

Abstract
Chiral diols and biphenols catalyze the multicomponent condensation reaction of phenols, aldehydes, and alkenyl or aryl boronates. The condensation products are formed in good yields and enantioselectivities. The reaction proceeds via an initial Friedel Crafts alkylation of the aldehyde and phenol to yield an ortho-quinone methide that undergoes an enantioselective boronate addition. A cyclization pathway was discovered while exploring the scope of the reaction that provides access to chiral 2,4-diaryl chroman products, the core of which is a structural motif found in natural products.
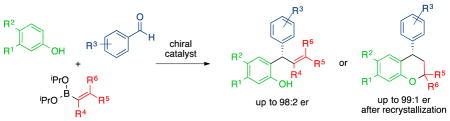
Ortho-quinone methides (oQMs) are reactive intermediates with wide-ranging applications in organic synthesis.1 As transient species, they have been exploited in biomimetic syntheses as hetero-diene partners in Diels-Alder reactions.2 The propensity to rearomatize prompts nucleophilic additions at the methide carbon, which has frequently been exploited asymmetrically within the past decade.3 The formation of metal-coordinated oQM complexes and bench stable conjugated oQMs have enabled their use in synthesis.4,5 More typically, however, oQMs are short-lived species, making their detection and utility in synthesis challenging.6 Therefore, in situ synthesis of the oQM would be an attractive approach to alleviate any issues involved in constructing a reactive intermediate.
Herein, we report a multicomponent condensation reaction to form oQMs through a bimolecular process involving a phenol and an aldehyde mediated by boronates and catalyzed by chiral phenols resulting in an enantioselective nucleophilic addition to the methide carbon. Enantioselective additions of boronates to oQMs catalyzed by chiral biphenols provide access to chiral motifs that appear in natural products and drugs.7 For instance, myristinin A is a potent inhibitor of DNA polymerase β and contains a chiral diaryl methane within its chroman core.8 Other notable examples include myristicyclin A, dracoflavans C and D, and cochinchinenins B and C.9–11 Methods to form oQMs typically require oxidation, acid/base chemistry, photolysis, or thermolysis.12 Asymmetric, multi-component reactions (MCR) provide access to structural complexity within a single transformation.13 A convergent approach in constructing this moiety leads to higher process efficiency.14 Despite these potential benefits, there are currently no examples of an asymmetric multicomponent reaction utilizing oQM chemistry.15 The efficiency of a possible MCR led us to investigate using boronates to mediate oQM formation.16 Our investigations to develop a multicomponent strategy began with an electron rich phenol, an aldehyde, and a styrenyl boronate. Mediated by the boronate, a Friedel-Crafts hydroxyalkylation reaction occurs to yield a dioxaborin intermediate.17 Nucleophilic attack by an activated and chiral boron ate complex forms the product in a stereoselective fashion.
Experiments were designed to evaluate chiral diol catalysts in the reaction (Table 1, entries 1–3). 1,1′-Bi-2-naphthol (BINOL) derived catalysts containing substituents at the 3,3′ positions led to higher enantioselectivities (Table 1, entries 4–6). In agreement with our previous studies, (R)-3,3′-Br2-BINOL was identified as the catalyst for further evaluation giving the best combination of yield and enantioselectivity.7 Higher concentrations improved the yield but resulted in lower enantioselectivity (Table 1, entries 7–9). A temperature of 80 °C was found to be ideal for promoting the condensation while maintaining enantioselectivity. As such, other high boiling solvents were examined, but did not yield product (Table 1, entries 10 and 11). Use of trifluorotoluene resulted in a higher yield, but the enantioselectivity was lower; likely due to an increase in a competing uncatalyzed background reaction rate (Table 1, entry 12). We rationalized that the boronate ester group would have an important influence on the selectivity of the reaction.18 It was found that changing the ethyl group to an isopropyl group led to a higher enantiomeric ratio (Table 1, entry 13). Adjusting the concentration and catalyst loading, we identified conditions resulting in 70% yield and a 95:5 enantiomeric ratio (Table 1, entry 14). These reaction conditions proved optimal to explore the scope of the reaction (Figure 1).
