

Abstract
Background
BAY 81-8973 is a new full-length human recombinant factor VIII product manufactured with technologies to improve consistency in glycosylation and expression to optimize clinical performance.
Objectives
To demonstrate superiority of prophylaxis vs. on-demand therapy with BAY 81-8973 in patients with severe hemophilia A.
Patients/Methods
In this multinational, randomized, open-label crossover study (LEOPOLD II; ClinicalTrials.gov identifier: NCT01233258), males aged 12–65 years with severe hemophilia A were randomized to twice-weekly prophylaxis (20–30 IU kg−1), 3-times-weekly prophylaxis (30–40 IU kg−1), or on-demand treatment with BAY 81-8973. Potency labeling for BAY 81-8973 was based on the chromogenic substrate assay or adjusted to the one-stage assay. Primary efficacy endpoint was annualized number of all bleeds (ABR). Adverse events (AEs) and immunogenicity were also assessed.
Results
Eighty patients (on demand, n = 21; twice-weekly prophylaxis, n = 28; 3-times-weekly prophylaxis, n = 31) were treated and analyzed. Mean ± SD ABR was significantly lower with prophylaxis (twice-weekly, 5.7 ± 7.2; 3-times-weekly, 4.3 ± 6.5; combined, 4.9 ± 6.8) vs. on-demand treatment (57.7 ± 24.6; P < 0.0001, anova). Median ABR was reduced by 97% with prophylaxis (twice-weekly, 4.0; 3-times-weekly, 2.0; combined, 2.0) vs. on-demand treatment (60.0). Median ABR was higher with twice-weekly vs. 3-times-weekly prophylaxis during the first 6-month treatment period (4.1 vs. 2.0) but was comparable in the second 6-month period (1.1 vs. 2.0). Few patients reported treatment-related AEs (4%); no treatment-related serious AEs or inhibitors were reported.
Conclusions
Twice-weekly or 3-times-weekly prophylaxis with BAY 81-8973 reduced median ABR by 97% compared with on-demand therapy, confirming the superiority of prophylaxis. Treatment with BAY 81-8973 was well tolerated.
Keywords: factor VIII, hemophilia A, prophylaxis, recombinant proteins, therapeutic equivalency
Introduction
Treatment of severe hemophilia A requires replacement of factor VIII (FVIII) with plasma-derived or recombinant FVIII (rFVIII) products. FVIII is administered on a regular basis to prevent bleeds (prophylaxis) or on demand when bleeding events occur. Several studies have demonstrated the benefits of prophylactic regimens compared with on-demand treatment. These benefits include a decrease in the frequency of bleeds, prevention of arthropathy development in children with no preexisting joint damage, and an improvement in health-related quality of life 1–5.
A new full-length rFVIII product, BAY 81-8973, has been developed for prophylaxis and on-demand use in patients with hemophilia A. BAY 81-8973 is manufactured with technologies to improve consistency in glycosylation and expression to optimize clinical performance compared with its predecessor, sucrose-formulated rFVIII (rFVIII-FS). Notably, BAY 81-8973 is manufactured without the addition of human or animal raw materials in the cell culture, purification, or formulation processes and has a nanofiltration step for an additional level of virus removal. Another advancement is the coexpression of human heat-shock protein 70 (HSP70), a molecular chaperone involved in the proper folding of proteins and prevention of protein aggregation, during the cell culture step of production. This step enhances the viability of the expression cell line by inhibiting apoptosis and possibly by increasing proper folding of the FVIII protein 6. As a result of the refinements in the BAY 81-8973 manufacturing process, the rFVIII product has low levels of protein aggregates, a high degree of N-terminal glycan sialylation, and high and consistent purity.
The current study is part of the Long-Term Efficacy Open-Label Program in Severe Hemophilia A Disease (LEOPOLD), a clinical trial program designed to evaluate the pharmacokinetics, efficacy, and safety of BAY 81-8973 in patients with severe hemophilia A. Results from LEOPOLD I demonstrated the noninferiority of BAY 81-8973 pharmacokinetics compared with rFVIII-FS (A. Shah, H. Delesen, S. Garger, S. Lalezari, submitted). LEOPOLD I also investigated the efficacy and safety of BAY 81-8973 for prophylaxis and treatment of bleeding episodes in patients with severe hemophilia A. The results of LEOPOLD I showed that a low annual bleed rate was achieved (median 1.0 bleed per yr) while undergoing twice- or 3-times-weekly prophylaxis treatment with BAY 81-8973 7.
Here we report the efficacy and safety results of the LEOPOLD II trial. The primary objective of LEOPOLD II was to demonstrate the superiority of prophylaxis vs. on-demand therapy with BAY 81-8973 in patients with severe hemophilia A previously receiving episodic treatment.
Methods
Patients
Patients were males aged 12–65 years with severe hemophilia A (< 1% FVIII:C) who were receiving episodic treatment with FVIII at screening and had not received regular prophylaxis for > 6 consecutive months in the previous 5 years. Additional key inclusion criteria were ≥ 150 exposure days to any FVIII product, no current FVIII inhibitors, and no history of FVIII inhibitors. Key exclusion criteria were the presence of bleeding disease other than hemophilia A, thrombocytopenia (platelet count < 100 000 mm−3), requirement for premedication to tolerate FVIII injections, abnormal renal function (serum creatinine > 2.0 mg dL−1), clinically relevant liver dysfunction, or CD4 count < 250 cells μL−1. Number of previous bleeds and joint bleeds occurring in the previous 12 months was collected retrospectively based on medical records.
