express mature endothelial markers were detected by real time RT-PCR. Representative data are present from 3 independent experiments. *, p < 0.05 compared with WT.
To assess the impact of TGF-β1, we treated wild type CACs with TGF-β1 and found impaired CAC differentiation into endothelial cells (Fig. 5G). Thus, high TGF-β1 in platelets from integrin β3 KO mice suppresses CAC differentiation into mature endothelial cells and then reduced the recovery of the endothelium following endothelial cell losses in an AVG.
Increased TGF-β1 in β3-null platelets inhibits CAC differentiation by stimulating Notch signaling
Results presented so far have suggested that poor endothelial regeneration in integrin β3 KO mice at an AVG site may be caused by 1) lack of interaction between β3−/− CACs and components of subendothelial matrix and 2) increased TGF-β1 in integrin β3−/− platelets. To explore the second possibility, we focus on Notch signaling in endothelial cells because TGF-β1 activates Notch by upregulating the Notch ligand Jagged1.12 This Notch signaling has also been linked to stem cell self-renewal and CAC differentiation. We detected enhanced expression of molecules critical for Notch signaling at the AVG site of integrin β3−/− mice as compared to WT (Fig. 6A). For example, the activated N1ICD (Notch 1 intracellular domain) was more prominently detected in AVGs isolated from β3−/− mice compared to WT mice (Fig. 6B & C). When treated with TGF-β1, CACs expressed the increasing levels of the Notch ligand Jagged 1 and its target Hes5 (Fig. 6D). In parallel with the increased Notch activation, CACs treated with -secretase inhibitor, DAPT, rescued the expressions of the endothelial cell markers VE-cadherin and CD31 that were reduced by treatment with TGF-β1 or co-cultured with integrin β3-null platelets (Fig. 6E). These results suggest that up-regulation of Notch signaling is associated with a poor differentiation of CACs at an AVG site. Consistent with the notion, when treated with soluble Jagged 1 (inhibits Notch signaling) or DAPT CACs from wild type and β3-null mice increased the rate of differentiation as demonstrated by increasing expression of VE-cadherin (Fig. 6F & G).
Figure 6. Integrin β3 KO inhibits CAC differentiation by increasing Notch signaling.
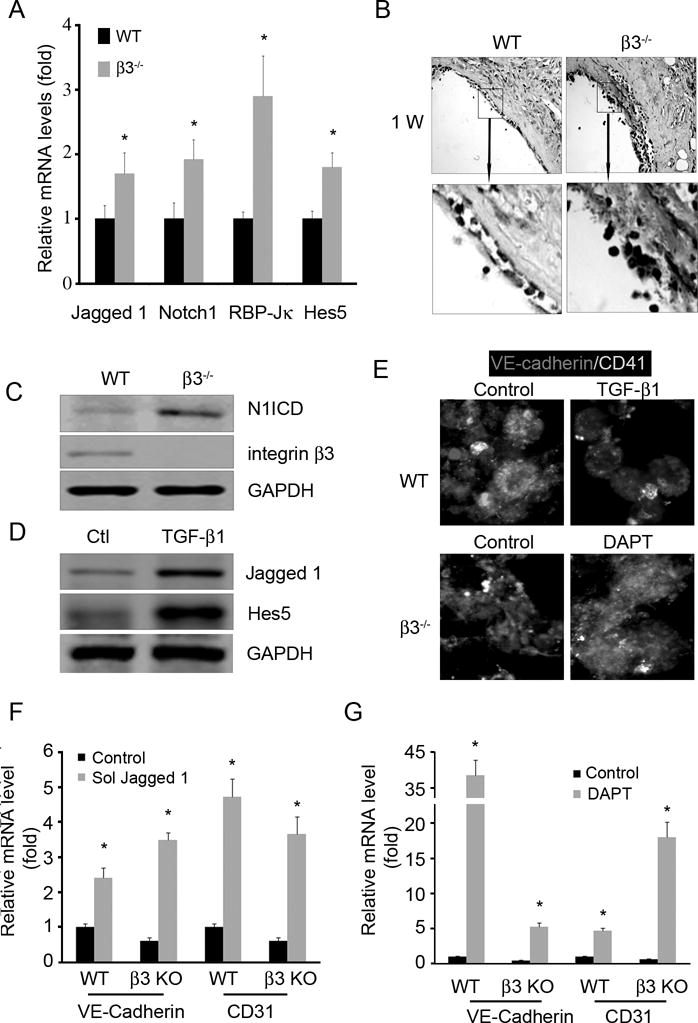