Table 1.
Chiral diols in the multicomponent boronate condensation reactiona

| ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | 6 (mol %) | concn [M] | R | yield [%]b | erc |
| 1 | 6a | 15 | 0.3 | Et | 77 | 50:50 |
| 2 | 6b | 15 | 0.3 | Et | 51 | 85:15 |
| 3d | 6c | 15 | 0.3 | Et | 96 | 57:43 |
| 4 | 6d | 15 | 0.3 | Et | 100 | 80:20 |
| 5 | 6e | 15 | 0.3 | Et | 77 | 91:9 |
| 6 | 6f | 20 | 0.2 | Et | 55 | 94:6 |
| 7 | 6f | 20 | 0.4 | Et | 78 | 92:8 |
| 8 | 6f | 20 | 0.8 | Et | 94 | 88:12 |
| 9e | 6f | 20 | 0.8 | Et | 50 | 86:14 |
| 10f | 6f | 20 | 0.2 | Et | - | - |
| 11g | 6f | 20 | 0.2 | Et | - | - |
| 12h | 6f | 20 | 0.2 | Et | 75 | 92:8 |
| 13 | 6f | 10 | 0.2 | i-Pr | 51 | 95:5 |
| 14 | 6f | 15 | 0.3 | i-Pr | 70 | 95:5 |
Reactions were run at 80 °C with 0.4 mmol phenol, 0.8 mmol aldehyde, and 0.8 mmol boronate for 24 h in toluene unless otherwise indicated.
Yield of isolated product.
Enantiomeric ratios (er) determined by HPLC analysis using a chiral stationary phase.
Reaction was run for 48 h
Reaction was run at 60 °C for 24 h.
Ethanol used as solvent.
1,4-Dioxane used as solvent.
Trifluorotoluene used as solvent.
Figure 1.

Multicomponent boronate condensation reactions
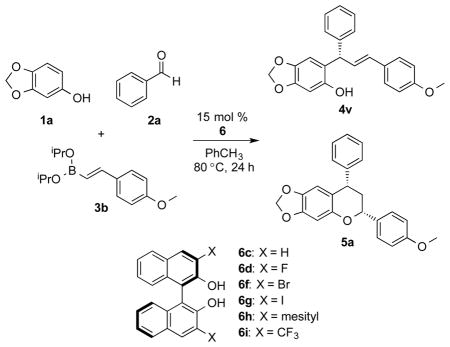
A wide range of aldehydes and boronates could be used in the reaction, but the phenol needed to be electron rich in order to promote the Friedel-Crafts alkylation reaction. Halogen substitution at the 4-position of the aldehyde was well tolerated, providing high enantioselectivity (4b, 4c). Electron rich aldehydes maintained selectivity, however, in lower yield (4d). Conversely, electron deficient substitution resulted in lower enantioselectivity, but higher yield (4e) supporting the hypothesis of a rate-determining Friedel-Crafts alkylation. Heterocyclic aldehydes could be used in the reaction; 2-thenaldehyde, gave a quantitative yield (4f). Comparison of (4f) to the previously reported compound confirmed the absolute stereochemistry of the product and is consistent with the reported oQM mechanistic model of enantioselectiv-ity.7 2-Naphthaldehyde was tolerated in the reaction, however, sterics limited the reactivity of 1-naphthaldehyde (4g, 4h). Notably, ortho-substitution improved the selectivity in the case of 2-bromobenzaldehyde (4i).
