

Abstract
Treatment of cyclopentanone and cyclobutanone-derived oximes with lead (IV) tetraacetate gives the bright blue acyloxy nitroso compounds, which upon basic hydrolysis yields the ring expansion product cyclic hydroxamic acids in 12–81% yield. Reactions of substituted cyclopentanones provide ring expanded products where the –NOH group regioselectively inserts to the more substituted position and gives a better yield compared to the treatment of the same ketone with a basic solution of Piloty’s acid. Reaction of phosphines with acyloxy nitroso compounds generally generates a ring-expanded Beckmann rearrangement product that can be hydrolyzed to the corresponding lactam. Acyloxy nitroso compounds that undergo rapid hydrolysis to HNO do not show this ring expansion reactivity. These results further demonstrate the versatility of acyloxy nitroso compound to yield structurally complex materials.
Keywords: Acyloxy nitroso compounds, Cyclic hydroxamic acids, Ring expansion, Organic phosphines, HNO donors
Graphical Abstract
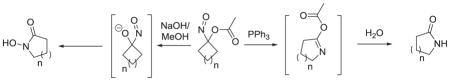
Introduction
The chemical properties of nitroxyl (HNO) biologically distinguish it from other nitrogen oxides including nitric oxide (NO).1–4 Nitroxyl readily dimerizes to ultimately form nitrous oxide (N2O) and condenses with protein thiols to generate disulfides and sulfinamides that mediate much of HNO’s distinct biology.3–5 This electrophilic reactivity demands the use of HNO donors in fundamental studies and such donors form potential therapeutics for congestive heart failure, cancer, alcoholism and hemolytic disorders.6–13 Angeli’s salt (Na2N2O3) and Piloty’s acid (PhSO2NHOH, N-hydroxy benzenesulfonamide) represent the most widely used HNO donors but numerous structurally and mechanistically diverse HNO donors have appeared, including acyloxy nitroso compounds.14–19 While stable acyloxy nitroso compounds react as N-O heterodienophiles,20 relatively little work describing their synthetic potential has appeared and we report two distinct ring expansions of acyloxy nitroso compounds.
Expansion to cyclic hydroxamic acids
Earlier work shows that 1-nitrosocyclohexyl acetate (1) hydrolyzes to an unstable α-hydroxy C-nitroso species that decomposes to cyclohexanone and HNO (Scheme 1).21 The reaction of Piloty’s acid under basic conditions with four and five-membered ring ketones forms cyclic hydroxamic acids through the intermediacy of the same α-hydroxy C-nitroso species.21 However, the basic hydrolysis of 1-nitroso cyclopentyl acetate (2a) yields a 6-membered ring cyclic hydroxamic acid (3a, 25 % yield) with little N2O (evidence of HNO) formation suggesting a strain-based ring expansion.21 Given this common α-hydroxy C-nitroso intermediate, we pursued the idea that other small (4–5) membered ring acyloxy nitroso compounds would rearrange to cyclic hydroxamic acids under basic conditions.
Scheme 1.

Pathways of acyloxy nitroso compound hydrolysis.
Condensation of a series of four and five-membered ring ketones (4a–g) with hydroxylamine gives the corresponding oximes (5a–g) in good yield (Scheme 2).
Scheme 2.

Conversion of ketones to cyclic hydroxamic acids via acyloxy nitroso compounds.
Treatment of these oximes with lead (IV) tetraacetate gives the bright blue acyloxy nitroso compounds (2a–g), which were quickly purified by passage through a short silica column and used without further purification. Slow addition of a methanol solution of 2a–g to a 2 M NaOH solution a 0 °C followed by work up generally produces the corresponding cyclic hydroxamic acid (3a–g) in good yield (12–81%, Scheme 2, Table 1).
Table 1.
The ring expansion products (3a–g) from the corresponding ketones (4a–g).
| Entry | Starting Ketone (4) | Acyloxy nitroso Compound (2) | Product (3) | % Yield |
|---|---|---|---|---|
| a |

