

Abstract
Genomes of all free-living organisms encode the enzyme dUTPase (dUTP pyrophosphatase), which plays a key role in preventing uracil incorporation into DNA. In the present paper, we describe the biochemical and structural characterization of DUT1 (Saccharomyces cerevisiae dUTPase). The hydrolysis of dUTP by DUT1 was strictly dependent on a bivalent metal cation with significant activity observed in the presence of Mg2+, Co2+, Mn2+, Ni2+ or Zn2+. In addition, DUT1 showed a significant activity against another potentially mutagenic nucleotide: dITP. With both substrates, DUT1 demonstrated a sigmoidal saturation curve, suggesting a positive co-operativity between the subunits. The crystal structure of DUT1 was solved at 2 Å resolution (1 Å = 0.1 nm) in an apo state and in complex with the non-hydrolysable substrate α,β-imido dUTP or dUMP product. Alanine-replacement mutagenesis of the active-site residues revealed seven residues important for activity including the conserved triad Asp87/Arg137/Asp85. The Y88A mutant protein was equally active against both dUTP and UTP, indicating that this conserved tyrosine residue is responsible for discrimination against ribonucleotides. The structure of DUT1 and site-directed mutagenesis support a role of the conserved Phe142 in the interaction with the uracil base. Our work provides further insight into the molecular mechanisms of substrate selectivity and catalysis of dUTPases.
Keywords: dITP, dUTP, dUTP pyrophosphatase (dUTPase), enzyme co-operativity, mutagenesis, protein structure
INTRODUCTION
Preserving DNA integrity is of vital importance for all organisms. Cellular metabolism constantly generates non-canonical nucleoside triphosphates, such as dUTP, dITP, dXTP, 8-oxo-dGTP or 2-oxo-dATP, which arise from oxidation, deamination or other modifications of canonical nucleotides [1]. Incorporation of non-canonical nucleotides into the nascent DNA results in increased mutagenesis and overloads the DNA excision repair system, leading to multiple DNA strand breaks and cell death [2,3]. The most common non-canonical nucleoside triphosphate is dUTP, which is continuously produced in the pyrimidine biosynthesis pathway by phosphorylation of dUDP or by deamination of dCTP [2,4]. Most DNA polymerases cannot distinguish between thymine and uracil (except for some archaeal enzymes), and the uracil/thymine incorporation ratio depends on the relative level of dUTP and dTTP [5].
Genomes of all free-living organisms and many viruses encode the enzyme dUTPase (dUTP pyrophosphatase) (EC 3.6.1.23), which cleaves dUTP into dUMP and pyrophosphate and plays a key role in preventing uracil incorporation into DNA [5,6]. dUTPase is essential for DNA integrity and viability in many prokaryotic and eukaryotic organisms including Escherichia coli, Saccharomyces cerevisiae, trypanosomes and human cancer cells [7–9]. The E. coli hypomorphic dUTPase mutant dut-1 retains less than 1% of wild-type dUTPase activity and is still viable. The dut-1 strain exhibits a spontaneous mutator phenotype, a high recombination frequency and SL (synthetic lethality) with mutations in the AP (apurinic/apyrimidinic) endonuclease gene (xth) or the homologous recombination gene recA [3]. In addition, a systematic study of genetic interactions of the dut-1 mutation with other E. coli genes revealed a single SL defect in the DNA precursor metabolism (thymidine kinase tdk), whereas the majority of the identified genetic interactions of dut were in DNA repair: uracil-DNA excision (ung, polA and xthA), repair of double-strand DNA breaks (recA, recBC and ruvABC) and chromosomal dimer resolution (xerCD and ftsK) [10]. Whereas the tdk–dut-1 interaction can be explained in terms of the functional redundancy for dUMP production, the interactions of dut with DNA repair genes were explained as ‘defect–damage–repair’ cycles linking unrelated pathways or as inactivation of compensating activity [10]. Similarly, the major role of DUT1 (S. cerevisiae dUTPase) involves preventing the incorporation of uracil into DNA [8,11]. The reduced viability of the yeast double mutants with deleted dut-1 and apn1 or apn2 (both of the latter encode the abasic DNA repair endonucleases) suggests that the phenotypes of the dut-1 mutant (increased division time, an abnormal FACS profile and high levels of large budded cells with the nucleus at the bud neck) are associated with the incorporation of dUMP into DNA and subsequent formation of endogenous abasic (AP) sites [11]. S. cerevisiae serves as a valuable model in cancer research for examining the action of anticancer agents, particularly to dissect the role of uracil incorporation into DNA as a mechanism contributing to cytotoxicity induced by antifolate drugs, which target the thymidylate synthase reaction (the reductive methylation of dUMP to dTMP) [12]. Recent biochemical and genetic studies have demonstrated that cytotoxicity of antifolates and the DNA-damaging agent oxaliplatin in yeast and human cancer cells is dependent on uracil misincorporation into DNA, and that dUTPase functions as the central regulator of the intracellular dUTP pool and modulates drug toxicity [1,13]. In addition, previous work on human cells has demonstrated the down-regulation of the dUTPase gene expression by the tumour-suppressor protein p53 in response to oxaliplatin, suggesting that the p53-mediated repression of dUTPase may increase DNA damage and therefore induce apoptosis [13]. Thus dUTPase represents an important target for anticancer, anti-retroviral and antimicrobial therapies, and dUTPase inhibitors could potentially fight infectious diseases such as malaria, tuberculosis and AIDS [5,14].
Three known dUTPase families include the monomeric, dimeric and trimeric enzymes, which have different subunit organization. The homotrimeric dUTPases represent the major and best characterized group of these enzymes, which have three identical active sites made up from the five highly conserved motifs, contributed from all three monomers [5,6]. The monomeric dUTPases contain the same five motifs, but organized in a different order [15,16]. In contrast, the homodimeric dUTPases from trypanosomes, Campylobacter jejunii and T4 bacteriophage function as physiological dimers and have no sequence similarity to the other two dUTPase families [9]. Crystal structures of dUTPases from all three families have been determined (PDB codes 1DUO, 1OGL and 2BSY) and revealed that trimeric and monomeric enzymes are composed primarily of β-pleated sheets (a jelly-roll fold), whereas the dimeric dUTPase from Trypanosoma cruzi has an all-α fold and a different active site, suggesting a dissimilar catalytic mechanism [5,17]. In trimeric dUTPases, the five conserved sequence motifs from each subunit are involved in the formation of three active sites located at the subunit interfaces, so all three subunits contribute to the formation of three identical active sites [5,6]. The residues from the first subunit are involved in base and sugar recognition, the second subunit provides residues for phosphate binding, and the extended flexible C-terminal end of the third subunit covers the active site with a bound substrate [6].
In the present paper, we describe the biochemical, structural and mutational studies of the S. cerevisiae dUTPase DUT1. Biochemical experiments have revealed that DUT1 exhibits significant activity against another non-canonical nucleotide, dITP. The crystal structure of DUT1 was solved both in the apo form and in complex with α,β-imido dUTP or dUMP. In combination with site-directed mutagenesis, these structures have provided insights into the molecular mechanisms of the substrate selectivity and catalysis by the yeast dUTPase.
EXPERIMENTAL
Gene cloning, overexpression and protein purification
The DUT1 gene (YBR252W) was amplified by PCR using S. cerevisiae genomic DNA and cloned into a modified pET15b vector (Novagen) using the In-Fusion Dry-Down PCR cloning kit (Clontech) as described by the manufacturer. The modified pET15b vector contains an N-terminal His6 tag followed by a tobacco etch virus protease cleavage site (ENLYFQ↓;G) [18]. DUT1 was overexpressed in the E. coli BL21-Gold(DE3) strain (Stratagene) and affinity purified using Ni2+-chelate chromatography on nickel-affinity resin (Qiagen) as described previously [18]. The oligomeric state of DUT1 was determined using gel-filtration analysis on a Superdex-200 10/300 GL column (GE Healthcare) as described previously [18].