A similar observation was made with 2-methylbenzaldehyde, suggesting a steric interaction during the enantio-determining event (4j). The boronate constituents were evaluated in the reaction, each were prepared from the boronic acid precursor.19 Both styrenyl and aryl boronates performed well in the reaction (4k, 4l, 4m). Despite being less nucleophilic, electron deficient boronate also afforded the product with good results (4n). The furanyl boronate was evaluated in the reaction proving to be highly reactive; it resulted in quantitative yield with low selectivity due to a competitive uncatalyzed background reaction (4o). Finally, the use of aliphatic hexenyl boronate resulted in excellent yield and enantioselectivity (4p). Phenol substitution was investigated; 3-methoxyphenol and 3,4-dimethoxyphenol both gave high yield and enantioselectivity (4q, 4r). Substrate combinations were tested and indicate this would likely work well for others (4s-4u). In the course of evaluating reaction components in the MCR, an unanticipated reaction pathway was identified. Using 4-methoxystyrenyl boronate afforded the 2,4-diarylchroman structure as the major product (5a). Identification of the chroman encouraged us to explore preferential chemoselectivity for the cyclization pathway. We initiated studies to evaluate catalysts that would promote chemo-, enantio-, and diastereoselective formation of the chroman (Table 2).
Table 2.
Chroman formationa
| |||||
|---|---|---|---|---|---|
| entry | catalyst 6 | product 4v yield [%] | product 4v er | product 5a yield [%] | product 5a er |
| 1 | 6c | 22 | 70:30 | 28 | 60:40 |
| 2 | 6d | 46 | 66:33 | 33 | 60:40 |
| 3 | 6f | 22 | 80:20 | 55 | 79:21 |
| 4 | 6g | 31 | 82:18 | 59 | 87:13 |
| 5b | 6h | 37 | 21:79 | 16 | 49:51 |
| 6 | 6i | 39 | 89:11 | 50 | 81:19 |
| 7c | 6g | 5 | - | 69 | 84:16 |
| 8d | 6g | - | - | 72 | 85:15 |
| 9d,e | 6g | - | - | 40 | 99:1 |
Reactions were run at 80 °C with 0.4 mmol phenol, 0.8 mmol aldehyde, and 0.8 mmol boronate for 24 h in toluene (0.3 M) unless otherwise indicated. Diastereomeric ratios were 2:1 as determined by 1H NMR unless otherwise indicated. Enantiomeric ratios are reported for the major diastereomer.
(S)-enantiomer of catalyst used. Mesityl = 2,4,6-trimethylbenzene.
Reaction was run in sealed tube at 120 °C for 24 h.
Reaction was run in a sealed tube at 80 °C for 24 h and then 150 °C for 1 h.
Product was recrystallized from hot hexanes. The diastereomeric ratio was >20:1 as determined by 1H NMR.
Of the chiral diol catalysts assessed, (R)-3,3′-I2BINOL (6g) was found to give the best combination of yield and enantioselectivity (Table 2, entry 4). Next, we turned our attention to the reaction conditions. We hypothesized that the electron rich nature of the styrene component under acidic conditions at higher temperatures facilitated the cyclization process. To test this hypothesis, the reaction was run in a sealed tube at 120 °C. A significant shift in chemoselectivity was observed, yielding the chroman product in 69% yield (Table 2, entry 7). We found that an increase in temperature for a brief time following the standard reaction conditions afforded the chroman product and maintained modest levels of selectivity (Table 2, entry 8). It was later determined that recrystallization from hot hexanes afforded the product with enhanced diastereo- and enantioselectivity (Table 2, entry 9). A few substrates are illustrated to show the potential of this reaction type (Figure 2).
Figure 2.

MCR Chroman Synthesis
As postulated, electron rich π-conjugation displayed a propensity to cyclize (5a–5e), while electron neutral styrene remained slow with regard to chroman formation, even at higher temperatures (5f). This observation led us to believe that the cyclization proceeds through protonation of the electron rich olefin with subsequent nucleophilic attack of the phenol oxygen. Experiments were designed to further investigate the nature of the cyclization. Enantioenriched addition product (S)-4v (81:19 er) was isolated and subjected to reaction conditions used to afford the chroman product; 15 mol % (R)-3,3′-I2-BINOL, benzaldehyde, and boronate 3b, omitting phenol 1a, at 150 °C for 24 hours. Chroman (S,S)-5a was isolated in a 90:10 enantiomeric ratio. The result demonstrates a stepwise pathway leading to the formation of the chromans and enhanced selectivity in the cyclization step facilitated by the catalyst; a matched cyclization to afford the syn product, confirmed by 1D NOE. We have developed a mechanistic model consistent with our observations (Scheme 1). Boron-mediated Friedel-Crafts hydroxyalkylation condensation of the phenol and aldehyde in complex 7 is most likely rate limiting. There is no product formation at lower temperatures, also consistent with observations made in the literature.17 Next, boronate complex 8 dissociates and ligand transfer occurs rapidly forming product 9, the enantiodetermining event. This is consistent with our earlier studies in which enantioselective additions of boronates to oQMs occur at low temperatures.7 The facile nature of this dissociation-ligand-transfer also explains why there is no observation of a dioxaborin intermediate during the reaction.20 The cyclization is initiated by protonation of the electron rich olefin. A resonance-stabilized intermediate 10 is produced and quickly trapped by subsequent attack of the phenol oxygen to provide 11.