|
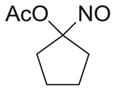
|
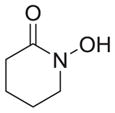
|
25 |
| b |
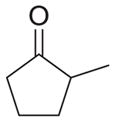
|
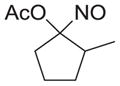
|
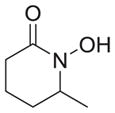
|
77 |
| c |
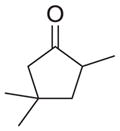
|
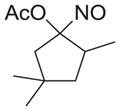
|
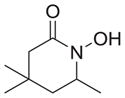
|
71 |
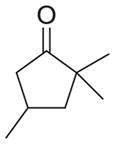
| d |
|
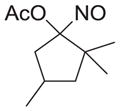
|
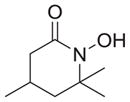
|
12 |
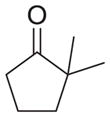
| e |
|
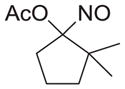
|
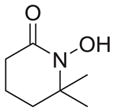
|
63 |
| f |
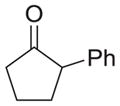
|
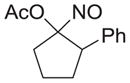
|
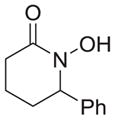
|
67 |
| g |
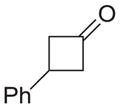
|
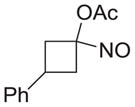
|
|
81 |
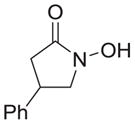
As shown in Table 1, basic hydrolysis of 2a yields 3a in 25% yield.20 Under these conditions, acyloxy nitroso compound (2a) disappears and both ketone (4a) and oxime (5a) form as byproducts. Similar treatment of the acyloxy nitroso compound derived from 2-methyl cyclopentanone (2b) gives the ring expanded cyclic hydroxamic acid (3b) in 77% yield. This result demonstrates two important features of this process: 1) the N-insertion occurs to the more hindered side of the ketone as shown by NMR chemical shift/splitting and integration analysis and 2) the yield vastly improves from the direct treatment of the ketone with basic solutions of Piloty’s acid (77% vs 5%).21 Examination of the crude reaction mixture by NMR spectroscopy shows the formation of only a single regioisomer. The yield improvement likely results from the lack of a requirement to generate the N-anion of Piloty’s acid under these reaction conditions in the presence of acidic ketone α-hydrogens. Other substituted acyloxy nitroso compounds (2c–f) smoothly rearrange to form the more hindered cyclic hydroxamic acids (3c–f). Single crystal X-ray crystallographic analysis clearly defines the structure of 3c and 3e and confirms rearrangement occurs to the most hindered side (Figure 1). Exposure of the cyclobutanone-derived acyloxy nitroso compound (2g) to basic conditions yields the five- membered ring hydroxamic acid (3g) in 81% yield (Scheme 2, Table 1).
Figure 1.

