

Abstract
Francisella tularensis is a facultative intracellular pathogen and potential biothreat agent. Evasion of the immune response contributes to the extraordinary virulence of this organism although the mechanism is unclear. Whereas wild-type strains induced low levels of cytokines, an F. tularensis ripA deletion mutant (LVSΔripA) provoked significant release of IL-1β, IL-18, and TNF-α by resting macrophages. IL-1β and IL-18 secretion was dependent on inflammasome components pyrin-caspase recruitment domain/apoptotic speck-containing protein with a caspase recruitment domain and caspase-1, and the TLR/IL-1R signaling molecule MyD88 was required for inflammatory cytokine synthesis. Complementation of LVSΔripA with a plasmid encoding ripA restored immune evasion. Similar findings were observed in a human monocytic line. The presence of ripA nearly eliminated activation of MAPKs including ERK1/2, JNK, and p38, and pharmacologic inhibitors of these three MAPKs reduced cytokine induction by LVSΔripA. Animals infected with LVSΔripA mounted a stronger IL-1β and TNF-α response than that of mice infected with wild-type live vaccine strain. This analysis revealed novel immune evasive mechanisms of F. tularensis.
Francisella tularensis is a Gram-negative, facultative intracellular pathogen that is the causative agent of the zoonotic disease tularemia. The organism can be transmitted to a host through insect bites, handling of infected carcasses, and inhalation of aerosolized bacteria (1–3). F. tularensis is a potential agent of biological warfare classified as a Select Agent by the Centers for Disease Control and Prevention (CDC) due to its highly infectious nature; ∼50% mortality is caused by an infectious dose of as few as 10 bacteria via inhalation (4). During the Cold War, both the former Soviet Union and the United States weaponized and stockpiled F. tularensis in their biological weapons programs (5). The CDC also estimated that an F. tularensis attack would cost $5.4 billion to the society for every 100,000 infected individuals (6). Four subspecies of F. tularensis exist, including tularensis, holarctica, mediasiatica, and novicida (7). The most virulent subspecies for humans is the subspecies tularensis, which is found primarily in North America. Subspecies holarctica is less virulent and was used to generate a live vaccine strain (LVS). LVS is attenuated in humans but remains highly virulent for mice making LVS a useful model to study F. tularensis pathogenesis.
Because exposure to the respiratory tract results in the most aggressive form of tularemia, several laboratories have developed mouse models of pulmonary tularemia to study this mode of infection (8–10). Using an intranasal delivery model, the LD50 of LVS is 103 CFU (9). By comparison, the LD100 for the highly virulent F. tularensis subsp. tularensis strain SchuS4 is less than 20 CFU (11). Upon inhalation, the bacteria are restricted to the lung for ∼36 to 48 h where the organism persists and multiplies (12). During this time, little host immune response occurs as evidenced by the lack of proinflammatory cytokines IL-1β, TNF-α, IL-6, and IFN-γ (13). Two to four days postinfection, inflammatory cells infiltrate the lung, and proinflammatory cytokines can be detected in vivo. By this time, bacteria have overwhelmed the lung and disseminated to other organs such as the spleen and liver. Mice eventually succumb to infection 5–7 d postinoculation. How F. tularensis circumvents the host immune response is not well understood.
F. tularensis is found in vivo within alveolar macrophages, dendritic cells, and lung epithelial cells (9). After entry into host cells, F. tularensis escapes the phagosome and enters the cytosol where it replicates. This process is apparently unhindered by innate immune defenses that typically detect and facilitate a response to eliminate foreign microbes. IFN-γ, TNF-α, and IL-1β are critical mediators of an effective defense against Francisella infection (14, 15). SchuS4 suppresses proinflammatory cytokine induction in mouse lung resident dendritic cells in early stages of the disease (13). Similarly, LVS dampens intracellular signaling and TNF-α in human monocytes and mouse macrophages (16). Administration of anti–IFN-γ and anti–TNF-α Abs greatly reduces the LD50 of LVS in an intradermal mouse model of mouse tularemia (17). IL-1β also is critical in innate defense against F. tularensis subsp. novicida (14). Disrupting the signaling pathway that leads to IL-1β processing greatly enhances host susceptibility to subspecies novicida infection.
IL-1β, one of the more potent proinflammatory cytokines, is tightly regulated by a two-step signaling process. The first step requires the transcriptional activation of pro–IL-1β followed by the translation of the proprotein. This is achieved by engagement of the TLRs and subsequent activation of MyD88-dependent signaling pathways, leading to NF-κB and MAPK induction. This leads to translocation of the NF-κB subunit p65 from the cytosol to the nucleus and activation of the MAPK phosphorylation cascade to cause the transcription of pro–IL-1β message. A second and separate signal is required after pro–IL-1β protein is produced, and this occurs in the cytosol, leading to the assembly of an inflammasome complex. There are several types of inflammasomes, including those that contain a nucleotide binding domain-leucine rich repeats containing (NLR) component, and those that contain a non-NLR component, such as absent in melanoma 2 or retinoic acid-inducible gene 2 (18–22). Procaspase-1 is a requisite member of the complex, and the adaptor pyrin-caspase recruitment domain (PYCARD)/apoptotic speck-containing protein with a CARD/target of methylation-induced silencing is found in most inflammasome complexes (23, 24). Upon inflammasome formation, procaspase-1 undergoes autocatalytic cleavage into an active, mature form that subsequently cleaves pro–IL-1β and pro–IL-18 into their active forms.
NLR family members share a conserved structure, with most of the members bearing an N-terminal pyrin or CARD domain followed by an nucleotide binding domain and a C-terminal leucine rich repeat domain that is homologous to those found in TLRs (25). A current working hypothesis is that upon induction, the NLR recruits PYCARD via a pyrin–pyrin homotypic interaction. PYCARD possesses its own CARD domain, which recruits caspase-1 into the inflammasome complex for IL-1β processing. Several NLRs that contribute to pathogen-induced IL-1β release in macrophages have been identified (26). The absence of NLRs or their adaptors renders hosts more susceptible to a variety of pathogens. The NLRP3 inflammasome senses a wide variety of intracellular pathogens including Gram-positive and Gram-negative bacteria, RNA viruses, DNA viruses, yeast, microbial toxins, and a host of damage-associated molecular patterns such as silica, asbestos, and alum (27–29). The NLRC4/Ipaf inflammasome responds to bacterial virulence factors from the type III and IV secretion systems from bacteria such as Salmonella typhi, Burkholderia pseudomallei, Escherichia coli (30–32), and Pseudomonas aeruginosa (33).
Unlike most Gram-negative bacterial pathogens, F. tularensis subsp. tularensis and F. tularensis subsp. holarctica do not provoke a substantial initial inflammatory response in vitro or in vivo (13, 33, 34). F. tularensis LPS does not stimulate significant signaling through TLR4 (35), but this property alone does not account for the muted host response to infection. The goal of this study was to identify mechanisms by which F. tularensis actively suppresses the host innate immune response. To achieve this goal, we used an attenuated F. tularensis mutant that elicited a robust inflammatory response to reveal signaling pathways that are normally suppressed by wild-type organisms and that dampen IL-1β, IL-18, and TNF-α responses to infection.
Materials and Methods
Cell lines and reagents
Bone marrow-derived macrophages (BMDMs) were harvested from 6- to 8-wk-old mice and cultured for 7 d in 30% M-CSF conditioned medium. THP-1 cells (American Type Culture Collection, Manassas, VA) were cultured as described (36). Anti–IL-1β Ab was obtained from R&D Systems (Minneapolis, MN); anti-IκBα, phospho-p65, phospho-ERK1/2, phospho-JNK, and phospho-p38 from Cell Signaling (Danvers, MA); and anti-GAPDH from Santa Cruz Biotechnology (Santa Cruz, CA). Detailed methods for preparation of retroviral shuttle vectors, transduction, and sorting to generate THP-1 cell lines stably expressing short hairpin RNA have been described (36).
