

Abstract
Despite a growing body of knowledge on the mechanisms underlying the onset and progression of cancer, treatment success rates in oncology are at best modest. Current approaches use statistical methods that fail to embrace the inherent and expansive complexity of the tumor/patient/drug interaction. Computational modeling, in particular mechanistic modeling, has the power to resolve this complexity. Using fundamental knowledge on the interactions occurring between the components of a complex biological system, large-scale in silico models with predictive capabilities can be generated. Here, we describe how mechanistic virtual patient models, based on systematic molecular characterization of patients and their diseases, have the potential to shift the theranostic paradigm for oncology, both in the fields of personalized medicine and targeted drug development. In particular, we highlight the mechanistic modeling platform ModCell™ for individualized prediction of patient responses to treatment, emphasizing modeling techniques and avenues of application.
Keywords: virtual patient models, virtual clinical trials, cancer, drug development, mechanistic models, ordinary differential equations
Introduction
Cancer as a single entity is the number one cause of deaths globally and currently the world’s biggest healthcare challenge. Due to the rapid population growth and aging populations, we are in the era of an impending cancer “tidal wave,” with the projected number of new cancer cases expected to increase by 70% over the next two decades to reach 24 million per year.1 Associated annual deaths are expected to nearly double in the same period from 8.2 to 14.6 million (http://www.globocan.iarc.fr).
Despite a long history of investment in the search for a “cure for cancer,” we are still facing a gloomy outlook when it comes to effectively treating the disease. Patient stratification based on the combination of selective molecular-based therapies with biomarkers has seen some improvements in the success rate of cancer treatment, representing a real paradigm shift in clinical practice; however, this current best practice still fails to help the majority of patients. At the basis of this often inadequate response is a lack of understanding of the high level of genetic (and epigenetic) heterogeneity exhibited by patients and their tumors, not only between patients but also often within tumors, a level of heterogeneity that current approaches to cancer treatment do not fully embrace.2,3
Recent advances in high-throughput sequencing technologies by initiatives such as the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA), focused on analyzing the genetic basis of cancer, are bringing to light a fuller appreciation of the molecular complexity of cancer and the realization that identification of key driver mutations may not be possible in many cancer types, an enhanced understanding that is ushering in a shift of focus from individual genes to a more expansive and heterogeneous cancer mutational landscape.4–7 With the advent of such progress, analysis of the generated “big data” becomes a major issue in biological research and translational medicine.8 Due to the availability of appropriate computational tools and computing power capabilities, systems-based computational approaches are being developed that can incorporate this emerging view of cancer and facilitate elucidation of pathways and networks involved in the response and resistance to treatment.
Here, we highlight how systems-based virtual patient models, in particular, the most relevant and influential techniques for the nascent field of mechanistic modeling, are shifting the theranostic paradigm for selection of individualized optimal therapy choices in cancer patients and targeted drug development.
The Evolving Landscape of Cancer Treatment
Over the past few years, large-scale sequencing initiatives (eg, ICGC and TCGA) have exponentially broadened our knowledge of the genetic and epigenetic alterations impacting an individual’s risk of cancer, the development of their tumor(s), and their prognosis. In tandem, associated research is further elucidating the function of an ever increasing number of genes and proteins. Fueled by this accumulating knowledge base, and the realization that human biology is extremely complex, the landscape of cancer treatment is also beginning to evolve.