Enzymatic screens and assays
HPLC analysis of the DUT1 reaction products was performed using a Varian ProStar HPLC system equipped with a 5 μm Varian Pursuit C18 column (15 cm×4.6 mm internal diameter) and a Varian MetaGuard guard column (flow rate 1 ml/min). The mobile phase contained two buffers: (A) 0.1 M KH2 PO4 (pH 6.0) with 8 mM TBH (tetrabutylammonium hydroxide); and (B) 0.1 M KH2 PO4 (pH 6.0) with 8 mM TBH and 30% methanol [19]. Purified DUT1 was screened for the presence of NTP pyrophosphatase activity against the commercially available canonical and non-canonical NTPs (purchased from Sigma, TriLink BioTechnologies or Jena Biosciences). NTP pyrophosphatase activity was determined by measuring the Pi release in a 160-μl assay in 96-well microplates containing 50 mM Hepes (pH 7.5), 5 mM MgCl2, 1 mM MnCl2, 0.5 mM NiCl2, 0.1 mM NTP, 10 m-units of inorganic pyrophosphatase (from baker’s yeast; Sigma), and 0.5 μg of DUT1. After a 30 min incubation at 30°C, the reaction was terminated by the addition of 40 μl of Malachite Green reagent [20] and the amount of Pi released was calculated on the basis of absorbance at 630 nm. The dependence of the DUT1 activity on bivalent metal cations was determined with dUTP (1 mM) as substrate without metal addition or in the presence of EDTA (5 mM) or various bivalent cations (5 mM MgCl2, or 0.1 mM MnCl2, CoCl2, NiCl2, CaCl2 or CuCl2, or 0.05 mM ZnCl2) using the HPLC-based assay described above. For the determination of Km and Vmax, pyrophosphatase activity was determined over a range of substrate concentrations (between 0.003 and 0.4 mM for dUTP and 0.025–0.4 mM for dITP) in the presence of 0.1 mM MgCl2 and 0.04 μg of DUT1 (for dUTP) or 0.1 μg of DUT1 (for dITP) using the pyrophosphatase-coupled assay (160 μl) described above. Kinetic parameters were calculated by non-linear regression analysis of raw data to fit to the Michaelis–Menten function using GraphPad Prism software (version 4.00 for Windows, GraphPad). For sigmoidal curve fitting, the Hill equation [21] was used to calculate kinetic parameters using GraphPad Prism:
where S is the substrate concentration, S0.5 is the half-saturating concentration of the substrate and h is the Hill coefficient.
Site-directed mutagenesis of DUT1
Selected residues of the wild-type DUT1 were mutated to alanine using PCR with mutagenic primers and the QuikChange® mutagenesis kit (Stratagene) as described previously [18]. The presence of mutations was confirmed by DNA sequencing using the T7 promoter primer. Plasmids containing the desired mutations were transformed into E. coli BL21(DE3) cells and mutated proteins were purified using the same protocol as for the wild-type protein.
Protein crystallization and structure determination
Crystals of apo-DUT1 and DUT1 in complex with dUMP were grown at 22°C by hanging-drop vapour diffusion with 2 μl of protein sample (25 mg/ml) and an equal volume of reservoir buffer using an optimized sparse matrix crystallization screen and small amounts of proteases according to the protocol of Dong et al. [22]. Crystals of apo-DUT1 were grown using a reservoir solution of 0.1 M ammonium sulfate, 0.1 M Bis-Tris (pH 5.5), and 25% PEG3350 [where PEG is poly(ethylene glycol)] with 0.02 mg/ml trypsin added to the protein solution (final concentration of 25 mg/ml). Crystals were cryoprotected with a solution of 7% glycerol, 7% ethylene glycol and 7% sucrose prior to freezing in liquid N2. Crystals of the DUT1–α,β-imido dUTP complex were grown in a reservoir solution containing 30% Jeffamine ED-2001 (pH 7.0), 0.1 M potassium Hepes (pH 7.0), 3 mM MgCl2 and 2.5 mM α,β-imido dUTP with the addition of trypsin (0.02 mg/ml). Crystals of the DUT1–dUMP complex were grown using a reservoir containing 0.2 M sodium acetate, 0.1 M Tris/HCl (pH 8.5), 30% PEG4000 and 10 mM dUTP, with 0.02 mg/ml trypsin added to the crystallization conditions. Crystals were cryoprotected with a solution containing 4% ethylene glycol, 4% glycerol and 4% sucrose prior to flash-freezing in liquid N2.
For the apo-DUT1 structure, diffraction data were collected at λ = 1.54178 Å (Cu Kα radiation) on a home-source Rigaku FR-E+ Superbright generator equipped with osmic mirrors and a Raxis4++ detector. Data were scaled and merged using HKL2000 [23]. The structure was solved by molecular replacement using the protein atoms of the DUT1–dUMP trimer (PDB code 3F4F; see below). The final model contained eight trimers in the unit cell (24 molecules of DUT1); the first six trimers were found using PHASER [24] and the final two trimers with MOLREP [25], both as part of the CCP4 suite [26]. Subsequent manual model building and automated water-picking using COOT [27] and refinement using Refmac [28] and TLS [29,30] were used to refine the model. The final model yielded Rwork and Rfree values of 17.8% and 23.6% respectively and contained 25 111 atoms, and each molecule of the asymmetric unit contains a single chain roughly spanning residues 6–133 of DUT1. The final model showed a high-quality stereochemistry [31], with 99.0% of residues occupying the most-favoured and additionally favoured regions of the Ramachandran plot.
For the DUT1–α,β-imido dUTP complex structure, datasets were collected at the APS ID19 beamline (Argonne) and reduced with XDS software [32]. The structure was solved by molecular replacement using the program MOLREP [25] with chain A of PDB structure 3HHQ as the model, and the refinement was performed using Refmac [28]. After several refinement cycles, water molecules were built iteratively by ARP/wARP [33]. At this stage, the densities of α,β-imido dUTP and magnesium were clearly highlighted. Three molecules of α,β-imido dUTP and magnesium atoms were added to the model using COOT [27]. The final refinement was performed using TLS and Phenix [29,30]. A total of 98.7% of residues of the final model were in the most-favoured regions and 1.3% were in allowed regions of the Ramachandran plot [34].
For the DUT1–dUMP complex structure, diffraction data were collected at λ = 1.54178 Å on a home-source Rigaku Micromax-007 generator equipped with osmic mirrors and a Raxis4++ detector. Data were also scaled and merged using HKL2000 [23]. The structure was solved by molecular replacement using a model of the protein generated by SWISS-MODELLER [35] based on PDB structure 2OKD. The final model contained one trimer in the unit cell (three molecules of DUT1); each of the three subunits was found using PHASER [24]. Following molecular replacement, electron density was observed in the active site for the product dUMP, not dUTP. Three molecules of dUMP were added to the model using COOT [27], and subsequent manual model building and automated water-picking using COOT and refinement using Refmac [28] and TLS [29,30] were used to refine the model. The final model, refined to 2.0 Å, yielded Rwork and Rfree values of 14.9% and 20.7% respectively and contained 3668 atoms, and each subunit of the asymmetric unit contained a single chain approximately spanning residues 6–143 of DUT1. The final model showed a high-quality stereochemistry [31] with 99.1% of residues occupying the most-favoured and additionally favoured regions of the Ramachandran plot. Data collection and refinement statistics are summarized in Table 1.