X-Ray Crystallography data for hydroxamic acids 3c and 3e.
Scheme 1 depicts a likely mechanism for these transformations. Basic hydrolysis of the acetate group would give the unstable α-hydroxy C-nitroso species that undergoes ring expansion to the cyclic hydroxamic acid as previously demonstrated suggesting that the electrophilic nitroso group coupled with the strain within the smaller rings induces rearrangement to the cyclic hydroxamic acid.21 These results show this pathway operates in four and five membered ring acyloxy nitroso compounds but not in the larger non-strained six membered ring compound. The reaction of substituted acyloxy nitroso compounds-derived from substituted cyclopentanones affords ring expanded products where the –NOH group regioselectively inserts to the more substituted position similar to other well-known ring expansion rearrangements (Baeyer-Villiger).21 This methodology provides a direct and rapid method to generate structurally diverse cyclic hydroxamic acids from four and five membered ring ketones that are not easily accessible other methods (including the reaction of N-hydroxybenzenesulfonamide with the ketone under basic conditions).22
Phopshine-Mediated Beckmann Rearrangement
During the investigation of acyloxy nitroso compounds as potential HNO donors, our group also examined the reactions of phosphines as new HNO traps through their formation of unique aza-ylide products.23,24 Phosphines react as nucleophiles with a variety of electrophilic nitroso compounds (C-nitroso, S-nitroso, and H-NO) to yield numerous products depending on the structure of the phosphine and the nitroso substrate.25,26 Given the known reactivity and the potential of acyloxy nitroso compounds to release HNO,14,16,27 the reaction of acyloxy nitroso compounds and triaryl phosphines was explored.
Treatment of 1-nitrosocyclohexyl acetate (1) with triphenylphosphine (TPP) in benzene or toluene at room temperature results in an exothermic reaction with the disappearance of the deep blue color. After 60 min, analysis of the reaction mixture by 31P NMR spectroscopy and mass spectrometry shows the generation of triphenyl phosphine oxide (δ = 26.09 ppm, m/z = 279.1, 95 % yield, Scheme 3), which was identical to a standard. In addition to phosphine oxide, a seven membered ring Beckmann rearrangement product (6, Scheme 3) forms in 55 % yield that was identified by GC-MS (m/z = 156) and both 1D and 2D NMR spectroscopy (Supporting Information). Acid-catalyzed hydrolysis of 6 (1M HCl) yields caprolactam (7, Scheme 3), which was also identical to a known standard by both MS and NMR analysis.
Scheme 3.

Reaction of 1-nitrosocyclohexyl acetate (1) with TPP.
The reaction of 1 with TPP was monitored using UV-Vis spectrometry following the disappearance of the absorbance at 667 nm corresponding to the nitroso group (Table 2). Incubation of 1 with TPP in benzene or toluene at room temperature shows an exponential decrease of 1 over time and the rate shows a linear dependence on the concentration of phosphine (Table 2) suggesting a bimolecular reaction. Under pseudo-first order conditions (10 equivalents of TPP) an observed rate constant of k = 3.5 min−1 with a half-life of t½ = 0.2 min is observed (Table 2). As expected, these results reveal that phosphines react faster with most acyloxy nitroso compounds than these compounds hydrolyze to HNO (for 1, t ½ = 800 min, pH 7.6 1:1 Tris buffer:MeOH).27
Table 2.
Kinetics of the reaction of compound 1 with TPP in benzene.
| Entry | Compound | TPP (equiv.) | Observed rate constant (kobs, min−1) | t1/2 (min) |
|---|---|---|---|---|
| 1 | 1 | 1 | 0.09 | 8 |
| 2 | 1 | 2 | 0.18 | 3.7 |
| 3 | 1 | 5 | 0.5 | 1.4 |
| 4 | 1 | 10 | 3.5 | 0.2 |
Scheme 4 shows the proposed mechanism for the conversion of 1 to caprolactam upon reaction with triphenyl phosphine.
Scheme 4.

Proposed mechanism for the reaction of 1-nitrosocyclohexyl acetate (1) with TPP.
Addition of the phosphine to the nitroso oxygen atom with simultaneous loss of the acetate group would yield the electrophilic phosphonium ion adduct (8, Scheme 4). Beckman rearrangement of this phosphonium ion results in ring expansion to 9 with the formation of triphenyl phosphine oxide (Scheme 4). Addition of acetate to 9 gives 6 and hydrolysis produces caprolactam (7, Scheme 4). Such a mechanism finds direct precedence in a similar ring expansion during the reaction of 1-chloro-1-nitrosocyclohexane during its reaction with triphenyl phosphine.28
Treatment of 1-nitrosocyclohexyl pivalate (10) with TPP in benzene also gives triphenyl phosphine oxide and the corresponding pivalate ester (11, Scheme 5). 1H and 13C NMR spectroscopy both confirm the structure of 11. Further support of Scheme 4 as a plausible rearrangement mechanism comes from the treatment of 1 with triphenyl phosphine in the presence of excess pivalic acid, which also yields 11 suggesting the trapping of 9 by the free carboxylic acid.
Scheme 5.