Western blotting and ELISA
Western blots were performed as described (37, 38). Quantification of Western blots by densitometry was performed using Adobe Photoshop (Adobe, San Jose, CA). For ELISAs, mouse and human cell supernatants were collected 24 h postinfection and assayed with BD OptEIA Mouse IL-1β, IL-18, and TNF-α ELISA Sets (BD Biosciences, San Diego, CA).
Cytotoxicity assays
Cytotoxicity assays were performed using the ToxiLight BioAssay kit (Lonza, Basel, Switzerland) following the manufacturer's protocol for detection from supernatants. The luminescence was read using a TECAN Infinite M200 and analyzed using Magellan v6 software (TECAN, Mannedorf, Switzerland).
Experimental animals
All studies were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee guidelines of the University of North Carolina, Chapel Hill. The generation of mice lacking functional Nlrp3, Nlrc4, Pycard, Caspase-1, and MyD88 has been previously described (38). C57BL/6 mice were purchased from The Jackson Laboratories (Bar Harbor, ME).
Bacteria preparation
F. tularensis LVS was obtained from the CDC (Atlanta, GA). F. tularensis subsp. novicida U112 was obtained from American Type Culture Collection. The LVSΔripA strain has been described (12). All Francisella strains were maintained on chocolate agar supplemented with 1% IsoVitaleX (BD Biosciences) and grown in Chamberlain's defined medium (39). Other bacteria were grown in lysogeny broth medium or brain–heart infusion medium. LVS and LVSΔripA were killed by incubation for 5 min at room temperature in 1 ml 2% paraformaldehyde in PBS, washed three times with PBS, and suspended in DMEM, 10% PBS. All cultures were grown at 37°C.
Francisella infection of mouse primary macrophages and human monocytic cell lines
LVS and LVSΔripA were grown overnight in Chamberlain's defined medium prior to infection, and U112 was grown overnight on chocolate agar prior to infection. Concentrations of bacteria were determined by Klett reading, and cells were exposed to the designated Francisella strain at the indicated multiplicity of infection (MOI). Infected cells were incubated at 37°C, and supernatants were harvested at select time points for cytokine analysis. For pharmacological assessments, cells were treated with Y-VAD-fmk (10 μM), U0126, SP-600125, and SB-202190 (0.5–25 μM) as described (36).
In vivo F. tularensis infection
F. tularensis LVS from an overnight culture in Chamberlain's medium was centrifuged, and the pellet was suspended in PBS. These suspensions were enumerated by Klett reading and diluted in Dulbecco's PBS for the inoculums. Wild-type 8- to 10-wk-old female C57BL/6 mice were anesthetized with Avertin(2,2,2-Tribromoethanaol, Sigma-Aldrich, St. Louis, MO), as determined by insensitivity to a toe pinch, and were then infected intranasally with bacterial inoculums in a 50-μl volume in Dulbecco's PBS. Mice were monitored for recovery from anesthesia.
Statistical analysis
Statistical significance in Figs. 1A–H, 2A–C, 3A–D, 5B–D was determined by two-way ANOVA followed by a Tukey post hoc test using GraphPad software (GraphPad, La Jolla, CA). Statistical significance in Fig. 6 was determined by two-tailed Mann–Whitney U tests using GraphPad software. The p values <0.05 were considered statistically significant. Unless otherwise specified, data are presented as the mean ± SD.
Figure 1.
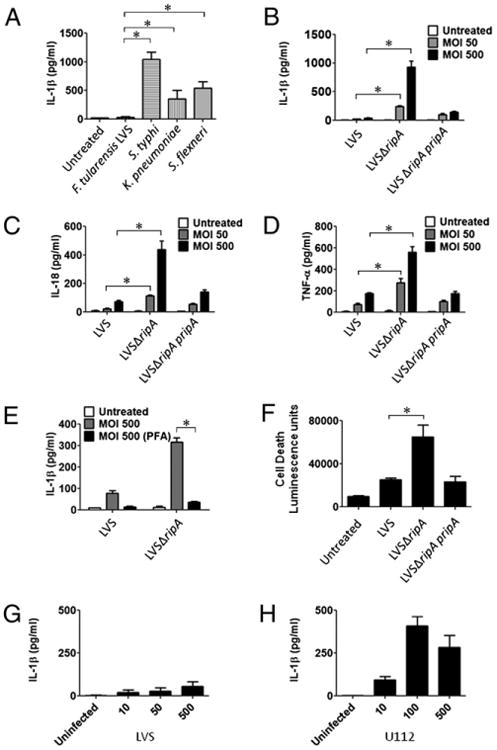
F. tularensis LVSΔripA fails to suppress proinflammatory cytokine release by primary mouse macrophages. A, IL-1β secretion levels in mouse macrophages after exposure to F. tularensis LVS, Salmonella typhi, K. pneumoniae, or Shigella flexneri at MOI 50 for 24 h as determined by ELISA. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. B, IL-1β secretion levels in mouse macrophages exposed to LVS, LVSΔripA, or LVSΔripA complementation strain at MOI 50 and 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. C, IL-18 secretion levels in mouse macrophages exposed to LVS, LVSΔripA, or LV S ΔripA complementation strain at MOI 50 and 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. D, TNF-α secretion levels in mouse macrophages exposed to LVS, LVSΔripA, or LVSΔripA complementation strain at MOI 50 and 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. E, IL-1β secretion levels in mouse macrophages exposed to live and PFA-treated LVS and LVSΔripA at MOI 50 and 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. F, Cytotoxicity of mouse macrophages exposed to LVS, LVSΔripA, or LVSΔripA complementation strain at MOI 500 for 24 h as measured by luminescence. Data represent mean ± SD for at least three independent experiments performed in duplicate or triplicate. A representative experiment is shown. G, IL-1β secretion levels in mouse macrophages exposed to LVS at MOI 10, 50, and 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. H, IL-1β secretion levels in mouse macrophages exposed to U112 at MOI 10, 100, and 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. *p < 0.05.
Figure 2.
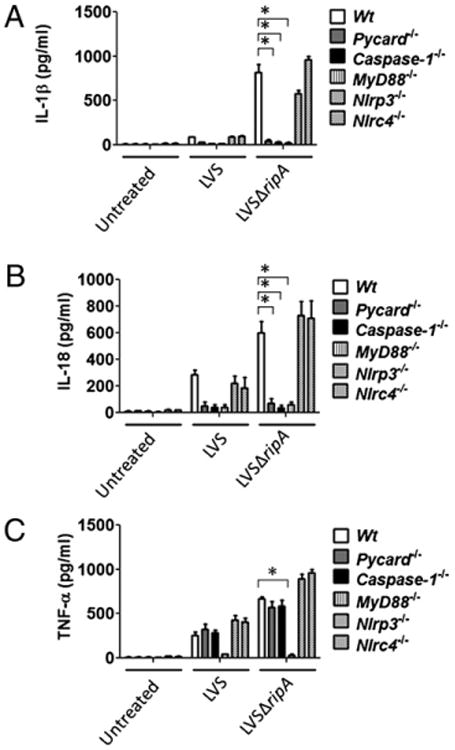
LVSΔripA induced proinflammatory cytokines are regulated by inflammasome components and MyD88. A, IL-1β secretion levels in mouse macrophages derived from Wt, Pycard−/−, Caspase-1−/−, MyD88−/−, Nlrp3−/−, and Nlrc4−/− mice exposed to LVS and LVSΔripA at MOI 500 for 24 h as assessed by ELISA of cell supernatants. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. B, IL-18 secretion levels in macrophages derived from Wt, Pycard−/−, Caspase-1−/−, MyD88−/−, Nlrp3−/−, and Nlrc4−/− mice exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. C, TNF-α secretion levels in mouse macrophages derived from Wt, Pycard−/−, Caspase-1−/−, MyD88−/−, Nlrp3−/−, and Nlrc4−/− exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. *p < 0.05.