Table 1.
Data collection and model refinement statistics
| DUT1 (apo structure) (PDB code 3HHQ) | DUT1 (+α,β-imido dUTP) (PDB code 3P48) | DUT1 (+dUMP) (PDB code 3F4F) | |
|---|---|---|---|
| Data collection | |||
| Space group | P1 | P212121 | P43212 |
| Cell dimensions | |||
| a,b,c (Å) | 95.5, 95.5, 97.6 | 51.7, 58.8, 112.3 | 124.5, 124.5, 51.7 |
| α,β,γ (°) | 98.8, 97.4, 107.1 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Wavelength | 1.541 | 0.979 | 1.541 |
| Resolution (Å) | 50.0–2.0 (2.07–2.0) | 28.47–1.67 (1.76–1.67) | 50–2.0 (2.07–2.0) |
| Reflections overall (outer shell) | 191 238 (19074) | 40 597 (5802) | 53 958 (5381) |
| R merge | 0.058 (0.441) | 0.043 (0.239) | 0.086 (0.386) |
| I/σ/ | 19.1 (3.4) | 28.4 (8.2) | 15.1 (4.9) |
| Completeness (%) | 89.2 (88.3) | 99.9 (99.8) | 99.9 (100) |
| Redundancy | 3.4 (3.2) | 6.3 (6.1) | 4.3 (4.0) |
| Refinement | |||
| Resolution (Å) | 40.62–2.0 | 28.47–1.67 | 47.7–2.0 |
| Number of reflections* | 181642 (9588) | 40 218 (2017) | 26 589 (1406) |
| Rwork/Rfree | 0.175/0.236 | 0.154/0.192 | 0.146/0.207 |
| Number of atoms | |||
| Protein | 23034 | 3326 | 3147 |
| Major ligand | – | 126 (42×3) | 60 |
| Solvent | 1765 | 405 | 437 |
| Other | 363 | 3 | 23 |
| Rmsd | |||
| Bond lengths (Å) | 0.021 | 0.017 | 0.015 |
| Bond angles (°) | 1.85 | 1.656 | 1.66 |
| Ramachandran plot | |||
| Most favoured (%) | 90.0 | 98.7 | 92.3 |
| Additionally allowed (%) | 8.1 | 1.3 | 6.9 |
| Disallowed (%) | 0.2 | 0.0 | 0.9 |
Values in parentheses are for the highest-resolution shell.
Number of reflections overall and in the test set.
RESULTS AND DISCUSSION
Enzymatic activity of DUT1
The yeast dUTPase DUT1 was overexpressed in E. coli and affinity purified using the N-terminal His6 tag with a high yield (>100 mg/l culture) and purity (>95% homogeneity). Gel-filtration experiments revealed that the native DUT1 has a molecular mass of 56.6 kDa, indicating that, like other characterized dUTPases, this protein exists as a trimer in solution (the sequence-based molecular mass of the His6-tagged DUT1 monomer is 18.1 kDa). Purified DUT1 hydrolysed dUTP over a broad pH range (7.0–9.0; results not shown) and required the addition of a bivalent metal cation for activity (shown using an HPLC-based assay) (Figure 1A). Mg2+ supported the highest rate of dUTP hydrolysis by DUT1, but significant activity was also observed in the presence of Co2+, Mn2+, Ni2+ or Zn2+ (Figure 1A). The pyrophosphatase-coupled assay also identified Mg2+ as the best metal cation for DUT1 following by Zn2+, Mn2+ and Co2+ (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/437/bj4370243add.htm). Mg2+ has been shown to support the activity of all characterized dUTPases, and several of them (e.g. from E. coli, Drosophila, or Bacillus subtilis phage SP-β) can also use Mn2+, Co2+ or Zn2+ [36]. DUT1 appeared to exhibit a higher affinity for Co2+, Zn2+ and Mg2+ (apparent Kd 23.9–37.6 μM), whereas the affinity for Mn2+ was lower (Kd 92.3 μM).
Figure 1. Metal ion and substrate profiles of DUT1.
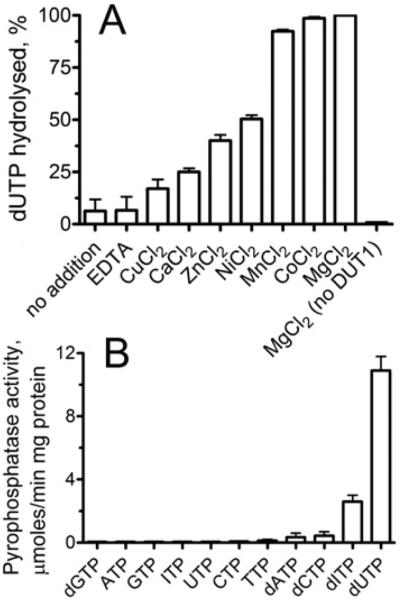
(A) Effect of bivalent metal cations on the hydrolysis of dUTP by DUT1. (B) Screening of DUT1 for nucleoside triphosphate pyrophosphatase activity against various nucleotides. The activity was measured in the presence of (A) 1 mM dUTP and 1 μg of DUT1 or (B) 0.1 mM substrate and 0.5 μg of DUT1 using HPLC (A) or a pyrophosphatase-coupled assay (B) as described in the Experimental section.
Prokaryotic and eukaryotic dUTPases exhibit exquisite substrate specificity for dUTP and show negligible hydrolysis of UTP or other nucleotides [37,38]. Under optimal assay conditions, the purified DUT1 showed very low activity against UTP (40–50 nmol/min per mg of protein), which is close to that of the rat liver dUTPase [39]. However, several biochemically characterized dUTPases from viruses or Thermococcus onnurineus also demonstrated a significant activity against dCTP, dTTP and UTP, suggesting a lower substrate selectivity of these enzymes [38,40]. To characterize the substrate specificity of the S. cerevisiae DUT1, the purified protein was screened for pyrophosphatase activity against an array of commercially available canonical and non-canonical nucleoside triphosphates. As shown in Figure 1(B), in addition to dUTP, DUT1 also exhibited significant activity against dITP, which represents another potentially mutagenic non-canonical nucleotide [2]. In cells, dITP can be produced by oxidative deamination of dATP or by reduction of ITP or IDP [1,2]. An HPLC analysis of the reaction products produced by DUT1 demonstrated that both dUTP and dITP were hydrolysed to the respective nucleoside monophosphate and pyrophosphate (see Supplementary Figure S2 at http://www.BiochemJ.org/bj/437/bj4370243add.htm). With both substrates, DUT1 showed sigmoidal saturation kinetics with the Hill coefficient (h) in the range 1.5–2.4, suggesting positive co-operativity between the DUT1 subunits in substrate binding (Figure 2 and Table 2). In contrast with DUT1, we found that the purified E. coli dUTPase exhibited a very low co-operativity (h 1.3±0.4) in the saturation experiments with dUTP (results not shown). Previous NMR experiments with the Drosophila dUTPase revealed a co-operative behaviour and ligand-induced co-operative conformational changes in this eukaryotic dUTPase too [41]. Our present results support the suggestion that eukaryotic dUTPases have acquired positive co-operativity during evolution [41]. In the presence of saturating Mg2+ concentrations (0.5 mM), DUT1 exhibited higher activity and affinity for dUTP resulting in 20 times higher catalytic efficiency compared with dITP (Table 2). Both the activity and affinity of DUT1 to dUTP and dITP fall within the range found for other biochemically characterized dUTPases (e.g. from E. coli, Drosophila or humans), which have been shown to have a Km for dUTP in the range 1–500 μM [36,42,43].
Figure 2. Phosphohydrolase activity of DUT1 as a function of substrate concentration.