A and B. AVGs were collected from wild type and integrin β3 KO mice and subjected to real time RT-PCR (A), immunostaining of activated Notch, N1ICD, reveal an increase in Notch signaling in AVGs created in integrin β3−/− mice (B). C. Western blot confirmed an increase in Notch1 cleavage in AVGs from integrin β3 KO mice. D. CACs were treated with TGF-β1 revealed an increase in Notch ligand and its target (Western blot). E–G. If Notch signaling is inhibited, integrin β3-null platelets will improve CAC differentiation. Bone marrow-derived CACs were cocultured with wild type or integrin β3-null platelets plus TGF-β1 or DAPT. Markers of endothelial cells and platelets were detected by immunostaining, demonstrating that integrin β3 null platelets suppress CAC differentiation. This occurs via Notch activation (E). To inhibit Notch, CACs were infected with an adenovirus expressing soluble Jagged 1 (F) or with the DAPT (G), an increase in endothelial markers (VE-cadherin and CD31) were detected by real time RT-PCR followed inhibition of Notch, *, p<0.05, compared with control.
Bone marrow cells from wild type mice rescue AVG failure in integrin β3−/− mice
The results so far have suggested two potential pathways through which integrin β3 deficiency results in poor homing of CACs and regeneration of the endothelium (differentiation) at an site of AVG: decreased interaction between CACs and the subendothelium (directly mediated through platelets) and enhanced Notch signaling in CACs mediated by platelet-derived TGF-β1. These possibilities were further investigated by the transplantation of bone marrow from a wild type mouse donor to an integrin β3−/− recipient. There was significant improvement in AVG patency in integrin β3−/− mice after transplantation of WT bone marrow cells vs results from integrin β3−/− mice (Fig. 7A). In a reciprocal experiment, AVGs in wild type mice transplanted with bone marrow cells from integrin β3−/− mice were clogged (Fig. 7A). The AVG from transplanted integrin β3−/− mice also had an intact vascular wall lined with mature CD31+-endothelial cells (Fig. 7B & C). There was ~75% of AVGs in the mice with the bone marrow transplantation had open lumens vs the AVGs generated in WT mice with bone marrow from integrin β3−/− mice (Fig. 7D). To further compare the CAC homing capability, a mixture of bone marrow cells from WT (GFP+) and integrin β3−/− mice were transplanted into integrin β3−/− mice before AVGs. The results showed that more WT (GFP+)-derived CACs incorporated into the lumen area of the AVGs (Fig. 7E), indicating that deficiency of integrin β3−/− limit CAC-mediated endothelium regeneration.
Figure 7. AVG failure in integrin β3 KO was rescued by wild type bone marrow cells.