Study design
LEOPOLD II was a multinational, open-label, randomized, crossover, phase 2/3 study (ClinicalTrials.gov identifier: NCT01233258) conducted between January 2011 and December 2012. The study was conducted at 30 centers in 11 countries in Europe, South Africa, North America, South America, and Asia. Patients were randomized to one of six treatment arms (two low-dose prophylaxis groups, two high-dose prophylaxis groups, and two on-demand treatment groups; Fig.1) by a system generated by the sponsor’s randomization management. Due to the expected high difference in bleeding rates between the prophylaxis and on-demand treatment regimens, a 3:1 ratio for enrollment of patients to the prophylaxis or on-demand treatment arms was adopted in order to put as many patients as possible on a prophylaxis regimen. The superiority testing of prophylaxis vs. on-demand treatment was planned for the total prophylaxis group, without taking into account the different prophylaxis treatment regimens.
Figure 1.

Study design for LEOPOLD II. CS/ADJ = labeled potency adjusted by a predefined factor to mimic results obtained with the one-stage assay; CS/EP = labeled potency determined by the chromogenic substrate assay per European Pharmacopoeia.
Patients received study medication labeled depending on the defined methods for measuring the content of FVIII in the vial. Study drug was labeled using the chromogenic substrate assay per European Pharmacopoeia (CS/EP) or adjusted by a predefined factor to mimic results obtained with the one-stage assay (CS/ADJ). Because of differences in the detection of FVIII activity between the two potency assays, the difference in the actual amount of FVIII received for prophylaxis injections in the CS/EP and CS/ADJ periods was ∼20–25%, with higher amounts received during the CS/ADJ period. Patients received treatment based on CS/EP or CS/ADJ for 6 months each with an intraindividual crossover after 6 months (Fig.1). Patient assignment was performed using a centralized telephone interactive voice response system or interactive web response system. Following a washout period of 2–3 days for the prophylaxis treatment arms, patients were crossed over within their respective treatment groups (prophylaxis or on-demand) to the other potency treatment regimen for another 6 months while maintaining the same nominal treatment dose (Fig.1). The nominal dose was based on the labeled potency (printed potency on the vial) in both periods. High-dose and low-dose prophylaxis regimens with no overlap in weekly dose range were chosen to address an authority request to assess the efficacy of two different prophylaxis dose regimens. Prophylaxis dosing was either 20, 25, or 30 IU kg−1 administered twice/week or 30, 35, or 40 IU kg−1 administered 3 times/week (Fig.1). The specific dose per injection was selected by the investigator within the predefined range and maintained throughout the study. The dosing for on-demand treatment and any breakthrough bleeds was dependent on the location and severity of the bleed. Major and minor surgeries were treated with BAY 81-8973 according to recommendations for rFVIII-FS 8. Assessment of hemostasis during surgery was based on a 4-point scale (excellent, good, moderate, poor) and was performed by the surgeon or investigator; local antifibrinolytics were allowed according to standard of care.
The injections for incremental recovery measurements were administered by the study staff in the clinical setting. All patients received training and information on administration for drug infusions. All patients provided written informed consent; the protocol was approved by each site’s independent ethics committee or institutional review board before the start of the study.
Efficacy assessments
The primary efficacy variable was the annualized number of all bleeding episodes (annualized bleeding rate [ABR]), defined as spontaneous bleeds, trauma-related bleeds, untreated bleeds, and unspecified events for which treatment was administered. Assessment of the efficacy of BAY 81-8973 CS/EP potency vs. CS/ADJ potency for treatment of bleeds in patients treated on demand was a secondary endpoint of the study. Other predefined efficacy variables included the ABR of spontaneous bleeds, trauma-related bleeds, joint bleeds, and all bleeds within 48 h of a prophylaxis injection as well as incremental recovery, hemostasis during surgery, and the proportion of all bleeds treated with ≤ 2 injections.
Incremental recovery assessments were conducted for the patients in the prophylaxis treatment arms at the beginning and end of each potency period following at least a 2- to 3-day washout. Patients were administered 50 IU kg−1 BAY 81-8973, and plasma FVIII concentrations were determined preinjection and 15 min postinjection using the one-stage and chromogenic assays. Incremental recovery was calculated as: (postinjection FVIII − preinjection FVIII) × weight/dose.
Subgroup and additional analyses
Additional assessments were ABR during the first and second 6-month treatment periods, as well as dose per infusion and FVIII consumption for prophylaxis and bleeds. Subgroup analyses for ABR were also performed based on patient age (14–16 yrs vs. ≥ 18 yrs [including 18–< 30 yrs and ≥ 30 yrs]) and geographical region (Asia vs. non-Asia [ie, South Africa, North America, Europe]).
Safety
All patients were monitored for adverse events (AEs). The development of FVIII inhibitors (defined as ≥ 0.6 Bethesda units [BU]), anti-HSP70, and anti–baby hamster kidney (BHK; host cell protein) antibodies was assessed in central laboratories. For FVIII inhibitor determination, the Nijmegen-modified Bethesda assay with a lower limit of detection of 0.2 Nijmegen BU mL−1 was used. For the measurement of anti-HSP70 antibodies, patient samples were assessed with a commercial anti-HSP70 ELISA (Assay Designs Inc., Ann Arbor, MI, USA). Bleeding events were not considered AEs unless the event was serious.