Figure 3.
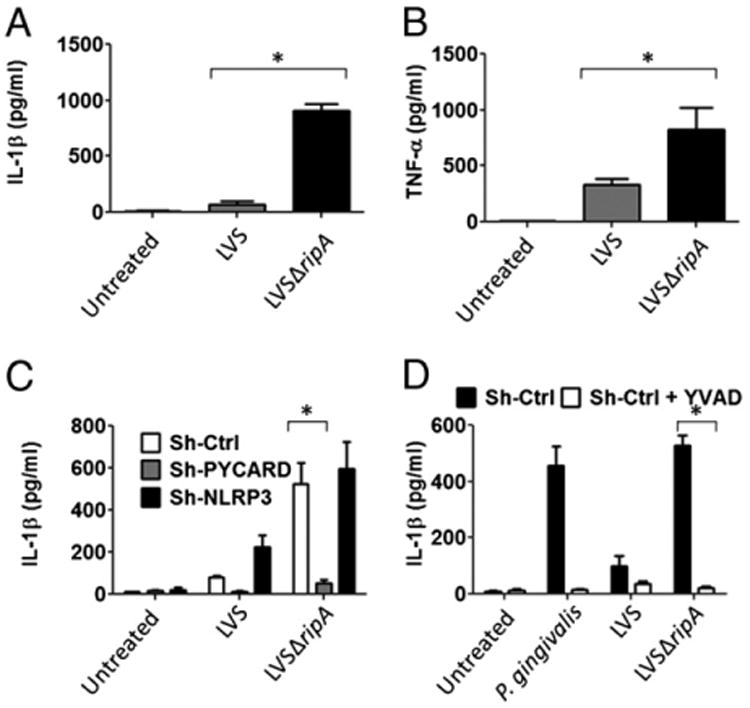
LVS ripA is necessary for suppression of proinflammatory cytokine release by human THP-1 cells. A, IL-1β secretion levels in THP-1 cells exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. B, TNF-α secretion levels in THP-1 cells exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. C, IL-1β secretion levels in THP-1 cells expressing stable Sh-Ctrl, Sh-PYCARD, and Sh-NLRP3 exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. D, IL-1β secretion levels in mock pretreated THP-1 cells and cells pretreated with Y-VAD and exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. *p < 0.05.
Figure 5.
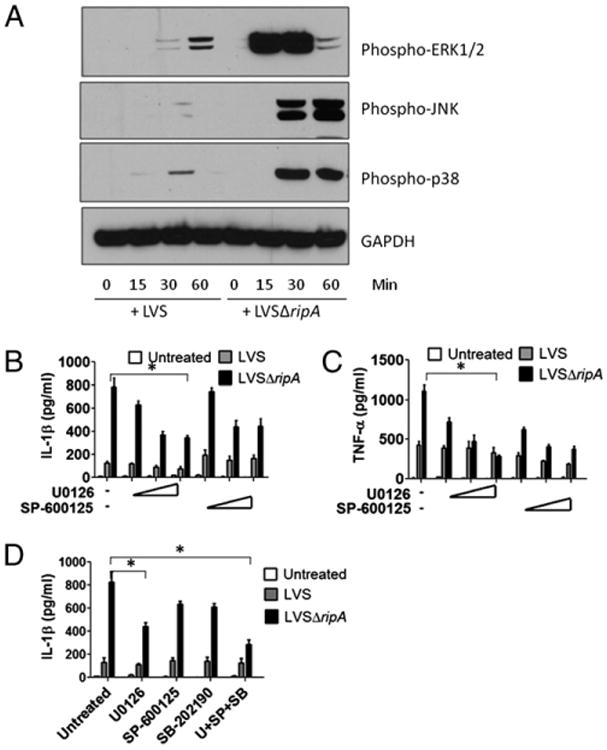
F. tularensis LVS suppresses proinflammatory cytokine release by disrupting MAPK signaling pathways A, Western blot analysis of phospho-ERK1/2, phospho-JNK, phospho-p38, and GAPDH in mouse macrophages exposed to LVS and LVSΔripA at MOI 500 for 0, 15, 30, and 60 min. At least three independent experiments were performed. A representative Western blot is shown. B, IL-1β secretion levels in mouse macrophages pretreated with ERK inhibitor U0126 and JNK inhibitor SP-600125 and exposed to LVS and LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. C, TNF-α secretion levels in mouse macrophages pretreated with U0126 or SP-600125 and exposed to LVS or LVSΔripA at MOI for 24 h. Data represent mean 6 SD for at least three independent experiments performed in triplicate. A representative experiment is shown. D, IL-1β secretion levels in mouse macrophages pretreated with U0126, SP-600125, p38 inhibitor SB-202190, or a combination of U0126, SP-600125, and SB-202190 and exposed to LVS or LVSΔripA at MOI 500 for 24 h. Data represent mean ± SD for at least three independent experiments performed in triplicate. A representative experiment is shown. *p < 0.05.
Figure 6.

LVSΔripA fails to suppress proinflammatory cytokine responses in a mouse respiratory tularemia model. A, Body weights of mice intranasally exposed to mock, LVS, and LVSΔripA at dose of 105 for 1–10 d. At least two independent experiments with seven animals per experimental condition (PBS, LVS, and LVSΔripA) were performed. A representative experiment is shown. B, Lung organ burdens of mice intranasally exposed to LVS and LVSΔripA at a dose of 105 for 6 and 24 h. At least three independent experiments with more than four animals per group were performed. Each symbol represents one animal. A representative experiment is shown. C, IL-1β levels in mouse BALF after intranasal exposure to LVSΔripA at increasing doses for 24 h. Data represent mean ± SD for at least two independent experiments with more than three animals per group. Each symbol represents one animal. A representative experiment is shown. D, IL-1β levels in mouse BALF after mock treatment or intranasal exposure to LVS or LVSΔripA (1010 bacteria per animal for both LVS and LVSΔripA) for 24 h. Data represent mean ± SD for at least three independent experiments with more than six animals per group. Each symbol represents one animal. All data are shown. E, TNF-α levels in mouse BALF after mock treatment or intranasal exposure LVS or LVSΔripA (1010 bacteria per animal for both LVS and LVSΔripA) for 24 h. Data represent mean ± SD for at least two independent experiments with more than six animals per group. Each symbol represents one animal. All data are shown. *p < 0.05.
Results
F. tularensis LVSΔripA fails to suppress the release of proinflammatory cytokines IL-1β, IL-18, and TNF-α by mouse primary macrophages
In response to foreign microbes, macrophages secrete proinflammatory cytokines including IL-1β, IL-18, and TNF-α, which activate neutrophils, fibroblasts, and endothelial cells to mount an antimicrobial response. F. tularensis can actively downregulate host immune cytokine production in vivo and in vitro (7, 15). To assess the ability of F. tularensis to induce proinflammatory cytokines, we first compared the induction profile for the cytokine IL-1β in response to various bacterial pathogens in macrophages. We selected IL-1β due to its central role in initiating a variety of host immune defense cascades (40). Compared with Salmonella typhi, Klebsiella pneumoniae, and Shigella flexneri, mouse primary macrophages exposed to F. tularensis LVS released significantly less IL-1β (Fig. 1A) suggesting that F. tularensis LVS represses this particular inflammatory response. One possible source of this difference is the LPS. LPS from Salmonella typhi, K. pneumoniae, and Shigella flexneri may be more immune-stimulatory than F. tularensis LPS as described by another report (33).