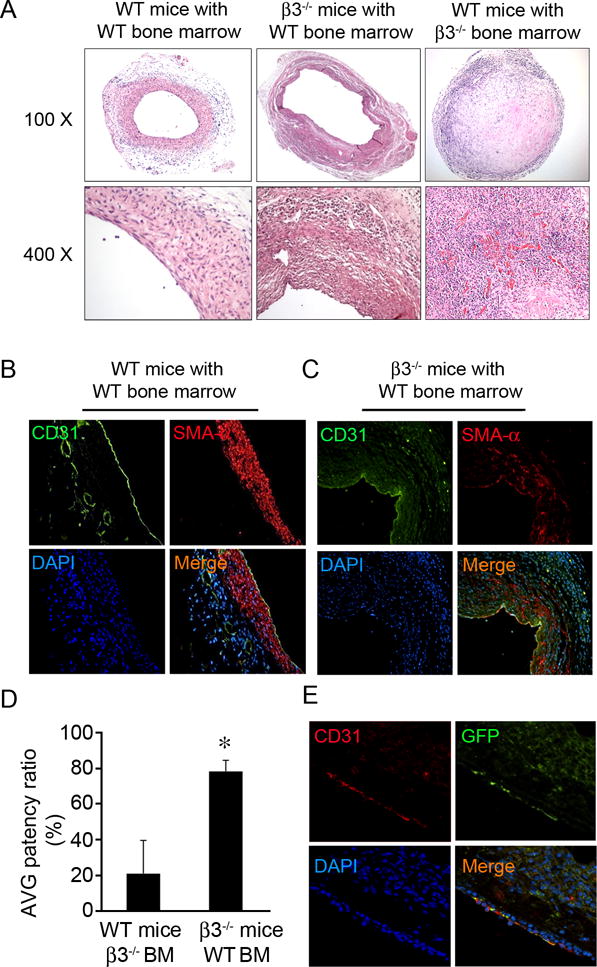
A. H & E staining of AVGs show the increased patency in integrin β3 KO mice. Each group had at least 6 mice for AVG surgery. B and C. Endothelial cell markers in AVGs of integrin β3 KO mice increased after transplantation with wild type bone marrow. D. Statistical analysis of the effect ofbone marrow transplantation on AVG patency. n = 7, *, compared with control WT mice, p < 0.01. E. Integrin β3 KO mice transplanted bone marrow cells from GFP transgenic mice demonstrate that cells from the transplanted bone marrow do contribute to regeneration of endothelial cells in an AVG. The GFP staining was detected after 5 days of AVGs. The endothelial marker and GFP costaining were shown.
Discussion
In our current study, we found that integrin β3−/− causes AVG occlusion. Several lines of evidence support that the dysfunctions of platelets and CACs in integrin β3−/− mice are the potential reasons for this failure. First, deficiency of integrin β3 in CACs impairs their adhesion to the exposed extracellular matrix. Integrin β3−/− platelets are deficient in mediating CAC homing and prevent CAC differentiation into mature endothelial cells due to an increased production of platelet-derived TGF-β1, which activates the Notch signaling pathway (Supplemental Fig. I).
AVG failure is a consequence of three related processes including early thrombosis formation, neointima formation, and graft atherosclerosis.13 About 40 percent of AVGs experience such a failure within 18 months of the operation.14 The failure of AVGs is attributable to adverse vascular remodeling, which remain poorly understood. Multiple events and cell types, including endothelial denude, platelet aggregation, smooth muscle cell proliferation, activated thrombocytes, and infiltration of monocytes, contributes to AVG failure. In mouse model of AVG, we found increased thrombosis and smooth muscle cell hyperplasia in AVGs at 2–4 weeks, and this process is accelerated in β3-integrin KO mice. Deficiency of integrin β3 delayed the endothelial cell regeneration in AVG in early time point, and led to AVG failure, which related with thrombosis. These findings are consistent with previous reports. The initial loss of venous endothelium after AVG is followed by platelet accumulation and the recruitment of CACs at a denuded AVG.15 CACs are BM-derived cells that are mobilized to the systemic circulation in response to tissue injury and incorporate into sites of neovascularization.16, 17 Following vascular injury, CACs have been shown to mobilize and home to the subendothelium at site of vessel injury.18 Peripheral blood CACs transplanted into balloon-injured arteries rapidly endothelialize the denuded vessels.19 In AVGs, the endothelium is denuded 72 hrs after surgery followed by on-site platelets aggregation and endothelial cell regeneration. There is approximately 30% of BM-derived CACs incorporation into newly formed endothelium.8, 15 An integrin β3 deficiency could potentially interfere with vascular repairs at an AVG site because the disruption of the integrin signal could result in a poor maturation of vessels6. Moreover, the neovascularization was restored in a DiYF β3-integrin mutant mouse by transplantation with WT BM.20