Statistical analysis
All patients who received study drug were assessed in the safety analysis; patients who also had any data on injections/bleedings were included in the intent-to-treat (ITT) population, which was used for the efficacy analysis. Summary statistics such as mean, SD, median, and quartiles were calculated for continuous data; frequencies were calculated for categoric data. Statistical tests were 2-sided and performed at the 0.05 significance level. The primary objective of the study was to demonstrate the superiority of prophylaxis over on-demand therapy with BAY 81-8973. Superiority was defined as a significant decrease in ABR for the combined prophylaxis groups during the total 12-month treatment period. An analysis of variance (anova) model was used with effect for treatment group only.
The percentage of bleeds in patients treated on demand that were controlled by ≤ 2 BAY 81-8973 injections was evaluated in both potency groups, and noninferiority of treatment of bleeds with CS/EP-labeled medication vs. treatment with CS/ADJ-labeled medication was determined. Noninferiority to a historical control of 88.7% was also tested. In both cases, the noninferiority margin was 10%. The CIs were calculated using the Hodges-Lehmann estimates using StatExact version 8 under SAS (Cary, NC, USA).
A sample size of 60 : 20 patients (prophylaxis:on-demand) was calculated using a 2-sided α level of 0.05, 90% power, an attrition rate of 15%, and a 3:1 ratio of patients (prophylaxis:on-demand), with the assumption that patients treated with prophylaxis would have an average of five bleeds per year and that patients treated on demand would have an average of 15 bleeds per year with a combined SD of 11 bleeds per year 2. Statistical analysis was performed using SAS 9.2 software.
Results
Patients
A total of 83 patients were randomized (Fig.2). Three patients withdrew before treatment initiation; therefore, 80 patients were included in the ITT and safety populations. Twenty-one patients were in the on-demand group, 28 in the twice-weekly prophylaxis group, and 31 in the 3-times-weekly prophylaxis group. The mean age of the patients was 29.6 years; the majority of patients were white (45.0%) or Asian (40.0%; Table1). The median number of bleeds in the previous year was 36.0 based on patient medical records. The mean ± SD total Gilbert score at baseline was 21.0 ± 14.1 (range, 2–61). There were no relevant differences in demographics or baseline characteristics between the on-demand and prophylaxis groups (Table1).
Figure 2.

Patient disposition. *Reason for termination was noncompliance with documentation of dosing. †Reasons for not being treated were withdrawal of consent (n = 2) and protocol violation (n = 1). CS/ADJ = labeled potency adjusted by a predefined factor to mimic results obtained with the one-stage assay; CS/EP = labeled potency determined by the chromogenic substrate assay per European Pharmacopoeia.
Table 1.
Demographic and baseline characteristics of the safety/ITT population
| On demand (n = 21) | Prophylaxis twice/wk (n = 28) | Prophylaxis 3 times/wk (n = 31) | Total (N = 80) | |
|---|---|---|---|---|
| Age, yrs | ||||
| Mean | 31.4 | 28.8 | 29.1 | 29.6 |
| Median (range) | 30.0 (14–53) | 27.0 (14–54) | 28.0 (14–59) | 28.5 (14–59) |
| < 18 yrs, n | 2 | 4 | 4 | 10 |
| Race, n (%) | ||||
| White | 6 (28.6) | 16 (57.1) | 14 (45.2) | 36 (45.0) |
| Asian | 9 (42.9) | 9 (32.1) | 14 (45.2) | 32 (40.0) |
| Black | 3 (14.3) | 0 | 1 (3.2) | 4 (5.0) |
| Hispanic | 3 (14.3) | 3 (10.7) | 2 (6.5) | 8 (10.0) |
| BMI, mean kg m−2 | 23.0* | 21.3 | 21.5 | 21.8 |
| Target joint, yes (%) | 19 (90.5) | 25 (89.3) | 28 (90.3) | 72 (90.0) |
| Number of target joints | ||||
| Mean | 3.2 | 3.0 | 2.8 | 3.0 |
| Median | 3.0 | 3.0 | 2.0 | 3.0 |
| Number of bleeds in last 12 mo | ||||
| Mean | 47.5 | 38.4 | 45.6 | 43.7 |
| Median | 41.0 | 35.0 | 38.5 | 36.0 |
| Number of joint bleeds in last 12 mo | ||||
| Mean | 33.5 | 30.3 | 32.7 | 32.1 |
| Median | 28.0 | 24.0 | 25.0 | 24.0 |
| Total Gilbert score | ||||
| Mean ± SD | 19.3 ± 13.3 | 20.9 ± 14.7 | 22.4 ± 14.5 | 21.0 ± 14.1 |
BMI, body mass index; ITT, intent-to-treat.
n = 20 patients.
The mean ± SD (median [range]) nominal dose was 33 ± 5.7 (32 [21–42]) IU kg−1 per prophylaxis injection in the combined prophylaxis group compared with 24 ± 6.8 (22 [11–35]) IU kg−1 per injection for the treatment of bleeds in the on-demand group. The mean ± SD (median [range]) nominal dose for the treatment of bleeds was 30 ± 6.9 (29 [19–49]) IU kg−1 per injection in the combined prophylaxis group. Mean ± SD (median [range]) total annual FVIII consumption based on nominal dose was higher in the combined prophylaxis group compared with the on-demand group (4621 ± 1421 [4783 (2305–6738)] vs. 1781 ± 852 [1728 (597–3529)] IU kg−1 per yr, respectively).