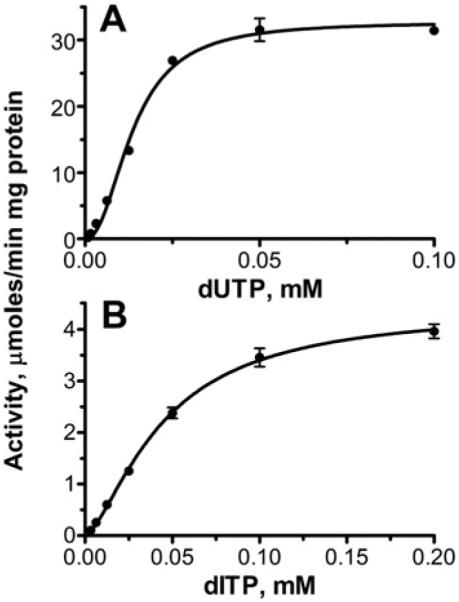
(A) dUTP; (B) dITP. Both kinetic profiles show a sigmoidal curve with positive co-operativity for substrate hydrolysis. Experimental details are described in the Experimental section. Reaction mixtures contained 0.02 μg (A) or 0.1 μg (B) of DUT1.
Table 2.
Kinetic parameters of the wild-type and mutant DUT1 proteins
| Protein | Variable substrate* | Km (μM) | h † | Vmax (units/mg)‡ | kcat (s−1) | kcat/Km (M−1 · s−1) |
|---|---|---|---|---|---|---|
| Wild-type | dITP | 44.7±3.8 | 1.5±0.1 | 4.4±0.2 | 1.3±0.06 | 2.9×104 |
| Wild-type | dUTP | 13.2±0.6 | 2.4±0.2 | 31.7±0.6 | 9.6±0.2 | 7.4×105 |
| Y88A | dUTP | 41.0±2.2 | 1.9±0.2 | 24.5±0.6 | 7.4±0.2 | 1.8×105 |
| R111A | dUTP | 31.5±6.3 | 1.3±0.2 | 10.9±0.8 | 3.3±0.2 | 1.0×105 |
| Q114A | dUTP | 14.2±2.6 | 1.1±0.2 | 4.4±0.2 | 1.3±0.06 | 0.9×105 |
| Y88A | UTP | 35.0±2.3 | 1.6±0.1 | 17.4±0.6 | 5.3±0.2 | 1.5×105 |
Units/mg, μmol/min per mg of protein.
Measured in the presence of 0.5 mM Mg2+.
Measured in the presence of 0.2 mM dUTP.
S. cerevisiae has been shown to contain the ITPase (ITP pyrophosphatase) family protein HAM1, which protects the cells from the mutagenic effect of HAP (6-N-hydroxyaminopurine) and 5-bromodeoxyuridine [44,45]. All ITPases characterized hydrolyse dITP, ITP and XTP, but show low activity against canonical NTPs [2]. The E. coli ITP pyrophosphatase RdgB has similar Km (5.6 and 11.3 μM) and kcat (18.9 and 13.2 s−1) values with both ITP and dITP as substrates [46]. Although the S. cerevisiae HAM1 protein has not yet been biochemically characterized, the partially purified protein showed the ability to hydrolyse ITP to IMP, implying that it might be able to hydrolyse dITP too [45]. This suggests that S. cerevisiae might potentially use two enzymes to detoxify dITP, DUT1 and HAM1.
Crystal structure of the ligand-free DUT1
The crystal structure of DUT1 in an apo state was solved to 2.0 Å resolution using a SAD (single-wavelength anomalous diffraction) method (PDB code 3HHQ; Table 1). The structure revealed a tightly packed homotrimeric protein (Figure 3), which is consistent with the results of our gel-filtration experiments. The DUT1 trimer has a wedge (triangular) form when viewed from the top or bottom point of the trimer (Figures 3A and 3C). The DUT1 monomer has a distorted six-stranded β-barrel fold, which is capped by a smaller five-stranded β-barrel with one α-helix (Figure 4A). The extended C-terminal tail contains one β-strand and wraps around the trimer bottom extending to the active site located between two other monomers (Figure 4B). In the DUT1 trimer, the subunits associate through interactions between the β-strands and α-helix of the cap domain at the top and the β-strands and loops of the core β-barrel domain at the bottom, creating a large cavity inside the trimer filled with solvent molecules. The overall fold of DUT1 shows a close resemblance to dUTPases from humans, E. coli and Mycobacterium tuberculosis, although these proteins share limited sequence similarity (31–55% sequence identity) [47,48]. A Dali search for DUT1 structural homologues identified several structures of dUTPases as the best matches, including the predicted dUTPase from Arabidopsis thaliana [PDB code 2P9O, Z-score 22.8, rmsd (root mean square deviation) 0.9 Å], dUTPase from the Chlorella virus IL-3A (PDB code 3C3I, Z-score 22.6, rmsd 1.2 Å), and human dUTPase (PDB code 3EHW, Z-score 22.5, rmsd 0.7 Å). In addition, structural comparisons also revealed a significant structural similarity between DUT1 and dCTP deaminases from E. coli (PDB codes 1XS4 and 2V9X, Z-score 9.8–10.6, rmsd 2.4 Å) and M. tuberculosis (PDB code 2QLP, Z-score 10.6, rmsd 2.5 Å).
Figure 3. Overall structure of DUT1.

(A–C) Three views (top, side and bottom respectively) of the DUT1 trimer related by a 90° vertical rotation. The subunits are shown as a surface presentation and in different colours. Small arrows (in A and C) indicate the position of the C-terminal domain.
Figure 4. Crystal structure of DUT1.

(A) DUT1 monomer. (B) Ribbon diagram showing the DUT1 trimer (top view) with three molecules of α,β-imido dUTP (shown as spheres) bound to the active sites located between the subunits. DUT1 subunits are shown in different colours. (C and D) Surface presentation of the DUT1 active site with the bound α,β-imido dUTP (C) or dUMP (D) (shown as sticks). The protein subunits are shown in different colours (pink, light green and light grey), and in (D) the surface has been rendered semi-transparent to show the bound nucleotide. Note that in (D) dUMP is almost completely covered by the C-terminal tail (grey), which is disordered in (C) and the active site with bound α,β-imido dUTP is open.
The location of the active sites in the DUT1 trimer is indicated by three sulfate molecules bound at the inter-subunit contacts (Figure 5A). Ammonium sulfate was used in the crystallization solution, and the sulfate molecule perhaps mimics the position of the β-phosphate of dUTP in the DUT1 active site. As shown in Figure 5(A), these sulfate molecules are co-ordinated by the side chain of the conserved Arg68 (2.7 and 3.1 Å) and the main-chain N atom of the conserved Gly70 (dUTPase motif 2). Similar to DUT1, three sulfate molecules have been found in the apo structure of the vaccinia virus dUTPase trimer (PDB code 2OKD) that occupy approximately the same position as the substrate β-phosphate.
Figure 5. DUT1 active site.

Close-up stereo view of the DUT1 active site with bound sulfate (A), α,β-imido dUTP and Mg2+ (B) or dUMP (C). The ligand molecules and amino acid residues are shown as sticks along a DUT1 ribbon coloured magenta (subunit A), cyan (subunit B) or blue (subunit C). In (B), the Mg2+ ion and catalytic water molecule are denoted by the grey and blue spheres respectively.