Our data suggest an underlying mechanism for how integrin β3−/− could impair CAC homing and differentiation. First, integrin β3 deficient CACs failed to efficiently adhere to the denuded AVGs and to the subendothelial matrix proteins, vitronectin, and fibronectin (Fig. 3). There are no differences in a total number of lineage negative c-Kit/Sca I double positive CACs (Fig. 4G), suggesting that this loss-of-function phenotype is not caused by changes in the number of circulating CACs. This finding is consistent with a previous observation that the loss function of integrin β3 did not change the numbers of ScaI+ cells.20
The activated platelets could be another factor that involved in CAC homing in AVGs.4, 10 Following vascular insult, platelets first tether and adhere to subendothelium exposed by injury to not only seal the wound, but also promote P-selectin-mediated CAC homing as previously demonstrated. 21,22 We have recently shown that platelets are involved in trapping and recruiting BM-derived fibroblast specific protein 1 positive cells.23 In current study, we further show that platelet aggregation was decreased in AVGs from integrin β3−/− mice, independent of platelet counts, which are normal in β3-null mice.24 The data is supported by previous finding that the integrin β3 partner, αIIb integrin, is required for the recruitment of bone morrow-derived CD34+ and Sca-1+ progenitor cells into injured vessel walls.5, 9, 11 More evidence showed that microparticles from Platelets promotes CAC function and tube formation through integrin β3.25
Second, β3 deficiency may also result in defective differentiation of CACs at sites of AVGs in a platelet-dependent manner. Human CD34+ CACs co-cultured with immobilized platelets form colonies and are capable of differentiating into mature endothelial cells.21, 26 This regulatory activity of platelets towards CAC differentiation is likely mediated by platelet-derived growth factors (e.g., TGF-β1, platelet-derived growth factor, vascular endothelial growth factor, epidermal growth factor, and insulin-like growth factor).27, 28 Among these factors, the TGF-β1 suppresses reprogramming of other type of cells into endothelial cells.29 A higher level of TGF-β1 from integrin β3 deficient platelets adversely affected CAC regeneration (Fig. 5B). The result is consistent with our recent report that integrin β3 deficiency increases TGF-β1 expression in muscle cells, impairing their capacity for muscle regeneration.30 Since integrin regulates outside-in signal through cytoskeleton, and a recently report showed that Wiskott-Aldrich syndrome protein, an actin accessory protein, through CDC42 to regulate TGF-β1 release.31 So the integrin β3-induced TGF-β1 release could be associated with cytoskeleton changes.
We further show that the TGF-β1-induced activation of Notch signaling, may play a critical role in accelerated AVG occlusion (Fig. 6A–C). Because Notch signal maintains multi-potential progenitor cells in a undifferentiated 32 and slows the differentiation of bone marrow-derived CAC.33, 34 Increased Notch signaling in endothelial cells upregulates mesenchymal marker expression leading to endothelial barrier dysfunction in arteriovenous fistula.35 TGF-β1 is significantly increased in β3 null platelets and could result in enhanced activation of Notch signaling through induction of Notch ligand Jagged1 (Fig. 6D). This finding was supported by an early report that endothelial repair is accelerated after arterial injury in B6 mice transplanted with the bone marrow cells from Notch1 knock down mice.33, 36 We also find that platelet-mediated CAC differentiation was suppressed by the excess presence of TGF-β1, leading to delayed endothelium regeneration at sites of AVGs (Fig. 5D).