Efficacy
The mean ± SD ABR was significantly lower with prophylaxis (twice-weekly, 5.7 ± 7.2; 3-times-weekly, 4.3 ± 6.5; combined, 4.9 ± 6.8) vs. on-demand treatment (57.7 ± 24.6; P < 0.0001, anova; Table2). Thus, prophylaxis treatment with BAY 81-8973 was superior to on-demand treatment, fulfilling the primary objective of the study. The median ABR (quartile 1 [Q1]; quartile 3 [Q3]) in the on-demand group (60.0 [41.7; 76.3]) was markedly higher than that of both prophylaxis groups (twice-weekly, 4.0 [0; 8.0]; 3-times-weekly, 2.0 [0; 4.9]; combined, 2.0 [0; 7.0]; Table2). This equates to a 97% reduction in the median bleeding rate with prophylaxis compared with on-demand treatment. ABRs during prophylaxis treatment assessed separately by CS/EP and CS/ADJ potency periods were also significantly lower compared with the on-demand group (P < 0.0001). There were no relevant differences in ABR between the potency periods (median difference, 0.0); therefore, only the ABRs for the total 1-year period are reported here.
Table 2.
Bleeding episode summary: ABR in the ITT population
| On demand (n = 21) | Prophylaxis twice/wk (n = 28) | Prophylaxis 3 times/wk (n = 31) | Prophylaxis combined (n = 59) | |
|---|---|---|---|---|
| All bleeds per yr | ||||
| Mean ± SD | 57.7 ± 24.6 | 5.7 ± 7.2* | 4.3 ± 6.5* | 4.9 ± 6.8* |
| Median (Q1; Q3) | 60.0 (41.7; 76.3) | 4.0 (0; 8.0) | 2.0 (0; 4.9) | 2.0 (0; 7.0) |
| Spontaneous bleeds per yr | ||||
| Mean ± SD | 45.3 ± 22.1 | 4.5 ± 7.1 | 2.6 ± 4.9 | 3.5 ± 6.1 |
| Median (Q1; Q3) | 42.1 (24.3; 61.3) | 2.0 (0; 6.5) | 0 (0; 3.0) | 1.0 (0; 4.0) |
| Trauma-related bleeds per yr | ||||
| Mean ± SD | 12.3 ± 16.4 | 0.9 ± 1.5 | 1.5 ± 2.8 | 1.3 ± 2.3 |
| Median (Q1; Q3) | 8.1 (1.0; 15.0) | 0 (0; 1.0) | 1.0 (0; 2.0) | 0 (0; 2.0) |
| Joint bleeds per yr | ||||
| Mean ± SD | 43.8 ± 24.7 | 5.2 ± 6.9 | 3.5 ± 6.2 | 4.3 ± 6.5 |
| Median (Q1; Q3) | 38.8 (24.3; 60.0) | 2.5 (0; 7.5) | 1.0 (0; 4.0) | 2.0 (0; 6.0) |
| Number of bleeds within 48 h after prophylaxis per yr | ||||
| Mean ± SD | – | 2.4 ± 2.4 | 3.2 ± 4.5 | 2.8 ± 3.7 |
| Median (Q1; Q3) | – | 2.0 (0; 4.5) | 1.0 (0; 4.0) | 2.0 (0; 4.1) |
ABR, annualized bleeding rate; ITT, intent-to-treat; Q1, quartile 1; Q3, quartile 3.
P < 0.0001 vs. on-demand group.
The median ABR for joint bleeds, spontaneous bleeds, and trauma-related bleeds was markedly lower in the combined prophylaxis group compared with the on-demand group (Table2). No patients in the on-demand group remained bleed-free during the study. In contrast, 27% of patients receiving prophylaxis remained bleed-free during the 1-year treatment period. In the combined prophylaxis group, 57% of the bleeds occurred within 48 h after a prophylaxis injection (median, 2.0), with a higher percentage in the 3-times-weekly than in the twice-weekly group (75.2% vs. 42.5%); this result was expected due to the more frequent injections in the 3-times-weekly group.
The total number of BAY 81-8973 injections administered for the treatment of bleeds was 1607 for the on-demand group (1204 bleeds) and 352 for the combined prophylaxis group (293 bleeds). The majority of bleeds were successfully treated with one or two injections (75.5% and 81.9% with one injection; 19.6% and 11.6% with two injections in the on-demand and prophylaxis groups, respectively).
In the on-demand group, 97% (median; lower limit 1-sided 95% CI, 95%) of bleeds per patient in the CS/EP potency period and 100% (median; lower limit 1-sided 95% CI, 93%) of bleeds per patient in the CS/ADJ potency period were treated with ≤ 2 injections, establishing the noninferiority of BAY 81-8973 to the historical control. The lower limit of the 1-sided 95% CI for the intraindividual difference for this variable was −4.9% (median, 0%), confirming noninferiority of BAY 81-8973 CS/EP potency vs. CS/ADJ potency for treatment of bleeds (P < 0.0001, exact permutation test).