Reaction of NCA and NCP with TPP.
Addition of 1-nitroscyclohexyl trifluoroacetate (12, Scheme 6), an acyloxy nitroso compound that rapidly hydrolyzes to HNO (t ½ = 121 ms),5 to a solution of TXPTS (tris(2,4-dimethyl-5-sulfophenyl)phosphine trisodium salt) in 1:1 acetonitrile:Tris buffer (0.1 M, pH 7.6) gives the corresponding phosphine oxide and aza-ylide. GC-MS analysis also reveals the formation of cyclohexanone from the reaction that further support hydrolysis of 12 to HNO. These results suggest that 12 rapidly hydrolyzes to HNO, which is trapped as the aza-ylide by TXPTS as previously described.26 Overall, these results show that acyloxy nitroso compounds directly react with phosphines to yield ring expanded products unless they first hydrolyze to HNO, which then reacts with the phosphine to give the expected products.
Scheme 6.

Reaction of 12 with TXPTS in 1:1 acetonitrile: Tris buffer (0.1 M, pH 7.6).
Conclusion
In summary, this work shows that acyloxy nitroso compounds undergo two separate ring expansion reactions. First, acyloxy nitroso compounds-derived from four and five membered ring ketones hydrolyze under basic conditions to give the cyclic hydroxamic acid in 12–81% yield. Reactions of acyloxy nitroso compounds derived from substituted cyclopentanones provide ring expanded products where the -NOH group regioselectively inserts to the more substituted position. These reactions show better yields than an earlier report of these products obtained by basic treatment of Piloty’s acid.21 Secondly, we also provide the first report of the reaction of acyloxy nitroso compounds with phosphines to yield Beckmann-type rearrangement products. Acyloxy nitroso compounds that rapidly hydrolyze to HNO do not directly react with the phosphine but instead the phosphine traps the nascent HNO to give a unique aza-ylide product. This work shows further versatility of acyloxy nitroso compounds, which are easily accessible from the corresponding oxime and thus ketone, to provide relatively complex products through simple transformations.
Supplementary Material
Acknowledgments
This project was supported by the National Institutes of Health (HL62198, SBK). We acknowledge the financial support of an Egyptian Government studentship to Mai E. Shoman through the Channel system between Minia University, Egypt and Wake Forest University. Dr. Cynthia Day (Wake Forest University) performed the X-ray crystallographic studies.
Footnotes
Includes experimental details of the synthetic procedures and characterization data for all compounds associated with this article.
Author Contributions
The manuscript was written through contributions of all authors. MBH, RM, RB and MES performed all of the experiments. SBK and OMA wrote and edited the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Samuni U, Samuni Y, Goldstein S. J Am Chem Soc. 2010;132:8428–8432. doi: 10.1021/ja101945j. [DOI] [PubMed] [Google Scholar]
- 2.Irvine JC, Ritchie RH, Favaloro JL, Andrews KL, Widdop RE, Kemp-Harper BK. Trends Pharmacol Sci. 2008;29:601–608. doi: 10.1016/j.tips.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Fukuto JM, Bartberger MD, Dutton AS, Paolocci N, Wink DA, Houk KN. Chem Res Toxicol. 2005;18:790–801. doi: 10.1021/tx0496800. [DOI] [PubMed] [Google Scholar]
- 4.Miranda KM. Coord Chem Rev. 2005;249:433–455. [Google Scholar]
- 5.Mitroka S, Shoman ME, DuMond JF, Bellavia L, Aly OM, Abdel-Aziz M, Kim-Shapiro DB, King SB. J Med Chem. 2013;56:6583–6592. doi: 10.1021/jm400057r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irvine JC, Favaloro JL, Widdop RE, Kemp-Harper BK. Hypertension. 2007;49:885–892. doi: 10.1161/01.HYP.0000259328.04159.90. [DOI] [PubMed] [Google Scholar]