Required for intracellular proliferation, factor A (RipA; FTL_1914) is a cytoplasmic membrane protein that is conserved among Francisella species and is required for adaptation of the bacteria to the host cell cytoplasm. Mutants lacking ripA (ΔripA strains) enter macrophages and escape from the phagosome at the same frequency and kinetics as wild-type F. tularensis but fail to replicate intracellularly in the host cell (12). In that study, we demonstrated that LVSΔripA was unable to replicate intracellularly in host epithelial cells or macrophages. We considered that ΔripA strains might also be affected in their ability to suppress host cell immune responses. To test this possibility, we monitored IL-1β release by macrophages infected with wild-type or LVSΔripA strains. Over a range of MOI, LVSΔripA induced ∼5- to 10-fold higher levels of IL-1β than that of wild-type–infected macrophages (Fig. 1B). Suppression of IL-1β was restored to LVSΔripA by in trans complementation with a ripA-containing construct, pripA (Fig. 1B), demonstrating a direct but inverse cause-and-effect relationship between F. tularensis RipA expression and IL-1β response.
We next asked whether the effect of RipA on cytokine expression was widespread or limited to IL-1β. IL-18 production was measured because the inflammasome is also involved in processing and release of this cytokine. Increased levels of IL-18 similar to that of IL-1β were released by the LVSΔripA-stimulated macrophages, and complementation reduced induction nearly to wild-type levels (Fig. 1C). Because F. tularensis-induced TNF-α is not regulated by inflammasome components (14) but is regulated by MyD88 (35), we sought to test whether these findings hold true for LVSΔripA-induced TNF-α release. LVSΔripA induced 2- to 3 fold more TNF-α than that of wild-type LVS (Fig. 1D). The magnitude of the difference between LVS and LVSΔripA-induced TNF-α release by macrophages was less than that of IL-1β. This suggests that ripA affects F. tularensis suppression of both inflammasome and noninflammasome cytokines, but that the impact on inflammasome cytokines is more significant. Neither paraformaldehyde (PFA)-fixed wild-type LVS nor LVSΔripA mutant strains provoked significant levels of IL-1β (Fig. 1E) demonstrating that the observed impact of RipA on cytokine expression was not due simply to lack of intracellular replication by the deletion mutant strain.
One possible mechanism for the different levels of IL-1β induced by LVS versus LVSΔripA is that the former might cause more cell death, thus interfering with IL-1β production and processing. To assess cell viability differences between LVS and LV S ΔripA, we measured cytotoxicity of primary mouse macro-phages postinfection with LVS, LVSΔripA, or the LVS ripA complementation strain. As shown in Fig. 1F, infection with LVSΔripA resulted in increased cytotoxicity compared with cells infected with wild-type LVS or the ripA complementation strain. These data together suggest that LVSΔripA is both hyper-inflammatory and hypercytotoxic compared with wild-type LVS. These results are consistent with the frequent association of inflammasome activation and cell death (41), as shown here for LVSΔripA, but inconsistent with the possibility that LVS causes more cell death, which interferes with IL-1β production and release.
To address further the potential contribution of MOI to the observed phenotypes, primary mouse macrophages were exposed to wild-type LVS at MOI 10, 50, or 500, and IL-1β secretion was assayed by ELISA at 24 h postinfection. As shown in Fig. 1G, IL-1β production increased with MOI; however, the levels of IL-1β produced at the lower MOI of 10 was near the limit of detection. Therefore, for the subsequent LVS infections in primary mouse macrophages, MOI 50 or 500 was used.
In contrast with the relatively low levels of IL-1β that we observe for wild-type LVS, several studies have shown that infection with subspecies novicida results in high levels of IL-1βsecretion and cell death, albeit the Mariathasan et al. (14) study used LPS-pretreated, thioglycolate-elicited peritoneal macrophages (42). To determine if the differences in IL-1β secretion are strain-specific, we infected resting BMDMs with F. tularensis subsp. novicida U112 at MOI 10, 100, or 500 and assayed for IL-1β secretion at 24 h postinfection (Fig. 1H). Similar to the results of Henry et al. (42) for subspecies novicida-induced IL-1β secretion in BMDMs, our data shows that subspecies novicida strain U112 induced higher levels of IL-1β secretion by primary mouse macrophages (Fig. 1H).
LVSΔripA-induced IL-1β is PYCARD, caspase-1, and MyD88 dependent
In the regulation of IL-1β release by macrophages, bacterial-derived pathogen-associated molecular patterns or microbe-associated molecular patterns first activate the TLR signaling pathway, thereby promoting the synthesis of pro–IL-1β transcript and later protein. A second signal initiates assembly of the inflammasome to process pro–IL-1β into IL-1β for release. To determine the level at which IL-1β release is regulated upon infection, we tested whether disruption of either TLR or NLR/inflammasome signaling pathways can abrogate LVSΔripA-induced IL-1β release. MyD88 was chosen for TLR signaling due to its central role in TLR/IL-1R pathways. To disrupt inflammasome formation, mice deficient in either Pycard or Caspase-1 were used because these two gene products are critical in subspecies novicida-induced IL-1β release by mouse macrophages and the ulceroglandular form of tularemia (14). Furthermore, two commonly interacting NLRs, NLRP3 and NLRC4, were tested because they are critical in the detection and response to several pathogens (26). In the absence of Pycard, Caspase-1, and MyD88, macrophages did not release IL-1β upon LVS or LVSΔripA exposure (Fig. 2A). However, absence of Nlpr3 or Nlrc4 did not impair the LVSΔripA-induced release of IL-1β suggesting that these NLRs are not involved in the sensing of F. tularensis.
Similar to IL-1β release, PYCARD, caspase-1, and MyD88 but not NLRP3 or NLRC4 were required for LVSΔripA-induced IL-18 release by macrophages, further confirming effects on an additional inflammasome cytokine (Fig. 2B). As expected, MyD88 but not PYCARD, caspase-1, NLRP3, or NLRC4 is required for LVSΔripA-induced release of the noninflammasome cytokine TNF-α by primary mouse macrophages (Fig. 2C). Taken together, LVSΔripA-induced IL-1β production is mediated by PYCARD, caspase-1, and MyD88, which suggests that F. tularensis LVS may target the PYCARD/caspase-1 axis as well as other TLR-dependent signaling pathways to suppress IL-1β release by macrophages.
F. tularensis LVSΔripA fails to suppress proinflammatory cytokine response by human monocytic THP-1 cells
To determine if the observed effects are also applicable to human cells, we measured cytokine expression by the human monocytic cell line THP-1 in response to wild-type LVS and LVSΔripA. THP-1 cells exposed to LVSΔripA released 10-fold more IL-1β (Fig. 3A) and 2- to 3-fold more TNF-α (Fig. 3B) than that of cells exposed to wild-type LVS, which is consistent with the mouse primary macrophage response. Also consistent, the difference in amount of TNF-α induced between LVS and LVSΔripA is of smaller magnitude than that of IL-1β.