Scheme 1.

Proposed mechanism for the asymmetric boronate MCR
In summary, an enantioselective multicomponent reaction was developed that provides access to chiral di- and triaryl methane products with high levels of yield and selectivity. We observed an unanticipated cyclization pathway, which yielded chiral 2,4-diaryl chromans. Further studies of this reaction are underway and involve optimizing the stereocontrol of the cyclization process as well as exploring its utility in natural product synthesis.
Supplementary Material
Acknowledgments
This research was supported by the NIH (R01 GM078240 and P50 GM067041).
Footnotes
Synthetic procedures, chiral HPLC analysis, characterization and spectral data for all compounds. The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.(a) Beaudry CM, Malerich JP, Trauner D. Chem Rev. 2005;105:4757. doi: 10.1021/cr0406110. [DOI] [PubMed] [Google Scholar]; (b) Rokita SE. Quinone Methides. WILEY; 2009. [Google Scholar]; c) Willis NJ, Bray CD. Chem Eur J. 2012;18:9160. doi: 10.1002/chem.201200619. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ungaro R, Arduini A, Bosi A, Pochini A. Tetrahedron. 1985;41:3095. [Google Scholar]; (b) Selenski C, Pettus TRR. J Org Chem. 2004;69:9196. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]; (c) Batsomboon P, Phakhodee W, Ruchirawat S, Ploypradith P. J Org Chem. 2009;74:4009. doi: 10.1021/jo900504y. [DOI] [PubMed] [Google Scholar]; (d) Zhao JJ, Sun SB, He SH, Wu Q, Shi F. Angew Chem Int Ed. 2015;54:5460. doi: 10.1002/anie.201500215. [DOI] [PubMed] [Google Scholar]; (e) Hsiao CC, Raja S, Liao HH, Atodiresei I, Rueping M. Angew Chem Int Ed. 2015;54:5762. doi: 10.1002/anie.201409850. [DOI] [PubMed] [Google Scholar]
- 3.(a) Zhang Y, Sigman MS. J Am Chem Soc. 2007;129:3076. doi: 10.1021/ja070263u. [DOI] [PubMed] [Google Scholar]; (b) Alden-Danforth E, Scerba MT, Lectka T. Org Lett. 2008;10:4951. doi: 10.1021/ol802029e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jensen KH, Panthak TP, Zhang Y, Sigman MS. J Am Chem Soc. 2009;131:17074. doi: 10.1021/ja909030c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Green JC, Pettus TRR. J Am Chem Soc. 2010;133:1603. doi: 10.1021/ja109925g. [DOI] [PubMed] [Google Scholar]; (e) Pathak TP, Sigman MS. J Org Chem. 2011;76:9210. doi: 10.1021/jo201789k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Green JC, Brown ER, Pettus TRR. Org Lett. 2012;14:2929. doi: 10.1021/ol301092w. [DOI] [PubMed] [Google Scholar]; (g) El-Sepelgy O, Haseloff S, Alamsetti SK, Schneider C. Angew Chem Int Ed. 2014;53:7923. doi: 10.1002/anie.201403573. [DOI] [PubMed] [Google Scholar]; (h) Hsiao CC, Liao HH, Rueping M. Angew Chem Int Ed. 2014;53:13258. doi: 10.1002/anie.201406587. [DOI] [PubMed] [Google Scholar]; (i) Yeung CS, Ziegler RE, Porco JA, Jacobsen EN. J Am Chem Soc. 2014;136:13614. doi: 10.1021/ja508523g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Wang Z, Ai F, Wang Z, Zhao W, Zhu G, Lin Z, Sun J. J Am Chem Soc. 2015;137:383. doi: 10.1021/ja510980d. [DOI] [PubMed] [Google Scholar]; (k) Saha S, Schneider C. Org Lett. 2015;17:648. doi: 10.1021/ol503662g. [DOI] [PubMed] [Google Scholar]; (l) Saha S, Schneider C. Chem Eur J. 2015;21:2348. doi: 10.1002/chem.201406044. [DOI] [PubMed] [Google Scholar]; (m) Saha S, Alamsetti SK, Schneider C. Chem Commun. 