Finally, the influence of integrin β3 was demonstrated by the drastic reduction in the rate of AVG failure by the transplantation of wild type bone marrow cells into integrin β3−/− mice (Fig. 7). Our results differ from those found in models of arterial injury, where the inhibition of integrin β3 reduces or blocks the neointima formation of vascular smooth muscle cells.37 We found that deficiency of integrin β3 delayed CAC regeneration, leading to thrombosis and adversely affect AVG function. Our results suggest that anti-platelet therapy in human should be careful considerate. For AVG, the αIIβ3 based anti-platelet therapy could worsen the thrombosis due to the delayed endothelium repairmen, like the case in current animal model, in this case P2Y12-based anti-platelet therapy may be evaluated and considered.38 For example, a report showed that a delayed anti-platelet therapy with clopidogrel and everolimus prevents progression of transplant arteriosclerosis in murine aortic allografts.39
It is known that in addition to a negative role of TGF β –signaling in CAC differentiation, other factors also regulate CAC adhesion and migration through β3-integrin. Pula et al. found that proangiogenic factor thymidine phosphorylase (TP)-induced CAC migration can be blocked by RGD peptides and inhibitory antibodies to integrin αVβ3.40 Without integrin β3 also impairs its interaction with VEGFR2 and hypoxia-induced angiogenesis.41 These results indicate that multiple factors could be involved in regulating integrin β3-impaired CAC function or differentiation.
In summary, we have uncovered a novel mechanism for causing AVG occlusion and failure. In addition to poor adhesion of β3 null endothelial progenitor cells to the subendothelium, β3 deficiency in platelet contributes to AVG occlusion 1) by defective as a carrier for CAC homing and 2) activating Notch signaling to suppress endothelial cell differentiation. Our results, therefore, suggest that strategies that block Notch signaling or reduce TGF-β1 could improve the patency of an AVG.
Supplementary Material
Key Points.
Integrin β3−/− causes failure of arteriovenous graft.
Dysfunction of platelets in integrin β3−/− mice suppresses homing and differentiation of endothelial progenitor cells into endothelial cells in arteriovenous graft.
Significance.
Neointima formation is associated with stenosis and subsequent thrombosis in arteriovenous grafts. Integrin β3 has been reported to regulate neointima formation in artery injury model. In a mouse model of arteriovenous graft, we found that deficiency of integrin β3 suppresses homing and differentiation of endothelial progenitor cells into mature endothelial cells in arteriovenous grafts, delays endothelial regeneration. These responses result in thrombosis and failure of arteriovenous graft. The increased TGF-β1 in platelets and Notch1 signaling are the underlying molecular mechanisms that cause accelerated failure of the grafts in integrin β3 deficient mice. Wild type bone marrow transplantation can rescue the arteriovenous graft patency in integrin β3 deficient mice. Our results suggest that boosting integrin β3 function in the endothelial cells and platelets could prevent neointima and thrombosis in AVGs.
Acknowledgments
We thank Dr. William E. Mitch for critical reading and editing of this manuscript. We thank Dr. Hongzhen Hu for helpful suggestions.
This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (NIAID P30AI036211, NCI P30CA125123, and NCRR S10RR024574) and the assistance of Joel M. Sederstrom.
Sources of Funding:
This study was supported by grants from NIH RO1DK095867 and American Heart Association Grant 10SDG2780009 (to J.C.), support from National Institutes of Health grants R37 DK37175 Grant, and a generous grant from Dr. and Mrs. Harold Selzman.
Abbreviations
- AVG
arteriovenous graft
- CAC
circulating angiogenic cells
- NICD
Notch intracellular domain
- DAPT
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine-t-butyl ester; γ-secretase inhibitor
Footnotes
Contribution: ML, JC, YW carried out experiments, study design, and data analysis. JC, ML, YW searched the literature and interpreted the data. JC, JD, and JFD were involved in writing the paper. All authors had final approval of the submitted and published versions.