We next investigated whether PYCARD/apoptotic speck-containing protein with a CARD was required for LVSΔripA-induced IL-1β release by THP-1 cells using cells stably expressing either a control scramble PYCARD retroviral ShRNA vector (Sh-Ctrl) that does not confer knock down, a specific PYCARD retroviral ShRNA vector (Sh-PYCARD) to knock down PYCARD, or a specific NLRP3 retroviral ShRNA vector (Sh-NLRP3) to knock down NLRP3 (36). In our previous studies, all pathogens investigated induced IL-1β in a PYCARD-dependent manner in a system where PYCARD expression was reduced by short hairpin RNA (36). Cells were infected with LVS and LVSΔripA, and IL-1β was monitored. 24 h postinfection, Sh-PYCARD–bearing THP-1 cells treated with either LVS or LVSΔripA did not release detectable IL-1β (Fig. 3C). In contrast, induction of IL-1β release was similar in Sh-NLRP3–bearing cells and Sh-Ctrl–bearing cells. This is consistent with our studies using gene-deficient mouse primary macrophages. To determine whether caspase-1 is required for LVSΔripA-induced IL-1β release by THP-1 cells, THP-1 cells were pretreated with a specific caspase-1 inhibitor (10 μM Y-VAD-fmk). Porphyromonas gingivalis, which induces a caspase-1–dependent IL-1β release in THP-1 cells (37), served as control. Y-VAD blocked IL-1β release in response to infection with P. gingivalis, and also with LVS and LVSΔripA (Fig. 3D). Based on this data, we concluded that PYCARD and caspase-1 are required in LVSΔripA-induced IL-1β release by human monocytic THP-1 cells.
F. tularensis LVSΔripA fails to suppress the processing and synthesis of IL-1β
To elucidate the mechanisms by which F. tularensis suppresses host macrophage release of proinflammatory cytokines, we first sought to determine kinetics of intracellular IL-1β synthesis and processing using Western blot analysis to detect pro–IL-1β (33 kDa) and processed IL-1β (17 kDa) present in LVSΔripA-infected mouse macrophages over a time course postinoculation. Pro–IL-1β levels increased starting at 30 min after LVSΔripA exposure (Fig. 4A), a time corresponding with the kinetics of phagosome escape by both wild-type F. tularensis and LVSΔripA (12). IL-1β processing occurred 30–45 min postinfection, and processed IL-1β accumulated over several hours (Fig. 4A). By 24 h post-infection, the intracellular level of processed IL-1β decreased, likely due to release of IL-1β into the extracellular space and death of macrophages. To determine whether the response to LVS and LVSΔripA differs in the synthesis and processing of IL-1β by macrophages, we repeated a time course of infection. By 60 min, LVS induced the synthesis of pro–IL-1β, and no processed IL-1β was evident. Pro–IL-1β increased significantly 120 min post-inoculation with LVS; however, only a small fraction of the pro-protein was processed to mature IL-1β at this point as shown by Western blotting for pro–IL-1β and processed IL-1β (Fig. 4B) and quantification of processed IL-1β by densitometry (Fig. 4C). In contrast, pro–IL-1β was present in LVSΔripA-infected macro-phages by 60 min postinfection, with the majority processed to mature IL-1β. Pro–IL-1β continued to increase by 120 min postinoculation, with half of the proprotein processed to mature IL-1β.
Figure 4.
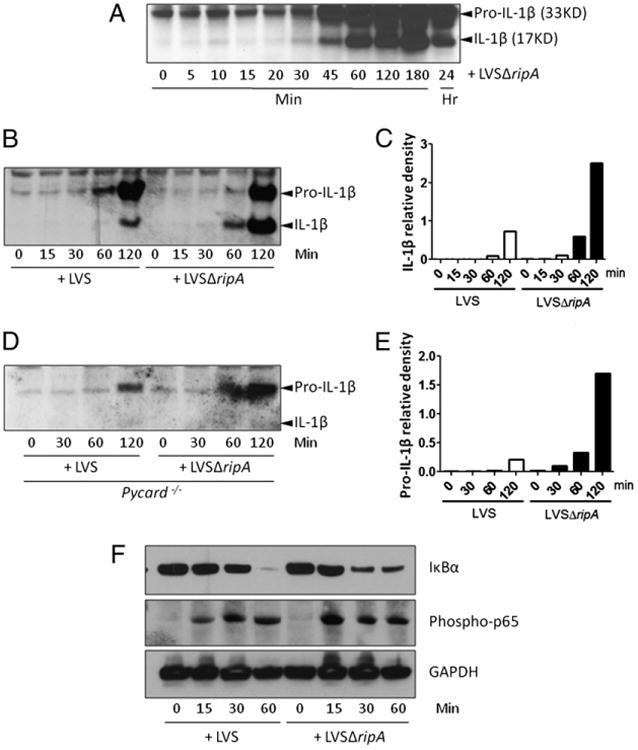
F. tularensis LVS suppresses IL-1β response in macrophages by interfering with IL-1β synthesis and processing. A, Western blot analysis of IL-1β expression in mouse macrophages exposed to LVSΔripA at MOI 500 for 0, 5, 15, 20, 30, 45, 60, 120, 180 min and 24 h. At least three independent experiments were performed. A representative Western blot is shown. B, Western blot analysis of IL-1β expression in mouse macrophages exposed to LVS and LVSΔripA at MOI 500 for 0, 15, 30, 60, and 120 min. At least three independent experiments were performed. A representative Western blot is shown. C, Densitometric quantification of the bands for processed IL-1β (17 kDa) shown in B. D, Western blot analysis of IL-1β expression in macrophages derived from Pycard−/− mice after exposure to LVS and LVSΔripA at MOI 500 for 0, 30, 60, and 120 min. At least three independent experiments were performed. A representative Western blot is shown. E, Densitometric quantification of the bands for pro–IL-1β (33 kDa) shown in D. F, Western blot analysis of IκBα and phospho-p65 in mouse macrophages exposed to LVS and LVSΔripA at MOI 500 for 0, 15, 30, and 60 min. At least three independent experiments were performed. A representative Western blot is shown.
Next, we sought to determine if F. tularensis LVS also disrupted the synthesis of pro–IL-1β. To accomplish this objective, we exposed Pycard−/− mouse primary macrophages to LVS and LVSΔripA and monitored the synthesis of pro–IL-1β. Because the absence of PYCARD abolishes the processing of pro–IL-1β into mature IL-1β, we can directly compare the macrophage synthesis of pro–IL-1β. In the absence of IL-1β processing, LVSΔripA-infected macrophages induced significantly more pro–IL-1β than LVS as shown by Western blot (Fig. 4D) and quantified by densitometry in Fig. 4E. Only pro–IL-1β is observed in LVS and LVSΔripA-infected Pycard−/− BMDMs because the absence of PYCARD prevents processing of IL-1β. Thus, F. tularensis LVS disrupts both the synthesis and processing of pro–IL-1β by macrophages. Collectively, these data suggest that the removal of RipA affected signaling pathways required for both the synthesis and processing of pro–IL-1β.
The effect of RipA on both pro–IL-1β and TNF-α synthesis led us to test the effect of RipA on pathways that can affect the production of both cytokines. Because NF-κB is a master transcriptional regulator that controls a wide range of host immune responses, including cytokine production, we sought to determine if F. tularensis LVS interfered with NF-κB signaling in macro-phages. Degradation of IκBα and phosphorylation of p65 were monitored because both events are involved in NF-κB activation. Degradation of IkBa allows phosphorylation and translocation of p65 into the nucleus, where p65 then binds to promoters of proinflammatory genes. LVS and LVSΔripA each induced degradation of IκBα 30–60 min postinfection and phosphorylation of p65 15–30 min postinfection (Fig. 4F). LVSΔripA appeared to induce slightly faster degradation of IκBα and phosphorylation of p65; however, these small differences alone are unlikely to account for the significant differences between LVSΔripA and LVS in the induction of TNF-α and pro–IL-1β.