Twice-weekly vs. 3-times-weekly prophylaxis treatment groups
In the prophylaxis groups, < 2% of the expected prophylaxis injections were skipped or replaced by other injections (ie, follow-up injections for surgeries or bleeds). The mean ± SD (median [range]) nominal dose was 29 ± 3.8 (30 [21–34]) IU kg−1 per prophylaxis injection in the twice-weekly group and 36 ± 4.5 (37 [30–42]) IU kg−1 per prophylaxis injection in the 3-times-weekly group. The mean ± SD (median [range]) nominal dose for the treatment of bleeds was 28 ± 5.4 (28 [19–39]) IU kg−1 per injection in the twice-weekly subgroup and 31 ± 7.6 (31 [20–49]) IU kg−1 per injection in the 3-times-weekly group. The mean ± SD (median [range]) total annual FVIII consumption based on the nominal dose was lower in the twice-weekly group compared with the 3-times-weekly group (3279 ± 589 [3282 (2305–4349)] vs. 5834 ± 622 [5961 (4620–6738)] IU kg−1 per yr, respectively).
For patients in the twice-weekly group, the ABR was higher during the first 6-month period of treatment (median, 4.1) compared with the second 6-month period (median, 1.1; Fig.3). However, in the second 6-month period, the median ABR for the twice-weekly group (1.1 bleeds per yr) was comparable to the 3-times-weekly group (2.0 bleeds per yr; Fig.3). No further improvement in bleeding rate was observed in the 3-times-weekly group in the second 6-month period (median ABR, 2.0 in both the first and second 6 months of treatment).
Figure 3.
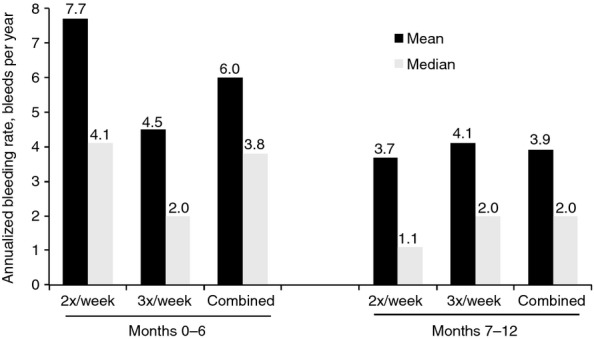
Annualized bleeding rate by time period for the twice-weekly and 3-times-weekly prophylaxis dosing groups.
Subgroup analysis: impact of age and geographical region
The number of adolescents treated with prophylaxis in this study was small (n = 8) and does not allow far-reaching conclusions; however, the ABR for these adolescents was in the same range as the ABR for adults treated with prophylaxis. Nevertheless, the group of young adults (age 18–< 30 yrs) tended to have higher ABRs in both prophylaxis regimens compared with older patients (≥ 30 yrs; Table3). The ABR during prophylaxis was similar in the Asian vs. the non-Asian (mainly European) regions (Table3).
Table 3.
Annualized number of bleeds for selected subgroups (ITT population)
| Stratification factor/Subgroup | On demand (n = 21) | Prophylaxis twice/wk (n = 28) | Prophylaxis 3 times/wk (n = 31) | Prophylaxis combined (n = 59) |
|---|---|---|---|---|
| Age group | ||||
| 14–16 yrs | ||||
| n | 2 | 4 | 4 | 8 |
| Mean ± SD | 20.1 ± 2.5 | 6.0 ± 7.2 | 2.5 ± 1.7 | 4.2 ± 5.2 |
| Median (range) | 20.1 (18.4–21.9) | 4.0 (0–15.8) | 2.0 (1.0–4.9) | 2.0 (0–15.8) |
| ≥ 18 yrs | ||||
| n | 19 | 24 | 27 | 51 |
| Mean ± SD | 61.7 ± 22.3 | 5.7 ± 7.3 | 4.5 ± 6.9 | 5.1 ± 7.1 |
| Median (range) | 61.3 (18.2–101.3) | 4.0 (0–33.1) | 2.0 (0–25.9) | 2.0 (0–33.1) |
| 18–< 30 yrs | ||||
| n | 6 | 14 | 12 | 26 |
| Mean ± SD | 70.3 ± 24.9 | 7.6 ± 8.9 | 6.0 ± 7.3 | 6.8 ± 8.1 |
| Median (range) | 63.1 (41.7–101.3) | 5.6 (0–33.1) | 3.0 (0–21.6) | 4.5 (0–33.1) |
| ≥ 30 yrs | ||||
| n | 13 | 10 | 15 | 25 |
| Mean ± SD | 57.7 ± 20.8 | 3.0 ± 3.1 | 3.3 ± 6.6 | 3.2 ± 5.4 |
| Median (range) | 60.0 (18.2–85.5) | 2.5 (0–8.1) | 1.0 (0–25.9) | 2.0 (0–25.9) |
| Region | ||||
| Asia | ||||
| n | 9 | 9 | 14 | 23 |
| Mean ± SD | 66.9 ± 21.7 | 5.7 ± 4.1 | 3.6 ± 6.6 | 4.4 ± 5.7 |
| Median (range) | 62.1 (37.1–101.3) | 5.1 (0–11.8) | 2.0 (0–21.6) | 2.0 (0–21.6) |
| Non-Asia* | ||||
| n | 12 | 19 | 17 | 36 |
| Mean ± SD | 50.8 ± 25.1 | 5.7 ± 8.3 | 4.8 ± 6.6 | 5.3 ± 7.5 |
| Median (range) | 48.0 (18.2–85.5) | 2.0 (0–33.1) | 2.0 (0–25.9) | 2.0 (0–33.1) |
ITT, intent to treat.