Disclosures
None
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. on behalf of the American Heart Association Statistics Committee and Stroke Statistics S Heart disease and stroke statistics–2011 update: A report from the american heart association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Group VAW. Clinical practice guidelines for vascular access. Am J Kidney Dis. 2006;48(Suppl 1):S176–247. doi: 10.1053/j.ajkd.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 3.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359:1261–1270. doi: 10.1056/NEJMra0800887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stellos K, Gawaz M. Platelet interaction with progenitor cells: Potential implications for regenerative medicine. Thromb Haemost. 2007;98:922–929. doi: 10.1160/th07-02-0147. [DOI] [PubMed] [Google Scholar]
- 5.Massberg S, Schurzinger K, Lorenz M, Konrad I, Schulz C, Plesnila N, Kennerknecht E, Rudelius M, Sauer S, Braun S, Kremmer E, Emambokus NR, Frampton J, Gawaz M. Platelet adhesion via glycoprotein iib integrin is critical for atheroprogression and focal cerebral ischemia: An in vivo study in mice lacking glycoprotein iib. Circulation. 2005;112:1180–1188. doi: 10.1161/CIRCULATIONAHA.105.539221. [DOI] [PubMed] [Google Scholar]
- 6.Watson AR, Pitchford SC, Reynolds LE, Direkze N, Brittan M, Alison MR, Rankin S, Wright NA, Hodivala-Dilke KM. Deficiency of bone marrow beta3-integrin enhances non-functional neovascularization. J Pathol. 2010;220:435–445. doi: 10.1002/path.2660. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds LE, Conti FJ, Lucas M, Grose R, Robinson S, Stone M, Saunders G, Dickson C, Hynes RO, Lacy-Hulbert A, Hodivala-Dilke K. Accelerated re-epithelialization in beta3-integrin-deficient- mice is associated with enhanced tgf-beta1 signaling. Nat Med. 2005;11:167–174. doi: 10.1038/nm1165. [DOI] [PubMed] [Google Scholar]
- 8.Xu Q, Zhang Z, Davison F, Hu Y. Circulating progenitor cells regenerate endothelium of vein graft atherosclerosis, which is diminished in apoe-deficient mice. Circ Res. 2003;93:e76–86. doi: 10.1161/01.RES.0000097864.24725.60. [DOI] [PubMed] [Google Scholar]
- 9.Langer H, May AE, Daub K, Heinzmann U, Lang P, Schumm M, Vestweber D, Massberg S, Schonberger T, Pfisterer I, Hatzopoulos AK, Gawaz M. Adherent platelets recruit and induce differentiation of murine embryonic endothelial progenitor cells to mature endothelial cells in vitro. Circ Res. 2006;98:e2–10. doi: 10.1161/01.RES.0000201285.87524.9e. [DOI] [PubMed] [Google Scholar]
- 10.Lev EI, Estrov Z, Aboulfatova K, Harris D, Granada JF, Alviar C, Kleiman NS, Dong JF. Potential role of activated platelets in homing of human endothelial progenitor cells to subendothelial matrix. Thromb Haemost. 2006;96:498–504. [PubMed] [Google Scholar]
- 11.Massberg S, Konrad I, Schurzinger K, Lorenz M, Schneider S, Zohlnhoefer D, Hoppe K, Schiemann M, Kennerknecht E, Sauer S, Schulz C, Kerstan S, Rudelius M, Seidl S, Sorge F, Langer H, Peluso M, Goyal P, Vestweber D, Emambokus NR, Busch DH, Frampton J, Gawaz M. Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med. 2006;203:1221–1233. doi: 10.1084/jem.20051772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of tgf-[beta]/smad and jagged1/notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23:1155–1165. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loscalzo J. Vascular matrix and vein graft failure: Is the message in the medium? Circulation. 2000;101:221–223. doi: 10.1161/01.cir.101.3.221. [DOI] [PubMed] [Google Scholar]