F. tularensis LVSΔripA fails to dampen the activation of MAPK pathways
Another common group of signaling pathways that contributes to cytokine activation by bacteria and bacterial components are the MAPK pathways. F. tularensis LVS initially activates MAPK signaling pathways but subsequently downregulates their activity (43). To determine whether the difference between the cytokine activation by LVS and LVSΔripA might be explained by differences in the induction of MAPKs, we profiled the phosphorylation of ERK1/2, JNK, and p38 in macrophages postinfection. LVS induced modest levels of ERK1/2 and p38 phosphorylation, which peaked at 30 and 60 min, but induced very little JNK activation (Fig. 5A). In contrast, LVSΔripA induced dramatic levels of ERK1/2 phosphorylation at 15–30 min and of JNK and p38 at 30– 60 min postinoculation. These data suggest that LVS dampened the induction of MAPK signaling pathways by a mechanism that is missing or ineffective in the ΔripA mutant strain.
To determine whether differences in MAPK activation might explain the increased inflammatory nature of LVSΔripA, we abrogated MAPK activity in ΔripA-infected cells with pharmacological inhibitors of ERK, JNK, and p38 signaling pathways. Primary mouse macrophages were treated with U0126, SP-600125, or SB-202190 to prevent the phosphorylation of ERK1/2, JNK, and p38, respectively. These cells were subsequently exposed to either LVS or LVSΔripA, and IL-1β release was measured 24 h postinfection. Both U0126 and SP-600125 reduced the release of IL-1β by macrophages by 1.5- to 2-fold postinfection with LVSΔripA over a range of doses (Fig. 5B). Thus, ERK and JNK pathways each partially contribute to IL-1β activation in ΔripA-infected cells. The addition of U0126 or SP-600125 also reduced the release of TNF-α by macrophages upon exposure to LVS and LVSΔripA (Fig. 5C). Finally, the combination of ERK, JNK, and p38 inhibitors blocked LVSΔripA-induced IL-1β release by macrophages more effectively than each inhibitor alone (Fig. 5D). Taken together, these data demonstrate that wild-type F. tularensis LVS interferes with the activation of multiple MAPK signaling pathways that are important for release of cytokines post-infection.
F. tularensis LVSΔripA fails to suppress proinflammatory cytokine responses in vivo
Our previous study showed that LVSΔripA is attenuated in mice as evidenced by a reduction in organ burden at days 1, 3, and 7 postintranasal infection compared with that of LVS (12). In this study, we confirm and expand this finding by monitoring mice morbidity, mortality, and cytokine responses postinfection. Mice were intranasally inoculated with LVS, LVSΔripA, and mock/PBS, and weight loss was monitored among the three experimental groups. When using a LD100 dose of LVS (105), LVS-inoculated animals exhibited severe weight loss, a measure of the severity of respiratory tularemia, and eventually succumbed to the disease (Fig. 6A). In contrast, LVSΔripA-inoculated animals only showed a modest weight loss compared with that of mock/PBS-treated animals. We also measured the bacterial burdens in the lungs of mice during the early stages of the disease (6 and 24 h postinfection). Similar lung burdens were observed 6 h post-infection when comparing LVS and LVSΔripA, indicating that the initial bacterial loads of LVS and LVSΔripA are indistinguishable (Fig. 6B). By 24 h postinoculation, LVS lung burdens rose to 106 CFU/organ. In contrast, the lung burden of LVSΔripA-inoculated animals did not increase beyond that of the 6-h time point. In our previous report, the ripA complementation strain of LVSΔripA-inoculated animals exhibited similar lung, spleen, and liver burdens compared with that of LVS-inoculated animals (12).
The in vitro analyses shown earlier indicate that LVSΔripA failed to suppress the release of IL-1β, IL-18, and TNF-α by primary mouse macrophages and human THP-1 cells. To determine if the absence of ripA similarly affected cytokine responses in vivo, we measured IL-1β and TNF-α in the lungs of infected mice. After testing multiple doses of LVSΔripA to determine the amount required to detect IL-1β and TNF-α in the lung (Fig. 6C), we found that mice inoculated with 1010 CFU LVSΔripA expressed on average 5- to 10-fold more IL-1β than that of animals inoculated with the same number of wild-type LVS (Fig. 6D). Notably, at that dose of bacteria, IL-1β levels in the bronchoalveolar lavage fluid (BALF) of animals infected with wild-type LVS was indistinguishable from mock/PBS-treated mice. We next examined levels of TNF-α in mouse lungs of mock/PBS, LVS, and LVSΔripA-inoculated animals. LVSΔripA induced more TNF-α than did LVS, but the difference was not as significant as that for IL-1β (Fig. 6E). These results are consistent with the in vitro results obtained with mouse and human cells (Figs. 1D, 3B) and support the effects of ripA deletion on both inflammasome and noninflammasome mediated cytokines. Taken together, these data suggest that the expression of ripA in LVS suppresses macrophage proinflammatory cytokine production during an infection both in vitro and in vivo.
Discussion
The innate immune response is indispensable to eradicate the invasion of microbial pathogens, and monocytes/macrophages are major arsenals in combating infectious diseases. Proper release of cytokines and chemokines by these cells is critical in the migration of polymorphonuclear cells and other effector cells to the site of infection. To establish a successful infection, a pathogen must either evade immune surveillance or modulate host antimicrobial response. Over time, pathogens have developed sophisticated strategies to manipulate host immune responses to their advantage. In this study, we demonstrated that F. tularensis actively suppresses the release of proinflammatory cytokines and that the F. tularensis gene ripA is critical in this process. In response to F. tularensis LVSΔripA, macrophages are able to mount an effective initial response by releasing higher amounts of IL-1β, IL-18, and TNF-α compared with that of LVS. This observation is also seen in vivo, which correlates with the reduced morbidity caused by LVSΔripA. These data suggest that ripA contributes to effective suppression of host immunity, and hence F. tularensis survival. Of equal importance is the role of IFN-γ and IFN-β during host response to F. tularensis, and it would be interesting to test whether the IFN-β and IFN-γ responses are altered in LVSΔripA-treated cells and animals during an infection.
An emerging theme in the literature is that a variety of pathogens can suppress IL-1β release by macrophages. P. aeruginosa induces the release of IL-1β by macrophages in a NLRC4-dependent manner (32). A subset of P. aeruginosa expresses the effector molecule ExoU, and this molecule inhibits caspase-1 activation thereby preventing the release of IL-1β and IL-18 by macro-phages. Mycobacterium tuberculosis circumvents both innate and adaptive immune responses. Similar to our observations with F. tularensis, the M. tuberculosis gene product zmp1 prevents the activation of the inflammasome to inhibit IL-1β processing (44). Among viruses, myxoma virus inhibits release of proinflammatory cytokines IL-1β, IL-18, TNF-α, IL-6, and MCP-1. Myxoma virus encodes a pyrin-containing protein m103, which has been shown to bind to PYCARD and thereby disrupt the activation of inflammasome (45). The m103 protein also binds NF-κB, which suppresses the degradation of IκBα and phosphorylation of IKK. Cowpox virus, via gene crmA, directly inhibits caspase-1 activity and suppresses IL-1β response to infection (46). Our study with F. tularensis LVS and LVSΔripA together with these and other reports exemplifies the multiple strategies employed by pathogens to disrupt host immune activation of the production of IL-1β and other cytokines by innate immune cells.