Including South Africa, North America, and Europe.
Impact of potency labeling
The ABR was determined for each potency period. The median intraindividual difference in the ABR between both periods was zero.
FVIII incremental recovery
Incremental recovery was measured approximately 15 min after the injection of 50 IU kg−1 BAY 81-8973 at the beginning and end of each period and described based on one-stage and chromogenic assay results. The median (Q1; Q3) recovery at the beginning of the CS/EP period was 2.1 (1.7; 2.4) IU kg−1 per IU dL−1 for the chromogenic assay and 2.1 (1.7; 2.3) IU kg−1 per IU dL−1 for the one-stage assay and did not change during the 6-month period.
Surgeries
One major (hemorrhoidectomy) and 20 minor surgeries, 16 of them dental extractions, were performed during the study. The hemostasis during surgery was assessed as good or excellent in all cases.
Safety
Treatment-emergent AEs were experienced by 44 (55%) patients. Only three patients (4%) experienced an AE assessed as treatment related. Two patients experienced serious AEs, neither of which was considered to be related to treatment.
No patients developed FVIII inhibitors. Changes in anti-HSP70 antibody levels during the study were unspecific and generally transient. The majority of patients had detectable levels of anti-HSP70 antibodies at baseline, which were below the 95th percentile validated in a normal population. The mean ± SD anti-HSP70 antibody level at baseline was 86.2 ± 99.0 (range, 25.0–861) ng mL−1. The lower limit of detection was 25 ng mL−1 (1:1000 diluted serum sample [25 μg mL−1 in undiluted serum]). Two patients had levels above this threshold at baseline; one patient maintained positive levels with some fluctuations during the study, and the second patient had a level below the threshold at the next determination at 3 months. Eight patients in the prophylaxis group had an increase in anti-HSP70 to levels above the 95th percentile of a normal population; five of these eight patients had increased levels at single visits but were below the threshold at the end of the study, and the titer was decreasing by the end of the study in the remaining three patients. No clinical symptoms were attributed to the presence of anti-HSP70 antibody (eg, the increased antibody levels did not coincide with an increased bleeding rate or hypersensitivity reactions). In some patients with elevated anti-HSP70 antibody levels, a recent infectious event could be identified. Two patients tested positive for anti-BHK antibodies during the study; however, for both patients the antibody levels at baseline were already borderline positive. Neither patient reported any hypersensitivity reactions, and they reported one and two bleeds per year, respectively.
Discussion
LEOPOLD II demonstrated the superiority of prophylaxis dosing vs. on-demand treatment with the newly developed rFVIII, BAY 81-8973, in patients with severe hemophilia A previously treated on demand. This superiority was observed with both twice-weekly (low-dose) and 3-times-weekly (high-dose) prophylaxis treatment. These results complement the efficacy of twice-weekly and 3-times-weekly prophylaxis dosing with BAY 81-8973 established by the LEOPOLD I study 7. The ABR in the on-demand group was slightly higher than the prestudy ABR. This result may be explained by the retrospective prestudy data collection and the lack of or incomplete patient diaries before the study, leading to an underestimation of the bleed rates. In addition, it is likely that only joint bleeds and treated bleeds that occurred before the study were captured in the medical records. Information on previous FVIII consumption was incomplete and was therefore not analyzed.
Patients in LEOPOLD II were randomized to the prophylactic dosing regimens (twice/week or 3 times/week) or on-demand treatment. Thus, the prophylactic dosing regimen assignments did not take into account a patient’s individual bleeding profile. Regardless of patient history, both twice-weekly and 3-times-weekly prophylaxis with BAY 81-8973 resulted in a significantly lower overall ABR compared with on-demand treatment, although the overall ABR was slightly higher in patients receiving twice-weekly treatment.
The duration of treatment with BAY 81-8973 had an impact on the efficacy of twice- vs. 3-times-weekly prophylaxis. Patients receiving twice-weekly dosing achieved an acceptable ABR in the first 6 months, although the results were less impressive than with 3-times-weekly dosing. However, in the second 6 months, bleeding rates for twice-weekly and 3-times-weekly dosing were comparable, demonstrating that continued improvement over time was seen with the twice-weekly dosing regimen. This result was not entirely unexpected in a patient population with severe hemophilia A in which patients had several target joints, a high number of joint bleeds before the study, and mean baseline Gilbert scores indicative of existing joint damage. This population may require a longer amount of time to achieve a robust response to a low-dose treatment. LEOPOLD II is the first prospective study to evaluate a low-dose prophylaxis and to report this type of improvement in the efficacy of prophylactic FVIII over time.
Among so-called ‘short-acting’ FVIII products (ie, those that have not been modified to extend half-life), effective prophylaxis at dosing regimens of <3 times/week has been demonstrated in few other prospective studies 9–12. Further studies with unmodified FVIII products are under way that will address effectiveness of less than 3-times-weekly dosing 13,14. Although recent studies have highlighted the high interpatient variability in FVIII pharmacokinetics following administration of comparable prophylactic doses 15,16, the pharmacokinetics of the administered drug may not be the only explanation for the effectiveness of a twice-weekly dosing regimen. Other factors such as the joint status (eg, the number of target joints and severity of chronic synovitis) and patient activity level may also play a major role in the bleeding tendency. The improvement in outcome over time may point to the impact of the severity of synovitis, which improves under prophylaxis treatment 9. The ability of a patient to receive only twice-weekly prophylaxis compared with standard 3-times-weekly prophylaxis is appealing from the perspectives of patient preference, cost, need for venous access, and rFVIII consumption.