DUT1 sequence and site-directed mutagenesis
As expected, DUT1 shows higher sequence similarity to eukaryotic dUTPases (54–56% sequence identity) than to prokaryotic enzymes (31–34% sequence identity). Structure-based sequence alignment of DUT1 with other structurally characterized dUTPases revealed the presence of five sequence motifs containing eight absolutely conserved polar or charged residues involved in substrate binding or catalysis (Figure 6). In the dUTPase active site, four of the motifs are contributed by two adjacent monomers, whereas motif 5 comes from the third subunit and is located on the C-terminal tail (Figure 6). In most available dUTPase structures, motif 5 is disordered and is only observed in a few structures with inhibitors (PDB codes 1F7P, 2HQU and 2PY4). The residues from motifs 1, 2, 4, and 5 have been shown to contribute to the co-ordination of the triphosphate moiety of dUTP, whereas the motif-3 residues are involved in catalysis and interaction with the deoxyribose ring [5].
Figure 6. Structure-based sequence alignment of DUT1 and other dUTPases with available crystal structures.

The secondary-structure elements derived from structures of DUT1 (PDB code 3HHQ) and M. tuberculosis dUTPase (PDB code 1MQ7) are shown above and below the alignment respectively. The DUT1 residues mutated to alanine in the present study are marked with black inverted triangles above the alignment and numbered. Absolutely conserved residues are shaded, and the five conserved sequence motifs of dUTPases are boxed and numbered. The proteins compared are S. cerevisiae DUT1 (Sc_DUT1; UniProt number P33317), human DUT (Hs_DUT; UniProt number P33316), E. coli DUT (Ec_DUT; UniProt number P06968) and M. tuberculosis DUT (Mt_DUT; UniProt number P0A552).
The conserved residues from the five DUT1 motifs were mutated to alanine, and the mutated proteins were overexpressed in E. coli and purified. Enzymatic assays with dUTP revealed that only three mutant proteins (Y88A, R111A, and Q114A from motifs 3 and 4) showed significant catalytic activity (Figure 7). Q114A showed essentially the wild-type affinity for dUTP and greatly reduced activity, whereas Y88A and R111A had reduced activity and lower substrate affinity (increased Km) (Table 2). In addition, the R111A and Q114A proteins showed reduced co-operativity (lower h) suggesting that these conserved residues from DUT1 motif 4 have a role in the co-operative behaviour of this enzyme (Table 2). All other mutant DUT1 proteins (D32A, R68A, S69A, D85A, D87A, R137A and F142A) exhibited negligible or very low activity, indicating that they are involved in substrate binding and/or catalysis (Figure 7). The important role of the residues homologous with DUT1 Ser69, Asp85, Arg137 and Phe142 has been recently demonstrated in the dUTPases from E. coli, humans and Epstein–Barr virus [16,49–51].
Figure 7. Alanine replacement mutagenesis of DUT1: hydrolysis of dUTP by purified mutant proteins.
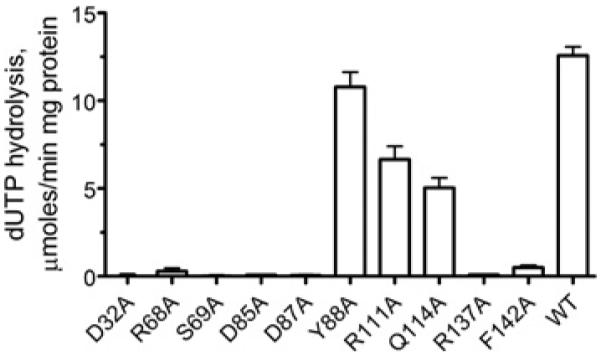
The reaction mixture contained 0.2 mM dUTP and 0.1 μg of purified protein.
Structure of DUT1 in complex with α,β-imido dUTP
DUT1 was crystallized in the presence of Mg2+ and α,β-imido dUTP, a non-hydrolysable dUTP analogue, and the structure was solved to 1.67 Å resolution using the SAD method (Table 1). The structure revealed the presence of three molecules of α,β-imido dUTP (all in the gauche conformation) per trimer bound to the DUT1 active sites located between the pairs of subunits (Figure 4B and Supplementary Figure S3A and Movie S1 at http://www.BiochemJ.org/bj/437/bj4370243add.htm). The deoxyuridine moiety is bound in the cavity formed by the α-helix of one subunit (dUTPase motif 2) and by the long loop between the β8 and β10 strands (motif 3) of another subunit (Figure 4C). In DUT1, as in human dUTPase, the uracil O2, N3, and O4 atoms make no direct contacts with the protein side chains, but they interact with the backbone N and O atoms of the conserved Gly82 and semi-conserved Lys93 (Figure 8). This is in contrast with the active site of prokaryotic dUTPases (E. coli or M. tuberculosis), where the side chain of a conserved asparagine residue (Asn77 in M. tuberculosis) makes a hydrogen bond to O4 [47]. In addition, prokaryotic dUTPases have two additional residues inserted into the uracil-binding pocket, suggesting that this would reduce its volume compared with eukaryotic dUTPases [47]. Modelling of the dITP molecule into the DUT1 active site revealed that with the deoxyribose and triphosphate moieties fixed as in dUTP the hypoxanthine base can be fitted in the active site with one steric clash (with the Lys93 carbonyl O atom) (see Supplementary Figure S4 at http://www.BiochemJ.org/bj/437/bj4370243add.htm). Since our co-crystallization experiments with DUT1 and dITP failed to produce well-diffracting crystals, it remains presently unclear how dITP can be accommodated in the DUT1 active site.
Figure 8. Co-ordination of dUTP in the active site of DUT1 and proposed catalytic mechanism of dUTP hydrolysis.

A bivalent metal cation (Me2+, usually Mg2+) co-ordinates the triphosphate moiety of the substrate, whereas the catalytic water molecule is proposed to be activated by the interaction with the side chains of the conserved Asp85 and Gln114. The hydroxy group of Ser69 promotes the hydrolysis of dUTP through the stabilization of the transition state and balancing the negative charge of the bridging α,β-O atom. The selectivity for the uridine nucleobase is provided by the hydrogen bonds with the main-chain amide and carbonyl groups of Gly82 and Lys93, whereas the side chain of the conserved Tyr88 discriminates against the 2′-hydroxyl (and UTP). The numbers represent interatomic distances in Å. MC, main chain.
The deoxyribose ring of α,β-imido dUTP is close to the side chain of the conserved Tyr88 (3.7 Å, Figure 5B), which probably prevents the binding of UTP through a steric clash with its 2′-hydroxy group. This role of Tyr88 in DUT1 activity is confirmed by the observation of high activity of the DUT1 Y88A mutant protein against UTP (17.4 μmol/min per mg of protein; ~70% of that against dUTP). Similar tyrosine-residue-based discrimination against ribonucleosides has been proposed for the human and E. coli dUTPases [48,49]. The O atom of the deoxyribose 3′-hydroxy group makes a weak hydrogen bond (3.6 Å) to the side-chain carbonyl group of the conserved Asp85, which is likely to function as a catalytic nucleophile in DUT1 (Figure 5B).
The triphosphate part of α,β-imido dUTP chelates the Mg2+ ion through a tridentate co-ordination by three O atoms (α, β, γ) and interacts mostly with the residues of the second DUT1 monomer (coloured cyan in Figure 5B). One of the α-phosphate O atoms is hydrogen bonded to the main-chain amino group of the conserved Ser69, whose side-chain hydroxy group points to the imido group of α,β-imido dUTP (2.7 Å) (Figure 5B). However, in one monomer of the DUT1–α,β-imido dUTP complex, as well as in the DUT1 apo structure, the Ser69 hydroxy group points in the opposite direction and can make a hydrogen bond with its main-chain carbonyl O atom (not shown). This serine residue is strictly conserved in dUTPases, and it has been proposed to be involved in the stabilization of the transition state and destabilization of the reactant ground state [50]. In the E. coli dUTPase, the second orientation of the homologous Ser72 side chain is stabilized by the formation of a hydrogen bond with the side chain of Asn84, which is absent from DUT1 and other eukaryotic dUTPases [50]. Two O atoms of the β-phosphate interact with the main-chain amino group of Gly70 (2.7 Å) and with the guanidinium group of conserved Arg68 (2.9 and 3.0 Å) (Figure 5B). The γ -phosphate shows no interactions with the DUT1 residues because its C-terminal tail is disordered in the DUT1–α,β-imido dUTP structure. This C-terminal loop was found to be disordered in most reported dUTPase structures (e.g. in E. coli or B. subtilis), whereas it was ordered in the dUTPases from humans and M. tuberculosis [47–49,52].