The prevailing approach to cancer treatment has been statistical, identifying the treatment that works best on large patient cohorts, ie, the first-line treatment. However, this approach fails to address the inherent individuality of each patient and their cancer, reflected by the low success rates of these treatments.9,10 This strategy has been supplanted by a more stratified approach, in which patient groups are subdivided on the basis of biomarkers, providing a crucial and necessary first step toward individualized medicine. Implementation of this biomarker-based approach has undoubtedly improved understanding of the molecular subtypes of cancer, with associated translational success. Of particular note is the use of human epidermal growth factor receptor 2 (HER2), a proto-oncogene encoding the HER2 tyrosine kinase receptor, as a biomarker for selecting treatment options. HER2-positive patients (between 15% and 20% of breast cancer patients) often exhibit an aggressive clinical phenotype associated with high metastatic potential and shortened survival.11 Targeting the HER2 receptor with trastuzumab, a monoclonal antibody therapy, as well as other HER2-directed agents, such as per-tuzumab, has dramatically improved patient outcomes in all stages of the disease12,13; however, despite these major steps forward, complete remission is still elusive.14 Similarly, the use of Kirsten rat sarcoma viral oncogene homolog (KRAS) mutational status can help determine which patients will respond to anti-epidermal growth factor receptor (EGFR) therapies (eg, cetuximab and panitumumab)15–17; in contrast to the HER2 biomarker, the response rate of combined EGFR inhibitor/irinotecan therapy in second-line colon cancer is only increased from 10% to 35% using the KRAS diagnostic test.18 Treatment approaches guided by a limited number of mutations within single genes fail to encompass the numerous associated alterations [eg, besides those in KRAS and B-Raf proto-oncogene, serine/threonine kinase (BRAF)] that may lead to differential responses. These current approaches are proving insufficient to provide the level of stratification required to improve therapy outcomes for the broader cancer patient pool.
Virtual Patient Models
If we view the onset and progression of a disease as a malfunction in the biological networks that act on multiple levels, spanning molecules, cells, tissues, whole body and populations and determined by various genetic and environmental factors, then a detailed modeling of the networks integral to a specific disease should deliver an enhanced understanding of that disease, as well as health. For the first time, we are now at a stage of understanding and technological development that enables the creation of more comprehensive computational models of cancer. A combination of decades of research on cancer relevant pathways,19,20 increased information on functional interaction networks, and improved technological and computational capabilities now makes it feasible to construct individualized computational models with predictive capacity. Implementation of such technology is set to drive a shift from the classical standpoint, in which there is insufficient information to accurately describe a complex biological system (eg, a cell) and predict its behavior to one that uses computational predictive modeling to identify any deregulated components within a system and predict its overall direction and behavior.
Various approaches for understanding the biological complexity of disease mechanisms have been employed, including multivariate analyses of variance, logistic regression models, and machine learning to identify intrinsic factors correlated with specific disease traits or treatment response. These approaches have been used to identify predictive biomarkers in the context of drug treatment in cancer cell lines or to automatically classify mutations identified in a given cancer sample as functionally important for drug treatment.21–23 The success of statistical as well as machine learning methods is very much dependent on the statistical model applied to a large set of data. Machine learning methodologies can be used for analyzing patient data; however, the large datasets generated today by international consortia such as the TCGA and ICGC can be seen as a compendium of small datasets with limited data/information available for each patient. The amount of training data for each patient is limited by the amount of somatic mutations, gene amplifications, deletions and/or differentially expressed genes, and other individual-specific information. Machine learning approaches, such as those implementing Bayesian models, build a statistical model based on available patient data, but they do this without acknowledging the biological, biomedical, and network context of that data. Nevertheless, multiple individualized statistical models can be coupled, potentially enabling patient classification into subgroups, eg, responders to certain forms of cancer treatment or long-term survivors of HIV infection,24 even if the statistical model has not been trained on a particular individual.
Mechanistic modeling
To investigate the dynamic behavior of such complex systems, mechanistic modeling approaches are also used, based on fundamental knowledge of the interactions occurring between components of a complex biological system, to generate in silico models with large-scale predictive capabilities. At least two distinct approaches are taken in mechanistic modeling: discrete and continuous. In discrete models, variables reflect either binary ON/OFF (Boolean) or multiple states, whereas continuous models, such as ordinary differential equation (ODE)-based models, can integrate continuous values. The timescales considered in these different modeling approaches also differ fundamentally, eg, a discrete, arbitrary, or continuous timescale can be implemented for updating species and parameter values present in the model (Table 1).25 Depending on the intended application and the available dataset to be reflected by the model, different combinations of approaches to model variables and time are used.25–30 An accurate description of the qualitative, semiquantitative, or even quantitative behavior of biological signaling systems can be gained. To generate quantitative predictions of signaling models and their associated gene regulatory networks, a combination of continuous variables that are simulated on a continuous timescale is required – an approach that can be taken using ODE systems. In particular, this applies to predictive mechanistic models implemented for simulation of direct (and indirect) effects of genetic alterations in a given patient and functional prediction of drug effects based on in silico modeling of drug action.