2015;51:1461. doi: 10.1039/c4cc08559k. [DOI] [PubMed] [Google Scholar]; (n) Li ML, Chen DF, Luo SW, Wu X. Tetrahedron: Asymmetry. 2015;26:219. [Google Scholar]; (o) Caruana L, Mondatori M, Corti V, Morales S, Mazzanti A, Fochi M, Bernardi L. Chem Eur J. 2015;21:6037. doi: 10.1002/chem.201500710. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kopach ME, Harman WD. J Am Chem Soc. 1994;116:6581. [Google Scholar]; (b) Amouri H, Besace Y, Le Bras J. J Am Chem Soc. 1998;120:6171. [Google Scholar]; (c) Amouri H, Vaissermann J. Organometallics. 2000;19:1740. [Google Scholar]; (d) MacIntosh AD, Yang H, Pike RD, Sweigart DA. J Organomet Chem. 2012;719:14. [Google Scholar]
- 5.(a) Jurd L. Tetrahedron. 1977;33:163. [Google Scholar]; (b) Roitman JN, Jurd L. Tetrahedron. 1978;34:57. [Google Scholar]
- 6.Van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367. [Google Scholar]
- 7.Luan Y, Schaus SE. J Am Chem Soc. 2012;134:19965. doi: 10.1021/ja309076g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Sawadjoon S, Kittakoop P, Kirtikara K, Vichai V, Tanticharoen M, Thebtaranonth Y. J Org Chem. 2002;67:5470. doi: 10.1021/jo020045d. [DOI] [PubMed] [Google Scholar]; (b) Deng JZ, Starck SR, Li S, Hecht SM. J Nat Prod. 2005;68:1625. doi: 10.1021/np058064g. [DOI] [PubMed] [Google Scholar]
- 9.Lu Z, Van Wagoner RM, Pond CD, Pole AR, Jensen JB, Blankenship D, Grimberg BT, Kiapranis R, Matainaho TK, Barrows LR, Ireland CM. Org Lett. 2014;16:346. doi: 10.1021/ol4022639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnone A, Nasini G, Vajna de Pava O. J Nat Prod. 1997;60:971. doi: 10.1021/np9702188. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y, Zhang P, Yu H, Wang MW, Zhao W. J Nat Prod. 2007;70:1570. doi: 10.1021/np070260v. [DOI] [PubMed] [Google Scholar]
- 12.(a) Cavitt SB, Sarrafizadeh H, Gardner J Org Chem. 1962;27:1211. [Google Scholar]; (b) Bolon DA. J Org Chem. 1970;35:715. [Google Scholar]; (c) Selenski C, Pettus TRR. Quinomethanes Science of Synthesis. 2006;28:4.28.12.1. [Google Scholar]; (d) Lumb JP, Choong KC, Trauner D. J Am Chem Soc. 2008;130:9230. doi: 10.1021/ja803498r. [DOI] [PubMed] [Google Scholar]; (e) Kumbaraci V, Ergunes D, Midilli M, Begen S, Talinli N. J Heterocycl Chem. 2009;46:226. [Google Scholar]; (f) Bai WJ, David JG, Feng ZG, Weaver MG, Wu KL, Pettus TRR. Acc Chem Res. 2014;47:3655. doi: 10.1021/ar500330x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yus M, Ramon DJ. Angew Chem Int Ed. 2005;44:1602. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]
- 14.Westermann B, Brauch S, van Berkel SS. Chem Soc Rev. 2013;42:4948. doi: 10.1039/c3cs35505e. [DOI] [PubMed] [Google Scholar]