To elucidate the pathways by which LVS suppresses the IL-1β release by macrophages, we have identified PYCARD, caspase-1, and MyD88 mediated signaling that the LVS strain can disrupt. Mariathasan and coworkers (14) have shown that PYCARD and caspase-1 mediated host IL-1β production is key in combating subspecies novicida infection. In their study, mice lacking Pycard or Caspase-1 were far more susceptible to challenge by F. tularensis subsp. novicida. Moreover, mice i.p. injected with IL-18 and IL-1β neutralization Ab prior to subspecies novicida challenge exhibited higher organ burdens than those treated with isotype control, further suggesting the importance of IL-1β and IL-18 in tularemia. Recently, absent in melanoma 2 has been identified as candidate proteins required in F. tularensis-induced inflammasome formation and subsequent IL-1β release (19, 47, 48). Thus far, TLR2, MyD88, and type I IFN-β have been implicated in F. tularensis-induced host cytokine response, including IL-1β by macrophages (14, 35, 49–51). Our data suggest that F. tularensis LVSΔripA-induced IL-1β is PYCARD, caspase-1, and MyD88 dependent and that LVS disrupts these signaling pathways.
In macrophages and epithelial cells, pathogen-associated molecular patterns or microbe-associated molecular patterns activate NF-κB and MAPK pathways to initiate the proinflammatory responses. There are three major families of MAPK including the ERKs, the JNKs, and the p38 kinases (52). Many pathogens have developed strategies to disrupt the MAPK pathway to subvert the immune response. Arbibe and coworkers (53) demonstrated that Shigella flexneri virulence factor OspF inactivates ERK1/2 and p38 by preventing their phosphorylation. YopJ, a type III effector protein of Yersinia species, has been shown to acetylate the serine and threonine residues on MAPK kinase thereby interrupting the downstream MAPK phosphorylation (54). Telepnev and co-workers (43) have demonstrated that LVS may interfere with MAPK signaling pathways and that iglC is critical in this process. Our data suggest that LVS may be able to interfere with both the synthesis and processing of IL-1β at multiple points and that ripA is critical in these processes. The degradation of IκBα and phosphorylation of p65 is not likely the major mechanism by which ripA mediates immune suppression. However, LVSΔripA induced a significant increase in phosphorylation of these three MAPKs compared with that of wild-type LVS. Moreover, our studies showed that the LVSΔripA-induced IL-1β and TNF-α release can be partially reduced by pretreating cells with inhibitors of ERK, JNK, and p38 alone, and the use of all three inhibitors resulted in an additive reduction of cytokine production that approaches the lower level caused by LVS infection. These results further validate that ripA contributes to the MAPK suppressive nature of F. tularensis. LVS synergistically downregulates the synthesis of pro–IL-1β and prevents the processing of IL-1β, thereby resulting in strong suppression of IL-1β release by macrophages.
A specific function for RipA protein has not yet been identified. Our experiments using PFA-killed LVS and LVSΔripA demonstrated that RipA-mediated suppression of the induction of IL-1β by LVS-infected host cells is an active process. In other words, the induction is not solely the result of gross morphological change in the bacterial cell or a cell surface marker that is present in LVSΔripA regardless of the bacterial viability. Preliminary data from our laboratory also indicate that the outer membrane profile and LPS profile of LVSΔripA is unchanged. Furthermore, our previous study demonstrated that RipA is localized to the cytoplasmic membrane of F. tularensis (12). This finding suggests that RipA does not mediate immune response suppression by directly interfering with or binding to innate immune receptors or sensors. More likely, RipA is interacting with another protein or proteins that interfere with the proinflammatory signaling pathways.
Others have also found that infection of macrophages with LVS blocks TLR-induced signaling (43); however, there are seemingly conflicting reports that F. tularensis subsp. novicida or LVS is a potent inducer of proinflammatory cytokines (14, 33, 55, 56). These apparent contradictions may be due to variations in the experimental designs, such as the differences in MOI, the activation state of the cell population used, or the different subspecies used. Ulland et al. (56) reported nanogram levels of IL-1β secretion from LPS-prestimulated BMDMs that were subsequently infected with LVS, whereas we infected naive BMDMs. We also have found that after prestimulation of BMDMs with E. coli LPS, LVS induced high levels of IL-1β (data not shown). Thus, the activation state of the cell likely affects the cytokine levels produced in response to F. tularensis. Consistent with this possibility, Cole et al. (55) found substantial LVS-induced IL-1β response by thioglycolate-elicited peritoneal macrophages. We believe that the use of nonstimulated macrophages, as in our study, might better reflect the host response during a primary infection.
An additional difference between our study and the latter is that our study primarily used LVS at MOIs of 50 to 500, whereas the study by Cole and colleagues (55) used MOIs of 5 to 10. We found that infection of resting macrophages with a low MOI of LVS induced little IL-1β, and the increased MOI is therefore unlikely to explain the difference in cytokine induction levels. Furthermore, the fact that LVSΔripA is both hyperinflammatory and hypercytotoxic compared with wild-type LVS suggests that differences in cell death induction in LVS cannot explain the differences in IL-1β production and release (Fig. 1F). Finally, it is possible that these differences are due to infection by different subspecies. F. tularensis subsp. novicida, a less virulent subspecies, caused substantial IL-1β and IL-18 production and caspase-1–dependent cell death at the low MOI (14). Likewise, we found that a similar range of doses of subspecies novicida U112 caused a significant amount of IL-1β release in resting BMDMs (Fig. 1H), whereas LVS did not (Fig. 1G). This suggests that the high levels of IL-1β in other studies relative to ours might also be explained by the use of different F. tularensis subspecies. Although we acknowledge that LVS may induce various levels of cytokines depending on the methods of preparing primary mouse macrophages, it is clear that the deletion of ripA alleviated the immunosuppressive nature of LVS with respect to IL-1β and TNF-α.
In conclusion, we elucidated a mechanism by which F. tularensis actively evades host immune response by an analysis of the attenuated LVSΔripA strain. These results indicate that the ripA gene product functions by targeting two prominent innate immune circuits: the circumvention of the host inflammasome response and the dampening of the activation of MAPK signaling pathways. During the review of this article, there was a report of another immune evasion gene in LVS that when removed resulted in elevated IL-1β and cell death accompanied by reduced in vivo bacterial replication (56). Thus, the phenomenon described in our study might be a common pathway for immune evasion by F. tularensis.
Acknowledgments
This work was supported by National Institutes of Health Grant U54 AI057157 from the Southeastern Regional Center of Excellence for Emerging Infections and Bio-defense.