- 14.Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC, Boehm M. Tgf-beta signaling mediates endothelial-to-mesenchymal transition (endmt) during vein graft remodeling. Sci Transl Med. 2014;6:227ra234. doi: 10.1126/scitranslmed.3006927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y, Davison F, Zhang Z, Xu Q. Endothelial replacement and angiogenesis in arteriosclerotic lesions of allografts are contributed by circulating progenitor cells. Circulation. 2003;108:3122–3127. doi: 10.1161/01.CIR.0000105722.96112.67. [DOI] [PubMed] [Google Scholar]
- 16.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–966. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 17.Kawamoto A, Losordo DW. Endothelial progenitor cells for cardiovascular regeneration. Trends Cardiovasc Med. 2008;18:33–37. doi: 10.1016/j.tcm.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM. Statin therapy accelerates reendothelialization: A novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- 19.Werner N, Junk S, Laufs U, Link A, Walenta K, Bohm M, Nickenig G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003;93:e17–24. doi: 10.1161/01.RES.0000083812.30141.74. [DOI] [PubMed] [Google Scholar]
- 20.Feng W, McCabe NP, Mahabeleshwar GH, Somanath PR, Phillips DR, Byzova TV. The angiogenic response is dictated by beta3 integrin on bone marrow-derived cells. J Cell Biol. 2008;183:1145–1157. doi: 10.1083/jcb.200802179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daub K, Langer H, Seizer P, Stellos K, May AE, Goyal P, Bigalke B, Schonberger T, Geisler T, Siegel-Axel D, Oostendorp RA, Lindemann S, Gawaz M. Platelets induce differentiation of human cd34+ progenitor cells into foam cells and endothelial cells. Faseb J. 2006;20:2559–2561. doi: 10.1096/fj.06-6265fje. [DOI] [PubMed] [Google Scholar]
- 22.Hidalgo A, Weiss LA, Frenette PS. Functional selectin ligands mediating human cd34(+) cell interactions with bone marrow endothelium are enhanced postnatally. J Clin Invest. 2002;110:559–569. doi: 10.1172/JCI14047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng J, Wang Y, Liang A, Jia L, Du J. Fsp-1 silencing in bone marrow cells suppresses neointima formation in vein graft. Circ Res. 2012;110:230–240. doi: 10.1161/CIRCRESAHA.111.246025. [DOI] [PubMed] [Google Scholar]
- 24.Hodivala-Dilke KM, McHugh KP, Tsakiris DA, Rayburn H, Crowley D, Ullman-Cullere M, Ross FP, Coller BS, Teitelbaum S, Hynes RO. Beta3-integrin-deficient mice are a model for glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest. 1999;103:229–238. doi: 10.1172/JCI5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prokopi M, Pula G, Mayr U, Devue C, Gallagher J, Xiao Q, Boulanger CM, Westwood N, Urbich C, Willeit J, Steiner M, Breuss J, Xu Q, Kiechl S, Mayr M. Proteomic analysis reveals presence of platelet microparticles in endothelial progenitor cell cultures. Blood. 2009;114:723–732. doi: 10.1182/blood-2009-02-205930. [DOI] [PubMed] [Google Scholar]
- 26.de Boer HC, Verseyden C, Ulfman LH, Zwaginga JJ, Bot I, Biessen EA, Rabelink TJ, van Zonneveld AJ. Fibrin and activated platelets cooperatively guide stem cells to a vascular injury and promote differentiation towards an endothelial cell phenotype. Arterioscler Thromb Vasc Biol. 2006;26:1653–1659. doi: 10.1161/01.ATV.0000222982.55731.f1. [DOI] [PubMed] [Google Scholar]
- 27.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115:3378–3384. doi: 10.1172/JCI27196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spencer EM, Tokunaga A, Hunt TK. Insulin-like growth factor binding protein-3 is present in the alpha-granules of platelets. Endocrinology. 1993;132:996–1001. doi: 10.1210/endo.132.3.7679986. [DOI] [PubMed] [Google Scholar]