Table 1.
Different approaches to computational modeling of biological networks.
| VARIABLES/TIME | ITERATIONS | DISCRETE | CONTINUOUS |
|---|---|---|---|
| Boolean | Boolean networks | Stochastic boolean networks | |
| Multi-valued | Generalized logic models | Discrete time piecewise linear differential equations | Stochastic multi valued gene networks/Piecewise linear differential equations |
| Continuous | Fuzzy logic models | Chemical kinetics |
Note: Adapted from Ref. 25.
Detailed mechanistic approaches to modeling cellular signaling events require a comprehensive assessment of the most important biological reactions underlying each event; however, this may not be directly perceived from the information available on a given pathway. Therefore, the actual process of model creation and improvement often requires several iterative steps, comprising (i) annotation of specific signaling events, based on the available scientific literature and/or pathway databases of signaling nodes (eg, protein species and their biochemical complexes, including any molecular modifications such as phosphorylation); (ii) identification and implementation of the biochemical reactions and molecular alterations with which a given network node influences the associated nodes (eg, activation/inhibition, phosphorylation, and transcriptional induction). This step ensures that the information flux within the interaction network follows a known and predetermined path given a priori by the known biology of the network; (iii) identification and implementation of appropriate kinetic laws along with their respective parameters and variables, including appropriate initial values (eg, species concentrations of the network nodes); and (iv) identification and implementation of reasonable values for parameters associated with molecular reaction kinetics, the latter reflecting the velocity and equilibrium of each individual reaction.
In recent years, a plethora of mechanistic computational models that aim to simulate disease processes have been established. These models effectively represent individual pathways (metabolic processes, individual signal transduction pathways, and cell cycle regulation) and have generated unprecedented biological insights, eg, into the oncogenic process.31–33 However, the insights gained may be limited, as these focused models do not effectively represent key cellular cross-talk mechanisms – a feature that is of particular significance in the context of predicting patient responses to drugs, as each drug perturbs multiple biological targets and is potentially involved in multiple biological processes. Overall, a larger scale approach is necessary.
Modeling of large-scale systems
Integral to the development of large-scale mechanistic models has been the steady increase (over the past 10–15 years) in the availability of public information sources, providing access to relevant data. Examples include PathGuide,34 a comprehensive list of databases and resources for molecular and cellular pathways and interaction networks, and the well-known pathway databases KEGG35 and Reactome.36 Consensus PathDB is a metadata-base that integrates different types of functional interactions from heterogeneous interaction data resources, as well as from other public databases.37 The current version comprises 32 different resources and can be used to generate generic cellular networks and perform enrichment or overrepresentation analysis, eg, of patient molecular data. BioModels38 and JWS39 are repositories for mathematical models of biological systems, while databases such as Brenda40 and SABIO-RK41 provide additional information on reaction kinetics and kinetic parameters. Detailed information on molecular species and drugs can be found in databases such as ChEMBL.42 An overview of the frequently used pathway interaction databases, kinetic repositories, and information resources for cellular molecules and drugs is given in Table 2.
Table 2.
Pathway and model data resources and databases.