One of the most serious complications in FVIII replacement therapy is the development of inhibitors. No patients developed FVIII inhibitors during the study. A few patients had an increase in the level of antibodies against HSP70, although the changes were transient, and two patients had levels at baseline above the prevalidated 95th percentile before start of treatment. The implications for development of anti-HSP70 antibodies are unclear; however, these antibodies are found in the general population 17,18 and in patients with certain inflammatory and infectious diseases 19–21. Thus, anti-HSP70 antibody levels do not appear to be drug specific but rather seem to be a reaction to inflammation.
The open-label design is a limitation of this study. Furthermore, results of the subgroup analyses should be interpreted with caution given the small number of patients in certain subgroups. In addition, patient self-reporting of bleeding is a subjective endpoint that might be imprecise. Nevertheless, the extreme difference in ABR between the on-demand and prophylaxis groups indicates that this difference would be evident in even a small number of enrolled patients.
Conclusions
Patients with severe hemophilia A receiving twice-weekly or 3-times-weekly BAY 81-8973 prophylaxis had a significantly lower annualized number of bleeds compared with on-demand therapy, confirming the superiority of prophylactic treatment. A low ABR was achieved with a 3-times-weekly regimen, as well as with a twice-weekly prophylaxis regimen. The response to prophylaxis treatment improved over 6 months in the low-dose (twice-weekly) prophylaxis group. BAY 81-8973 also exhibited good tolerability and no anti-FVIII inhibitor development.
Addendum
K. Kavakli was the principal investigator for the LEOPOLD II study. The LEOPOLD II steering committee was responsible for overseeing the conduct of the study. K. Kavakli was a member of the steering committee and contributed to the study design and data acquisition, interpretation, and analysis. R. Yang and L. Rusen contributed to the acquisition of data. H. Beckmann and D. Tseneklidou-Stoeter contributed to data analysis and data interpretation. M. Maas Enriquez contributed to the study design, data analysis, and data interpretation. Each author contributed to the development of the manuscript, reviewed and commented on each draft, and approved the final draft.
Acknowledgments
This study was funded by Bayer HealthCare AG, Leverkusen, Germany. Medical writing assistance was provided by Erin P. Scott for Complete Healthcare Communications, Inc. (Chadds Ford, PA) and was fully funded by Bayer HealthCare Pharmaceuticals.
Appendix
LEOPOLD II Study Investigators
China: Renchi Yang, Yongqiang Zhao, Jing Sun, Xuefeng Wang, Depei Wu; Czech Republic: Antonin Hlusi; Japan: Katsuyuki Fukutake, Hideji Hanabusa, Teruhisa Fujii; Mexico: Oscar Pérez Ramírez, Blanca Salazar Alvarado; Romania: Margit Serban, Luminita Rusen, Valentina Uscatescu, Cristina Truica; Serbia: Gordana Kostic, Nada Konstantinidis, Zoran Igrutinovic; Russia: Farida Perina, Tatiana Andreeva; Turkey: Kaan Kavakli, Bulent Antmen, Ilgen Sasmaz, Alphan Kupesiz, Mehmet Akif Yesilipek; Taiwan: Ching-Tien Peng; United States: James French II, Miguel Escobar; South Africa: Johnny Mahlangu, Roger Pool.
Disclosure of Conflict of Interests
K. Kavakli has received research funding, honoraria, and scientific congress travel expenses from Bayer. R. Yang received honoraria and scientific congress travel expenses from Bayer China. L. Rusen has received fees for the trial from Bayer. H. Beckmann, D. Tseneklidou-Stoeter, and M. Maas Enriquez are employees of Bayer Pharma AG.