Crystal structure of DUT1 in complex with dUMP
The wild-type DUT1 was also co-crystallized with dUTP, and this structure was solved at 2.0 Å resolution (Table 1). However, the analysis of the electron density bound to the protein active site revealed the presence of dUMP, a reaction product of the dUTPase reaction (Figure 5C and Supplementary Figure S3B). This structure showed no presence of the pyrophosphate product, suggesting that it has already been released from the active site. In the structure of the DUT1–α,β-imido dUTP complex (Figure 5B), the last 14 residues of the C-terminal tail including the conserved Arg137 and semi-conserved Phe142 of the dUTPase motif-5 were disordered and not modelled. Surprisingly, in the DUT1–dUMP complex these residues were ordered, although it has been suggested that both the bivalent metal cation and the triphosphate moiety are required to order the C-terminal tail [47]. The DUT1–dUMP structure shows that the C-terminal tail completely closes the nucleoside moiety of dUMP in the active site, so only the dUMP phosphate group is exposed to the bulk solvent (Figures 4D and 5C). The side chain of Phe142 is stacked over the uracil ring of dUMP (3.3 Å) (Figure 5C). The aromatic stacking between the uracil base and dUTPase has been proposed to increase the reaction rate in dUTPases and possibly in other enzymes too [51]. This is supported by the very low activity of the F142A mutant DUT1 protein, suggesting that this residue might contribute to catalysis (Figure 7). The DUT1–dUMP structure also implies that, in the DUT1–dUTP complex, the side chain of the conserved Arg137 probably interacts with the α-phosphate oxygen of dUTP and potentially with the γ -phosphate.
In S. cerevisiae cells, the semi-conserved Thr89 of DUT1 has been shown to be phosphorylated [53]. On the basis of the DUT1 structure, this residue is located near the catalytically important Asp85 and Asp87 (8.7–10.4 Å) and the bound substrate (9.3–11.1 Å). Therefore its phosphorylation will probably affect substrate binding and hydrolysis, as well as the binding of the C-terminal strand containing motif 5, the Phe142 of which is essential for activity. It has been shown that the single G82S substitution in DUT1 produced a viable allele (dut1-1) due to the low residual dUTPase activity of the mutated protein (determined in crude extracts) [11]. This absolutely conserved glycine residue is part of motif 3 (Figure 6) and its main-chain amido group is involved in the binding of the uracil base (2.8 Å to the C4 carbonyl O atom) (Figure 8). Therefore the insertion of a small serine side chain into the base-binding pocket will probably have a negative effect on the dUTP binding and activity of DUT1.
Role of active-site residues and proposed catalytic mechanism of DUT1
The structure of DUT1 revealed that most of the catalytically important residues interact (directly or indirectly) with the triphosphate moiety of dUTP, whereas the side chains of only two residues contribute to the binding of the uracil base (Phe142) and deoxyribose ring (Tyr88) (Figure 8). Our site-directed experiments also confirmed an important role of the side chain of the conserved Asp32 (motif 1) for the activity of DUT1 (Figure 7). In the structure of the DUT1–α,β-imido dUTP complex (Figure 4B), the two carboxy group O atoms of Asp32 co-ordinate two water molecules (#248 and #249, 2.7 Å and 2.8 Å), which together with another water molecule (#250) form the co-ordination sphere of the Mg2+ ion bound to the triphosphate moiety of dUTP. Therefore, as in the E. coli dUTPase [49], the replacement of Asp32 with an alanine residue will probably disturb the binding of Mg2+ and activity of DUT1. In addition to Mg2+, the leaving pyrophosphate group might be stabilized by hydrogen bonds to the main-chain N atom of Gly70 and the side chains of Arg68 and Arg137 (Figure 8). Our mutational studies also revealed an important role of the conserved Asp87 for the activity of DUT1 (Figure 7), but this residue shows no direct interactions with the substrate in the enzyme structure (Figure 5). In the structure of the DUT1–dUMP complex (Figures 5C and 9), the Asp87 side chain interacts with the side chain of the conserved Arg137 (2.9 Å), which is close to the catalytic Asp85 (3.3 Å). In the active sites of various enzymes, the carboxy group–arginine residue interactions stabilize the charged forms of interacting residues and are often found where an unprotonated carboxy group is required for catalysis [54]. Thus the DUT1 structure suggests that the triad of the absolutely conserved interacting residues Asp87/Arg137/Asp85 (Figure 9) is important for the stabilization of the unprotonated state of Asp85 (required for the activation of the catalytic water molecule) and the charged state of Arg137 (required for the neutralization of the negative charge of the dUTP triphosphate).
Figure 9. Close-up view of the Asp87/Arg137/Asp85 triad in the active site of DUT1 with bound dUMP.
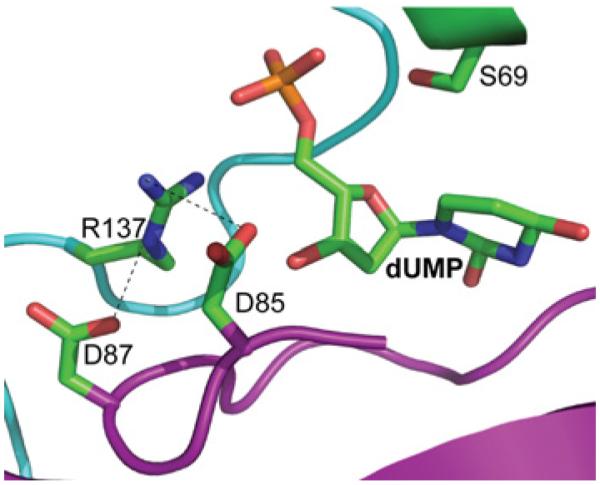
Amino acid residues and dUMP are shown as sticks, with the protein ribbon coloured magenta (subunit A) or cyan (subunit C).
Previous kinetic studies of the E. coli dUTPase suggested that the dUTPase catalytic mechanism involves Mg2+ ion binding to the α-phosphate followed by a rate-limiting hydrolysis of dUTP by a shielded and activated water molecule and a fast ordered desorption of the products [43]. The structure of DUT1 suggests that the catalytic water molecule is co-ordinated by the side chains of the conserved Asp85 (3.0 Å) and Gln114 (3.8 Å). As in the dUTPases from E. coli and M. tuberculosis [47,49], this water molecule is located 2.9–3.6 Å away from the α-phosphorus atom and is completely shielded by the substrate molecule from bulk solvent. The Asp85 carboxy group (also shielded from solvent) is proposed to deprotonate the catalytic water molecule generating a nucleophilic OH− ion (Figure 8). The important role of Asp85 in DUT1 activity is supported by the negligible activity of the D85A mutant protein, whereas the Q114A protein retained approximately 30% of the wild-type DUT1 activity (Figure 7). The non-bridging α-phosphate O atoms are co-ordinated through interactions with Mg2+, the side chain of the conserved Arg137 (2.9 Å in the DUT1–dUMP complex structure), and the main-chain N atom of Ser69, whereas the bridging αβ-O atom is likely to interact with the Ser69 side-chain hydroxy group (Figure 8). This hydroxy group is proposed to stabilize the developing negative charge at the bridging αβ-oxygen of dUTP [47,50], and the important role of Ser69 in DUT1 activity is supported by almost complete loss of activity in the S69A mutant protein (Figure 7). After dUTP hydrolysis, dUMP remains closed in the active site by the side chain of Phe142 of motif 5, whereas the pyrophosphate product perhaps leaves the active site first.