Abbreviations used in this paper
- BMDM
bone marrow-derived macrophage
- BALF
bronchoalveolar lavage fluid
- CARD
caspase recruitment domain
- CDC
Centers for Disease Control and Prevention
- LVS
live vaccine strain
- MOI
multiplicity of infection
- NLR
nucleotide binding domain-leucine rich repeats containing
- PFA
paraformaldehyde
- PYCARD
pyrin-CARD
- RipA
required for intracellular proliferation, factor A
- Z-VAD-fmk
Z-Tyr-Val-Ala-Asp-fluoromethylketone
Footnotes
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Keim P, Johansson A, Wagner DM. Molecular epidemiology, evolution, and ecology of Francisella. Ann N Y Acad Sci. 2007;1105:30–66. doi: 10.1196/annals.1409.011. [DOI] [PubMed] [Google Scholar]
- 2.Vorou RM, Papavassiliou VG, Tsiodras S. Emerging zoonoses and vector-borne infections affecting humans in Europe. Epidemiol Infect. 2007;135:1231–1247. doi: 10.1017/S0950268807008527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 4.Rotz LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. Public health assessment of potential biological terrorism agents. Emerg Infect Dis. 2002;8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pechous RD, McCarthy TR, Zahrt TC. Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol Mol Biol Rev. 2009;73:684–711. doi: 10.1128/MMBR.00028-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufmann AF, Meltzer MI, Schmid GP. The economic impact of a bioterrorist attack: are prevention and postattack intervention programs justifiable? Emerg Infect Dis. 1997;3:83–94. doi: 10.3201/eid0302.970201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sjöstedt A. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect. 2006;8:561–567. doi: 10.1016/j.micinf.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Duckett NS, Olmos S, Durrant DM, Metzger DW. Intranasal interleukin-12 treatment for protection against respiratory infection with the Francisella tularensis live vaccine strain. Infect Immun. 2005;73:2306–2311. doi: 10.1128/IAI.73.4.2306-2311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, Frelinger JA, Kawula TH. Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect Immun. 2008;76:5843–5852. doi: 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiavolini D, Rangel-Moreno J, Berg G, Christian K, Oliveira-Nascimento L, Weir S, Alroy J, Randall TD, Wetzler LM. Bronchus-associated lymphoid tissue (BALT) and survival in a vaccine mouse model of tularemia. PLoS ONE. 2010;5:e11156. doi: 10.1371/journal.pone.0011156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirimanjeswara GS, Olmos S, Bakshi CS, Metzger DW. Humoral and cell-mediated immunity to the intracellular pathogen Francisella tularensis. Immunol Rev. 2008;225:244–255. doi: 10.1111/j.1600-065X.2008.00689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, Kawula TH. RipA, a cytoplasmic membrane protein conserved among Francisella species, is required for intracellular survival. Infect Immun. 2008;76:4934–4943. doi: 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chase JC, Celli J, Bosio CM. Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect Immun. 2009;77:180–195. doi: 10.1128/IAI.00879-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–1049. doi: 10.1084/jem.20050977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Metzger DW, Bakshi CS, Kirimanjeswara G. Mucosal immunopathogenesis of Francisella tularensis. Ann N Y Acad Sci. 2007;1105:266–283. doi: 10.1196/annals.1409.007. [DOI] [PubMed] [Google Scholar]
- 16.Telepnev M, Golovliov I, Grundström T, Tärnvik A, Sjöstedt A. Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell Microbiol. 2003;5:41–51. doi: 10.1046/j.1462-5822.2003.00251.x. [DOI] [PubMed] [Google Scholar]
- 17.Leiby DA, Fortier AH, Crawford RM, Schreiber RD, Nacy CA. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect Immun. 1992;60:84–89. doi: 10.1128/iai.60.1.84-89.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 19.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, Hannesschläger N, Schlee M, Rothenfusser S, Barchet W, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol. 2010;11:63–69. doi: 10.1038/ni.1824. [DOI] [PubMed] [Google Scholar]
- 22.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 23.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–33838. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 24.Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000;60:6236–6242. [PubMed] [Google Scholar]
- 25.Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- 26.Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol. 2009;21:10–16. doi: 10.1016/j.coi.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 28.Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010;22:28–33. doi: 10.1016/j.coi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol. 2009;21:194–198. doi: 10.1016/j.smim.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abdul-Sater AA, Said-Sadier N, Ojcius DM, Yilmaz O, Kelly KA. Inflammasomes bridge signaling between pathogen identification and the immune response. Drugs Today (Barc) 2009;45(Suppl B):105–112. [PMC free article] [PubMed] [Google Scholar]
- 31.Miao EA, Warren SE. Innate immune detection of bacterial virulence factors via the NLRC4 inflammasome. J Clin Immunol. 2010;30:502–506. doi: 10.1007/s10875-010-9386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cole LE, Elkins KL, Michalek SM, Qureshi N, Eaton LJ, Rallabhandi P, Cuesta N, Vogel SN. Immunologic consequences of Francisella tularensis live vaccine strain infection: role of the innate immune response in infection and immunity. J Immunol. 2006;176:6888–6899. doi: 10.4049/jimmunol.176.11.6888. [DOI] [PubMed] [Google Scholar]
- 34.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 35.Abplanalp AL, Morris IR, Parida BK, Teale JM, Berton MT. TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS ONE. 2009;4:e7920. doi: 10.1371/journal.pone.0007920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taxman DJ, Zhang J, Champagne C, Bergstralh DT, Iocca HA, Lich JD, Ting JP. Cutting edge: ASC mediates the induction of multiple cytokines by Porphyromonas gingivalis via caspase-1-dependent and -independent pathways. J Immunol. 2006;177:4252–4256. doi: 10.4049/jimmunol.177.7.4252. [DOI] [PubMed] [Google Scholar]
- 37.Huang MT, Taxman DJ, Holley-Guthrie EA, Moore CB, Willingham SB, Madden V, Parsons RK, Featherstone GL, Arnold RR, O'Connor BP, Ting JP. Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J Immunol. 2009;182:2395–2404. doi: 10.4049/jimmunol.0800909. [DOI] [PubMed] [Google Scholar]
- 38.Willingham SB, Bergstralh DT, O'Connor W, Morrison AC, Taxman DJ, Duncan JA, Barnoy S, Venkatesan MM, Flavell RA, Deshmukh M, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–159. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chamberlain RE. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl Microbiol. 1965;13:232–235. doi: 10.1128/am.13.2.232-235.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 41.Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES. The pyroptosome: a supra-molecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590–1604. doi: 10.1038/sj.cdd.4402194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007;204:987–994. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Telepnev M, Golovliov I, Sjöstedt A. Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mono-nuclear cells. Microb Pathog. 2005;38:239–247. doi: 10.1016/j.micpath.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Master SS, Rampini SK, Davis AS, Keller C, Ehlers S, Springer B, Timmins GS, Sander P, Deretic V. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe. 2008;3:224–232. doi: 10.1016/j.chom.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnston JB, Barrett JW, Nazarian SH, Goodwin M, Ricciuto D, Ricuttio D, Wang G, McFadden G. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity. 2005;23:587–598. doi: 10.1016/j.immuni.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 46.Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, Salvesen GS, Pickup DJ. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell. 1992;69:597–604. doi: 10.1016/0092-8674(92)90223-y. [DOI] [PubMed] [Google Scholar]
- 47.Gavrilin MA, Mitra S, Seshadri S, Nateri J, Berhe F, Hall MW, Wewers MD. Pyrin critical to macrophage IL-1beta response to Francisella challenge. J Immunol. 2009;182:7982–7989. doi: 10.4049/jimmunol.0803073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi: 10.1038/ni.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cole LE, Shirey KA, Barry E, Santiago A, Rallabhandi P, Elkins KL, Puche AC, Michalek SM, Vogel SN. Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect Immun. 2007;75:4127–4137. doi: 10.1128/IAI.01868-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Nookala S, Bina XR, Bina JE, Re F. Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J Leukoc Biol. 2006;80:766–773. doi: 10.1189/jlb.0406294. [DOI] [PubMed] [Google Scholar]
- 51.Collazo CM, Sher A, Meierovics AI, Elkins KL. Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 2006;8:779–790. doi: 10.1016/j.micinf.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 52.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 53.Arbibe L, Kim DW, Batsche E, Pedron T, Mateescu B, Muchardt C, Parsot C, Sansonetti PJ. An injected bacterial effector targets chromatin access for transcription factor NF-kappaB to alter transcription of host genes involved in immune responses. Nat Immunol. 2007;8:47–56. doi: 10.1038/ni1423. [DOI] [PubMed] [Google Scholar]
- 54.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 55.Cole LE, Santiago A, Barry E, Kang TJ, Shirey KA, Roberts ZJ, Elkins KL, Cross AS, Vogel SN. Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. J Immunol. 2008;180:6885–6891. doi: 10.4049/jimmunol.180.10.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ulland TK, Buchan BW, Ketterer MR, Fernandes-Alnemri T, Meyerholz DK, Apicella MA, Alnemri ES, Jones BD, Nauseef WM, Sutterwala FS. Cutting edge: Mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J Immunol. 2010;185:2670–2674. doi: 10.4049/jimmunol.1001610. [DOI] [PMC free article] [PubMed] [Google Scholar]