- 15.(a) Kaim LE, Gimaud L, Oble J. Org Biomol Chem. 2006;4:3410. doi: 10.1039/b610229h. [DOI] [PubMed] [Google Scholar]; (b) Lawrence AL, Adlington RM, Baldwin JE, Lee V, Kershaw JA, Thompson AL. Org Lett. 2010;12:1676. doi: 10.1021/ol100138k. [DOI] [PubMed] [Google Scholar]; (c) Rama V, Kanagaraj K, Pitchumani K. Tetrahedron Lett. 2012;53:1018. [Google Scholar]; (d) Yoshioka E, Kohtani S, Miyabe H. Molecules. 2014;19:863. doi: 10.3390/molecules19010863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Wu TR, Shen L, Chong JM. Org Lett. 2004;6:2701. doi: 10.1021/ol0490882. [DOI] [PubMed] [Google Scholar]; (b) Wu TR, Chong JM. J Am Chem Soc. 2005;127:3244. doi: 10.1021/ja043001q. [DOI] [PubMed] [Google Scholar]; (c) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308. [DOI] [PubMed] [Google Scholar]; (d) Wu TR, Chong JM. J Am Chem Soc. 2007;129:4908. doi: 10.1021/ja0713734. [DOI] [PubMed] [Google Scholar]; (e) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398. doi: 10.1021/ja075204v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Barnett DS, Moquist PN, Schaus SE. Angew Chem Int Ed. 2009;48:8679. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Moquist PN, Kodama T, Schaus SE. Angew Chem Int Ed. 2010;122:7250. doi: 10.1002/anie.201003469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Barnett DS, Schaus SE. Org Lett. 2011;13:4020. doi: 10.1021/ol201535b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Lundy BJ, Jansone-Popova S, May JA. Org Lett. 2011;13:4958. doi: 10.1021/ol2020847. [DOI] [PubMed] [Google Scholar]; (j) Turner HM, Patel J, Niljianskul N, Chong JM. Org Lett. 2011;13:5796. doi: 10.1021/ol202391r. [DOI] [PubMed] [Google Scholar]
- 17.(a) Peer HG. Recl Trav Chim Pays-Bas. 1960;79:825. [Google Scholar]; (b) Nagata W, Okada K, Aoki T. Synthesis. 1979:365. [Google Scholar]; (c) Lau CK, Williams HWR, Tardiff S, Defresne C, Scheigetz J, Belanger PC. Can J Chem. 1989;67:1384. [Google Scholar]; (d) Lau CK, Chambers JD, Crawford J, Williams HWR, Dufresne C, Scheigetz J, Bernstein MA. Can J Chem. 1992;70:1717. [Google Scholar]; (e) Olson BS, Trauner D. Synlett. 2005;4:700. [Google Scholar]; (f) Lumb JP, Trauner D. J Am Chem Soc. 2005;127:2870. doi: 10.1021/ja042375g. [DOI] [PubMed] [Google Scholar]; (g) Zheng H, Hall DG. Tetrahedron Lett. 2010;51:4256. [Google Scholar]; (h) Sharifi A, Abaee MS, Mirzaei M, Naimi-Jamal MR. Asian J Chem. 2010;22:6519. [Google Scholar]
- 18.(a) Roy CD, Brown HC. J Organomet Chem. 2007;692:784. [Google Scholar]; (b) Roy CD, Brown HC. Monatsh Chem. 2007;138:879. [Google Scholar]
- 19.Hall DG. Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials. 2. WILEY-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 20.A model for enantioselectivity has been proposed based on the calculation of a cyclic BINOL-boronate structure; however, the transition state energy required to access the seven-membered reactive boronate nucleophile has never been calculated. Grayson MN, Goodman JM. J Org Chem. 2015;80:2056–2061. doi: 10.1021/jo502616a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.