Thus the S. cerevisiae dUTPase DUT1 exhibits a remarkable ability to hydrolyse two non-canonical and potentially mutagenic nucleotides: dUTP and dITP. It will be interesting to determine whether other dUTPases also show dITPase activity, and whether this activity is physiologically significant. Since the ability to hydrolyse dCTP, dTTP and UTP has been demonstrated previously for the T. onnurineus and viral dUTPases [38,40], these studies and our present results imply that, in some organisms, the substrate specificity of dUTPase is not limited to dUTP. With both dUTP and dITP, DUT1 displayed a positive co-operativity, making it the second dUTPase (in addition to the Drosophila enzyme) showing a co-operative behaviour in substrate hydrolysis. Given that no positive co-operativity has been revealed in prokaryotic dUTPases, our results suggest that the presence of positive co-operativity might be a distinctive property of eukaryotic dUTPases. Future comparative studies of prokaryotic and eukaryotic dUTPases are needed to elucidate the functional differences of these enzymes, which can be used for the development of novel anticancer or anti-microbial therapies. In addition, the mutational studies of DUT1 revealed an important role of the Asp87/Arg137/Asp85 triad, which is absolutely conserved in dUTPases. Overall, the results of the present study provide further insights into the substrate specificity and activity of a eukaryotic dUTPase, enhancing our understanding of the dUTPase mechanism.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Aiping Dong and all members of the Ontario Centre for Structural Proteomics in Toronto (SPiT) for help with conducting experiments and discussions.
FUNDING
The project was funded by the Government of Canada (through Genome Canada and the Ontario Genomics Institute) [grant number 2009-OGI-ABC-1405] and by the Protein Structure Initiative of the National Institutes of Health (Midwest Center for Structural Genomics) [grant number GM074942]. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, and the use of Structural Biology Center beamlines was supported by the Office of Biological and Environmental Research [contract number DE-AC02-06CH11357].
Abbreviations used
- AP
apurinic/apyrimidinic
- dUTPase
dUTP pyrophosphatase
- DUT1
Saccharomyces cerevisiae dUTPase
- ITPase
ITP pyrophosphatase
- PEG
poly (ethylene glycol)
- rmsd
root mean square deviation
- SAD
single-wavelength anomalous diffraction
- SL
synthetic lethality
- TBH
tetrabutylammonium hydroxide
Footnotes
The co-ordinates for the apo-DUT1 and the DUT1 complexes with α,β-imido dUTP or dUMP have been deposited in the PDB under accession codes 3HHQ, 3P48 and 3F4F respectively.
AUTHOR CONTRIBUTION
Anatoli Tchigvintsev, Robert Flick and Greg Brown carried out the biochemical studies. Elena Evdokimova performed the crystallization experiments. Alexander Singer, Pierre Petit, and Alexei Savchenko solved and refined the crystal structures. Alexander Yakunin designed the research, analysed the data and wrote the manuscript.
REFERENCES
- 1.Kamiya H. Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: approaches using synthetic oligonucleotides and nucleotides: survey and summary. Nucleic Acids Res. 2003;31:517–531. doi: 10.1093/nar/gkg137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galperin MY, Moroz OV, Wilson KS, Murzin AG. House cleaning, a part of good housekeeping. Mol. Microbiol. 2006;59:5–19. doi: 10.1111/j.1365-2958.2005.04950.x. [DOI] [PubMed] [Google Scholar]
- 3.Kouzminova EA, Kuzminov A. Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair. Mol. Microbiol. 2004;51:1279–1295. doi: 10.1111/j.1365-2958.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- 4.Mathews CK. DNA precursor metabolism and genomic stability. FASEB J. 2006;20:1300–1314. doi: 10.1096/fj.06-5730rev. [DOI] [PubMed] [Google Scholar]
- 5.Vertessy BG, Toth J. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 2009;42:97–106. doi: 10.1021/ar800114w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Persson R, Cedergren-Zeppezauer ES, Wilson KS. Homotrimeric dUTPases; structural solutions for specific recognition and hydrolysis of dUTP. Curr. Protein Pept. Sci. 2001;2:287–300. doi: 10.2174/1389203013381035. [DOI] [PubMed] [Google Scholar]
- 7.el-Hajj HH, Zhang H, Weiss B. Lethality of a dut (deoxyuridine triphosphatase) mutation in Escherichia coli. J. Bacteriol. 1988;170:1069–1075. doi: 10.1128/jb.170.3.1069-1075.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gadsden MH, McIntosh EM, Game JC, Wilson PJ, Haynes RH. dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. EMBO J. 1993;12:4425–4431. doi: 10.1002/j.1460-2075.1993.tb06127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hidalgo-Zarco F, Gonzalez-Pazanowska D. Trypanosomal dUTPases as potential targets for drug design. Curr. Protein Pept. Sci. 2001;2:389–397. doi: 10.2174/1389203013381026. [DOI] [PubMed] [Google Scholar]
- 10.Ting H, Kouzminova EA, Kuzminov A. Synthetic lethality with the dut defect in Escherichia coli reveals layers of DNA damage of increasing complexity due to uracil incorporation. J. Bacteriol. 2008;190:5841–5854. doi: 10.1128/JB.00711-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillet M, Van Der Kemp PA, Boiteux S. dUTPase activity is critical to maintain genetic stability in Saccharomyces cerevisiae. Nucleic Acids Res. 2006;34:2056–2066. doi: 10.1093/nar/gkl139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tinkelenberg BA, Hansbury MJ, Ladner RD. dUTPase and uracil-DNA glycosylase are central modulators of antifolate toxicity in Saccharomyces cerevisiae. Cancer Res. 2002;62:4909–4915. [PubMed] [Google Scholar]
- 13.Wilson PM, Fazzone W, LaBonte MJ, Lenz HJ, Ladner RD. Regulation of human dUTPase gene expression and p53-mediated transcriptional repression in response to oxaliplatin-induced DNA damage. Nucleic Acids Res. 2009;37:78–95. doi: 10.1093/nar/gkn910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koehler SE, Ladner RD. Small interfering RNA-mediated suppression of dUTPase sensitizes cancer cell lines to thymidylate synthase inhibition. Mol. Pharmacol. 2004;66:620–626. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 15.McGeoch DJ. Protein sequence comparisons show that the ‘pseudoproteases’ encoded by poxviruses and certain retroviruses belong to the deoxyuridine triphosphatase family. Nucleic Acids Res. 1990;18:4105–4110. doi: 10.1093/nar/18.14.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman L, Buisson M, Tarbouriech N, Van der Heyden A, Labbe P, Burmeister WP. The flexible motif V of Epstein–Barr virus deoxyuridine 5’-triphosphate pyrophosphatase is essential for catalysis. J. Biol. Chem. 2009;284:25280–25289. doi: 10.1074/jbc.M109.019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harkiolaki M, Dodson EJ, Bernier-Villamor V, Turkenburg JP, Gonzalez-Pacanowska D, Wilson KS. The crystal structure of Trypanosoma cruzi dUTPase reveals a novel dUTP/dUDP binding fold. Structure. 2004;12:41–53. doi: 10.1016/j.str.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 18.Kuznetsova E, Xu L, Singer A, Brown G, Dong A, Flick R, Cui H, Cuff M, Joachimiak A, Savchenko A, Yakunin AF. Structure and activity of the metal-independent fructose-1,6-bisphosphatase YK23 from Saccharomyces cerevisiae. J. Biol. Chem. 2010;285:21049–21059. doi: 10.1074/jbc.M110.118315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stocchi V, Cucchiarini L, Canestrari F, Piacentini MP, Fornaini G. A very fast ion-pair reversed-phase HPLC method for the separation of the most significant nucleotides and their degradation products in human red blood cells. Anal. Biochem. 1987;167:181–190. doi: 10.1016/0003-2697(87)90150-3. [DOI] [PubMed] [Google Scholar]