- 29.Ginsberg M, James D, Ding BS, Nolan D, Geng F, Butler JM, Schachterle W, Pulijaal VR, Mathew S, Chasen ST, Xiang J, Rosenwaks Z, Shido K, Elemento O, Rabbany SY, Rafii S. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ets factors and tgfbeta suppression. Cell. 2012;151:559–575. doi: 10.1016/j.cell.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Dong Y, Dong Y, Cheng J, Du J. Role of integrin-beta3 protein in macrophage polarization and regeneration of injured muscle. J Biol Chem. 2012;287:6177–6186. doi: 10.1074/jbc.M111.292649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim H, Falet H, Hoffmeister KM, Hartwig JH. Wiskott-aldrich syndrome protein (wasp) controls the delivery of platelet transforming growth factor-beta1. J Biol Chem. 2013;288:34352–34363. doi: 10.1074/jbc.M113.459750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurlbut GD, Kankel MW, Lake RJ, Artavanis-Tsakonas S. Crossing paths with notch in the hyper-network. Current opinion in cell biology. 2007;19:166–175. doi: 10.1016/j.ceb.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 33.Ii M, Takeshita K, Ibusuki K, Luedemann C, Wecker A, Eaton E, Thorne T, Asahara T, Liao JK, Losordo DW. Notch signaling regulates endothelial progenitor cell activity during recovery from arterial injury in hypercholesterolemic mice. Circulation. 2010;121:1104–1112. doi: 10.1161/CIRCULATIONAHA.105.553917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon SM, Eguchi M, Wada M, Iwami Y, Hozumi K, Iwaguro H, Masuda H, Kawamoto A, Asahara T. Specific jagged-1 signal from bone marrow microenvironment is required for endothelial progenitor cell development for neovascularization. Circulation. 2008;118:157–165. doi: 10.1161/CIRCULATIONAHA.107.754978. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Liang A, Luo J, Liang M, Han G, Mitch WE, Cheng J. Blocking notch in endothelial cells prevents arteriovenous fistula failure despite ckd. J Am Soc Nephrol. 2014;25:773–783. doi: 10.1681/ASN.2013050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vooijs M, Ong CT, Hadland B, Huppert S, Liu Z, Korving J, van den Born M, Stappenbeck T, Wu Y, Clevers H, Kopan R. Mapping the consequence of notch1 proteolysis in vivo with nip-cre. Development. 2007;134:535–544. doi: 10.1242/dev.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi ET, Khan MF, Leidenfrost JE, Collins ET, Boc KP, Villa BR, Novack DV, Parks WC, Abendschein DR. {beta}3-integrin mediates smooth muscle cell accumulation in neointima after carotid ligation in mice. Circulation. 2004;109:1564–1569. doi: 10.1161/01.CIR.0000121733.68724.FF. [DOI] [PubMed] [Google Scholar]
- 38.Hollopeter G, Jantzen H-M, Vincent D, Li G, England L, Ramakrishnan V, Yang R-B, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet adp receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 39.Heim C, Eckl S, Preidl R, Ramsperger-Gleixner M, Koch N, Goldmann K, Spriewald BM, Weyand M, Ensminger SM. Delayed therapy with clopidogrel and everolimus prevents progression of transplant arteriosclerosis and impairs humoral alloimmunity in murine aortic allografts. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2014 doi: 10.1093/ejcts/ezu098. [DOI] [PubMed] [Google Scholar]
- 40.Puri TS, Shakaib MI, Chang A, Mathew L, Olayinka O, Minto AWM, Sarav M, Hack BK, Quigg RJ. Chronic kidney disease induced in mice by reversible unilateral ureteral obstruction is dependent on genetic background. Am J Physiol Renal Physiol. 2010;298:F1024–F1032. doi: 10.1152/ajprenal.00384.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Di Q, Cheng Z, Kim W, Liu Z, Song H, Li X, Nan Y, Wang C, Cheng X. Impaired cross-activation of beta3 integrin and vegfr-2 on endothelial progenitor cells with aging decreases angiogenesis in response to hypoxia. Int J Cardiol. 2013;168:2167–2176. doi: 10.1016/j.ijcard.2013.01.240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.