- 7.Fukuto JM, Bianco CL, Chavez TA. Free Rad Biol Med. 2009;47:1318–1324. doi: 10.1016/j.freeradbiomed.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Paolocci N, Katori T, Champion HC, St John ME, Miranda KM, Fukuto JM, Wink DA, Kass DA. Proc Natl Acad Sci USA. 2003;100:5537–5542. doi: 10.1073/pnas.0937302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Favaloro JL, Kemp-Harper BK. Cardiovasc Res. 2007;73:587–596. doi: 10.1016/j.cardiores.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 10.Johnson GM, Chozinski TJ, Gallagher ES, Aspinwall CA, Miranda KM. Free Radical Biol Med. 2014;76:299–307. doi: 10.1016/j.freeradbiomed.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flores-Santana W, Salmon DJ, Donzelli SS, CH, Basudhar D, Ridnour L, Cheng R, Glynn SA, Paolocci N, Fukuto JM, et al. Antioxid Redox Signal. 2011;14:1659–1674. doi: 10.1089/ars.2010.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson MI, Han TH, Serbulea L, Dutton A, Ford E, Miranda KM, Houk KN, Wink DA, Fukuto JM. Free Rad Biol Med. 2009;47:1130–1139. doi: 10.1016/j.freeradbiomed.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miranda KM, Paolocci N, Katori T, Thomas DD, Ford E, Bartberger MD, Espey MG, Kass DA, Feelisch M, Fukuto JM, et al. Proc Natl Acad Sci USA. 2003;100:9196–9201. doi: 10.1073/pnas.1430507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DuMond JF, Wright MW, King SB. J Inorg Biochem. 2013;118:140–147. doi: 10.1016/j.jinorgbio.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sha X, Isbell TS, Patel RP, Day CS, King SB. J Am Chem Soc. 2006;128:9687–9692. doi: 10.1021/ja062365a. [DOI] [PubMed] [Google Scholar]
- 16.DuMond JF, King SB. Antioxid Redox Sign. 2011;14:1637–1648. doi: 10.1089/ars.2010.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aizawa K, Nakagawa H, Matsuo K, Kawai K, Ieda N, Suzuki T, Miyata N. Bioorg Med Chem Lett. 2013;23:2340–2343. doi: 10.1016/j.bmcl.2013.02.062. [DOI] [PubMed] [Google Scholar]
- 18.Guthrie DA, Ho A, Takahashi CG, Collins A, Morris M, Toscano JP. J Org Chem. 2015;80:1338–1348. doi: 10.1021/jo502330w. [DOI] [PubMed] [Google Scholar]
- 19.Guthrie DA, Nourian S, Takahashi CG, Toscano JP. J Org Chem. 2015;80:1349–1356. doi: 10.1021/jo5023316. [DOI] [PubMed] [Google Scholar]
- 20.Calvet G, Dussaussois M, Blanchard N, Kouklovsky C. Org Lett. 2004;6:2449–2451. doi: 10.1021/ol0491336. [DOI] [PubMed] [Google Scholar]
- 21.Renz M, Meunier B. Eur J Org Chem. 1999;73:7–750. [Google Scholar]
- 22.Banerjee R, King SB. Org Lett. 2009;11:4580–4583. doi: 10.1021/ol9018198. [DOI] [PubMed] [Google Scholar]
- 23.Bechtold E, Reisz JA, Klomsiri C, Tsang AW, Wright MW, Poole LB, Furdui CM, King SB. ACS Chem Biol. 2010;5:405–414. doi: 10.1021/cb900302u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reisz JA, Zink CN, King SB. J Am Chem Soc. 2011;133:11675–11685. doi: 10.1021/ja203652z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reisz JA, Klorig EB, Wright MW, King SB. Org Lett. 2009;11:2719–2721. doi: 10.1021/ol900914s. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Wang H, Xian M. Org Lett. 2009;11:477–480. doi: 10.1021/ol802663q. [DOI] [PubMed] [Google Scholar]
- 27.Shoman ME, DuMond JF, Isbell TS, Crawford JH, Brandon A, Honovar J, Vitturi DA, White CR, Patel RP, King SB. J Med Chem. 2011;54:1059–1070. doi: 10.1021/jm101432z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakai I, Kawabe N, Ohno M. Bull Chem Soc Jpn. 1979;52:3381–3383. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.