- 20.Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 21.Hill AV. A new mathematical treatment of changes of ionic concentration in muscle and nerve under the action of electric currents, with a theory as to their mode of excitation. J. Physiol. 1910;40:190–224. doi: 10.1113/jphysiol.1910.sp001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong A, Xu X, Edwards AM, Chang C, Chruszcz M, Cuff M, Cymborowski M, Di Leo R, Egorova O, Evdokimova E, et al. In situ proteolysis for protein crystallization and structure determination. Nat. Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 25.Vaguine AA, Richelle J, Wodak SJ. SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:191–205. doi: 10.1107/S0907444998006684. [DOI] [PubMed] [Google Scholar]
- 26.Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 29.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 30.Winn MD, Murshudov GN, Papiz MZ. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 2003;374:300–321. doi: 10.1016/S0076-6879(03)74014-2. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 33.Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Crystallogr. Sect. D Biol. Crystallogr. 1993;49:129–147. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 34.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 36.Giroir LE, Deutsch WA. Drosophila deoxyuridine triphosphatase. Purification and characterization. J. Biol. Chem. 1987;262:130–134. [PubMed] [Google Scholar]
- 37.Ingraham HA, Goulian M. Deoxyuridine triphosphatase: a potential site of interaction with pyrimidine nucleotide analogues. Biochem. Biophys. Res. Commun. 1982;109:746–752. doi: 10.1016/0006-291x(82)92003-4. [DOI] [PubMed] [Google Scholar]
- 38.Bjornberg O, Nyman PO. The dUTPases from herpes simplex virus type 1 and mouse mammary tumour virus are less specific than the Escherichia coli enzyme. J. Gen. Virol. 1996;77:3107–3111. doi: 10.1099/0022-1317-77-12-3107. [DOI] [PubMed] [Google Scholar]
- 39.Hokari S, Sakagishi Y, Tsukada K. Enhanced activity of deoxyuridine 5′-triphosphatase in regenerating rat liver. Biochem. Biophys. Res. Commun. 1982;108:95–101. doi: 10.1016/0006-291x(82)91836-8. [DOI] [PubMed] [Google Scholar]
- 40.Cho Y, Lee HS, Kim YJ, Kang SG, Kim SJ, Lee JH. Characterization of a dUTPase from the hyperthermophilic archaeon Thermococcus onnurineus NA1 and its application in polymerase chain reaction amplification. Mar. Biotechnol. 2007;9:450–458. doi: 10.1007/s10126-007-9002-8. [DOI] [PubMed] [Google Scholar]
- 41.Dubrovay Z, Gaspari Z, Hunyadi-Gulyas E, Medzihradszky KF, Perczel A, Vertessy BG. Multidimensional NMR identifies the conformational shift essential for catalytic competence in the 60-kDa Drosophila melanogaster dUTPase trimer. J. Biol. Chem. 2004;279:17945–17950. doi: 10.1074/jbc.M313644200. [DOI] [PubMed] [Google Scholar]
- 42.Williams MV, Cheng Y. Human deoxyuridine triphosphate nucleotidohydrolase. Purification and characterization of the deoxyuridine triphosphate nucleotidohydrolase from acute lymphocytic leukemia. J. Biol. Chem. 1979;254:2897–2901. [PubMed] [Google Scholar]
- 43.Larsson G, Nyman PO, Kvassman JO. Kinetic characterization of dUTPase from Escherichia coli. J. Biol. Chem. 1996;271:24010–24016. doi: 10.1074/jbc.271.39.24010. [DOI] [PubMed] [Google Scholar]
- 44.Noskov VN, Staak K, Shcherbakova PV, Kozmin SG, Negishi K, Ono BC, Hayatsu H, Pavlov YI. HAM1, the gene controlling 6-N-hydroxylaminopurine sensitivity and mutagenesis in the yeast Saccharomyces cerevisiae. Yeast. 1996;12:17–29. doi: 10.1002/(SICI)1097-0061(199601)12:1%3C17::AID-YEA875%3E3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 45.Takayama S, Fujii M, Kurosawa A, Adachi N, Ayusawa D. Overexpression of HAM1 gene detoxifies 5-bromodeoxyuridine in the yeast Saccharomyces cerevisiae. Curr. Genet. 2007;52:203–211. doi: 10.1007/s00294-007-0152-z. [DOI] [PubMed] [Google Scholar]
- 46.Savchenko A, Proudfoot M, Skarina T, Singer A, Litvinova O, Sanishvili R, Brown G, Chirgadze N, Yakunin AF. Molecular basis of the antimutagenic activity of the house-cleaning inosine triphosphate pyrophosphatase RdgB from Escherichia coli. J. Mol. Biol. 2007;374:1091–1103. doi: 10.1016/j.jmb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Chan S, Segelke B, Lekin T, Krupka H, Cho US, Kim MY, So M, Kim CY, Naranjo CM, Rogers YC, et al. Crystal structure of the Mycobacterium tuberculosis dUTPase: insights into the catalytic mechanism. J. Mol. Biol. 2004;341:503–517. doi: 10.1016/j.jmb.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 48.Mol CD, Harris JM, McIntosh EM, Tainer JA. Human dUTP pyrophosphatase: uracil recognition by a β hairpin and active sites formed by three separate subunits. Structure. 1996;4:1077–1092. doi: 10.1016/s0969-2126(96)00114-1. [DOI] [PubMed] [Google Scholar]
- 49.Barabas O, Pongracz V, Kovari J, Wilmanns M, Vertessy BG. Structural insights into the catalytic mechanism of phosphate ester hydrolysis by dUTPase. J. Biol. Chem. 2004;279:42907–42915. doi: 10.1074/jbc.M406135200. [DOI] [PubMed] [Google Scholar]
- 50.Palmen LG, Becker K, Bulow L, Kvassman JO. A double role for a strictly conserved serine: further insights into the dUTPase catalytic mechanism. Biochemistry. 2008;47:7863–7874. doi: 10.1021/bi800325j. [DOI] [PubMed] [Google Scholar]
- 51.Pecsi I, Leveles I, Harmat V, Vertessy BG, Toth J. Aromatic stacking between nucleobase and enzyme promotes phosphate ester hydrolysis in dUTPase. Nucleic Acids Res. 2010;38:7179–7186. doi: 10.1093/nar/gkq584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Nafria J, Burchell L, Takezawa M, Rzechorzek NJ, Fogg MJ, Wilson KS. The structure of the genomic Bacillus subtilis dUTPase: novel features in the Phe-lid. Acta Crystallogr. Sect. D Biol. Crystallogr. 66:953–961. doi: 10.1107/S0907444910026272. [DOI] [PubMed] [Google Scholar]
- 53.Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10364–10369. doi: 10.1073/pnas.0701622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gutteridge A, Thornton JM. Understanding nature’s catalytic toolkit. Trends Biochem. Sci. 2005;30:622–629. doi: 10.1016/j.tibs.2005.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.