References
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ML, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357:535–44. doi: 10.1056/NEJMoa067659. [DOI] [PubMed] [Google Scholar]
- Manco-Johnson MJ, Kempton CL, Reding MT, Lissitchkov T, Goranov S, Gercheva L, Rusen L, Ghinea M, Uscatescu V, Rescia V, Hong W. Randomized, controlled, parallel-group trial of routine prophylaxis vs. on-demand treatment with sucrose-formulated recombinant factor VIII in adults with severe hemophilia A (SPINART) J Thromb Haemost. 2013;11:1119–27. doi: 10.1111/jth.12202. J Thromb Haemost [published correction appears in. 2014;12:119-122.]. [DOI] [PubMed] [Google Scholar]
- Mondorf W, Kalnins W, Klamroth R. Patient-reported outcomes of 182 adults with severe haemophilia in Germany comparing prophylactic vs. on-demand replacement therapy. Haemophilia. 2013;19:558–63. doi: 10.1111/hae.12136. [DOI] [PubMed] [Google Scholar]
- Noone D, O’Mahony B, van Dijk JP, Prihodova L. A survey of the outcome of prophylaxis, on-demand treatment or combined treatment in 18–35-year old men with severe haemophilia in six countries. Haemophilia. 2013;19:44–50. doi: 10.1111/j.1365-2516.2012.02934.x. [DOI] [PubMed] [Google Scholar]
- Royal S, Schramm W, Berntorp E, Giangrande P, Gringeri A, Ludlam C, Kroner B, Szucs T. European Haemophilia Economics Study Group. Quality-of-life differences between prophylactic and on-demand factor replacement therapy in European haemophilia patients. Haemophilia. 2002;8:44–50. doi: 10.1046/j.1365-2516.2002.00581.x. [DOI] [PubMed] [Google Scholar]
- Ishaque A, Thrift J, Murphy JE, Konstantinov K. Over-expression of Hsp70 in BHK-21 cells engineered to produce recombinant factor VIII promotes resistance to apoptosis and enhances secretion. Biotechnol Bioeng. 2007;97:144–55. doi: 10.1002/bit.21201. [DOI] [PubMed] [Google Scholar]
- Saxena K, Lalezari S, Oldenburg J, Delesen H, Shah A, Tseneklidou-Stoeter D, Maas-Enriquez M. Pharmacokinetics, efficacy, and safety of BAY 81-8973, a full-length plasma-protein-free recombinant factor VIII product: results from the LEOPOLD trial. J Thromb Haemost. 2013;11:928. [Google Scholar]
- Kogenate®. Tarrytown, NY: Bayer HealthCare, LLC; 2013. FS (antihemophilic factor [recombinant] formulated with sucrose). Full Prescribing Information, [Google Scholar]
- Aznar JA, Garcia-Dasi M, Perez-Alenda S, Marco A, Jaca M, Moret A, Querol F. Secondary prophylaxis vs. on-demand treatment to improve quality of life in severe adult haemophilia A patients: a prospective study in a single centre. Vox Sang. 2014;106:68–74. doi: 10.1111/vox.12066. [DOI] [PubMed] [Google Scholar]
- Valentino LA, Mamonov V, Hellmann A, Quon DV, Chybicka A, Schroth P, Patrone L, Wong WY. Prophylaxis Study Group. A randomized comparison of two prophylaxis regimens and a paired comparison of on-demand and prophylaxis treatments in hemophilia A management. J Thromb Haemost. 2012;10:359–67. doi: 10.1111/j.1538-7836.2011.04611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman BM, Pai M, Rivard GE, Israels S, Poon MC, Demers C, Robinson S, Luke KH, Wu JK, Gill K, Lillicrap D, Babyn P, McLimont M, Blanchette VS. Association of Hemophilia Clinic Directors of Canada Prophylaxis Study Group. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia Primary Prophylaxis Study. J Thromb Haemost. 2006;4:1228–36. doi: 10.1111/j.1538-7836.2006.01953.x. [DOI] [PubMed] [Google Scholar]
- Vdovin VV, Andreeva TA, Chernoua TA, Perina FG, Shiller EEM, Svirin PV, Maas EnriquezM, Rauchensteiner S. Prophylaxis with once, twice or three-times weekly dosing of rFVIII-FS Prevents joint bleeds in a previously treated pediatric population with moderate/severe hemophilia A. J Coagul Disord. 2011;3:1–8. [Google Scholar]
- Valentino LA, Negrier C, Kohla G, Tiede A, Liesner R, Hart D, Knaub S. The first recombinant FVIII produced in human cells - an update on its clinical development programme. Haemophilia. 2014;20:1–9. doi: 10.1111/hae.12322. [DOI] [PubMed] [Google Scholar]
- Ragni MV. Rationale for a randomized controlled trial comparing two prophylaxis regimens in adults with severe hemophilia A: the Hemophilia Adult Prophylaxis Trial. Expert Rev Hematol. 2011;4:495–507. doi: 10.1586/ehm.11.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C. Importance of pharmacokinetics in the management of hemophilia. Pediatr Blood Cancer. 2013;60:S27–9. doi: 10.1002/pbc.24339. [DOI] [PubMed] [Google Scholar]
- Collins PW, Fischer K, Morfini M, Blanchette VS, Bjorkman S. Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. Haemophilia. 2011;17:2–10. doi: 10.1111/j.1365-2516.2010.02370.x. [DOI] [PubMed] [Google Scholar]
- Pockley AG, Shepherd J, Corton JM. Detection of heat shock protein 70 (Hsp70) and anti-Hsp70 antibodies in the serum of normal individuals. Immunol Invest. 1998;27:367–77. doi: 10.3109/08820139809022710. [DOI] [PubMed] [Google Scholar]
- Rea IM, McNerlan S, Pockley AG. Serum heat shock protein and anti-heat shock protein antibody levels in aging. Exp Gerontol. 2001;36:341–52. doi: 10.1016/s0531-5565(00)00215-1. [DOI] [PubMed] [Google Scholar]
- Mattei D, Scherf A, Bensaude O, da Silva LP. A heat shock-like protein from the human malaria parasite Plasmodium falciparum induces autoantibodies. Eur J Immunol. 1989;19:1823–8. doi: 10.1002/eji.1830191010. [DOI] [PubMed] [Google Scholar]
- Pockley AG, De Faire U, Kiessling R, Lemne C, Thulin T, Frostegard J. Circulating heat shock protein and heat shock protein antibody levels in established hypertension. J Hypertens. 2002;20:1815–20. doi: 10.1097/00004872-200209000-00027. [DOI] [PubMed] [Google Scholar]
- Tishler M, Shoenfeld Y. Anti-heat-shock protein antibodies in rheumatic and autoimmune diseases. Semin Arthritis Rheum. 1996;26:558–63. doi: 10.1016/s0049-0172(96)80043-6. [DOI] [PubMed] [Google Scholar]