| DATABASE/DATA RESOURCE | DESCRIPTION | REFERENCE |
|---|---|---|
| Pathway databases | ||
| PathGuide | Comprehensive reference list of pathway-related databases and resources | http://www.pathguide.org/ 34 |
| STRING | (Meta-)database of physical and functional protein-protein interactions | http://string-db.org/ 60 |
| iHOP | Exploring gene/protein interaction networks by directly navigating through scientific literature | http://www.ihop-net.org/ 61 |
| GeneOntology (GO) | Comprehensive biological ontology database | http://geneontology.org 62 |
| KEGG – Kyoto Encyclopedia of Genes and Genomes | Provides pathway maps for biological interpretation | http://www.genome.jp/kegg/ 35 |
| Reactome | Manually curated open-data resource of human pathways and reactions | http://reactome.org 36 |
| ConsensusPathDB | Meta-database integrating functional interaction data from heterogeneous interaction data resources | http://consensuspathdb.org/ 37 |
| PID (Pathway Interaction Database) | Collection of curated and peer-reviewed pathways of human molecular signaling and cell processes | http://pid.nci.nih.gov/ 63 |
| Kinetic databases | ||
| Brenda | Information systems for functional and molecular properties of enzymes | http://www.brenda-enzymes.org/ 40 |
| Sabio-RK | Comprehensive information about biochemical reactions and their kinetic properties | http://sabio.villa-bosch.de/ 41 |
| Model databases | ||
| BioModels | Repository for mathematical models of biological processes | http://biomodels.org 38 |
| JWS online | Repository for kinetic models of biological systems that can be simulated and interrogated online. | http://jjj.biochem.sun.ac.za/ 39 |
| Chemical entity and drug databases | ||
| ChEBI | Database of chemical entities of biological interest | http://www.ebi.ac.uk/chebi/ 64 |
| ChEMBL | Open access large-scale bioactivity database | https://www.ebi.ac.uk/chembl/ 42 |
| PubChem | Public repository of biological activity data on small molecules and RNAi reagents | http://pubchem.ncbi.nlm.nih.gov 65 |
| Guide to pharmacology | Open access resource on pharmacological, chemical, genetic, functional and pathophysiological targets of approved and experimental drugs | http://www.guidetopharmacology.org/ 66 |
Major progress has also been achieved in the development of systems biology software to facilitate the establishment of detailed mechanistic models and to study the behavior of complex systems. Examples include CellDesigner43 and COPASI44; CellDesigner acts as a structured diagram editor for drawing biochemical and gene regulatory networks and enables simulations using an internal ODE solver or COPASI as a backend. In contrast, COPASI does not have a graphic diagram editor, but features stochastic and deterministic time course simulation functions and provides advanced methods for steady-state analysis, metabolic control analysis, parameter scanning, and parameter estimation.
More than 10 years ago, we started developing a web-based modeling system called PyBioS (http://pybios.molgen.mpg.de).45,46 PyBioS uses an object-oriented design, which allows the generation of large-scale models of cellular interaction networks comprising “objects” (eg, genes, proteins, protein modifications, and small molecules). Using PyBioS, we have established a large and comprehensive model of cancer-related signal transduction pathways and related processes leading to the development of the predictive ModCell™ systems biology modeling platform.46–48 ModCell™ is based on a mechanistic model of cancer-related cellular pathways and processes and currently integrates 45 different signal transduction pathway “modules” that represent the flow of information through the cellular signaling network. The model’s modular structure enables flexible extension of the network through integration of relevant information, eg, loss- or gain-of-function effects triggered by specific mutations in oncogenes/tumor suppressor genes, or drug action-specific effects. At present, 284 mutation-specific modules covering 116 different genes (a total of 728 genes are implemented in the model) and 108 drug- specific modules are part of the ModCell™ network, representing many of the known signaling pathways related to cancer.19,20 Updates to the network are made as part of an ongoing process as and when relevant information becomes available. ModCell™ incorporates only a fraction (~2.5%) of all human protein-encoding genes and, as yet, it does not include aspects of the immune system, metabolism, and other factors, such as the microbial milieu, that may influence the response to drugs. However, the approach taken does provide a large-scale signaling network and has been robust enough to identify patient-specific responses to miRNA-based treatments.48 Further work is ongoing to evaluate the validity and accuracy of the predictions (including within clinical scenarios) generated by ModCell™ and to address sensitivity and uncertainty issues. Examples of the signaling pathways, receptors, and ligands implemented within the model are described in detail in the study by Röhr et al.48
To carry out simulations, the model is converted into a system of ODEs, which can be solved numerically, providing predictions regarding the functional consequences of the molecular changes observed within a particular model. The system is seeded with known initial values of all model components as well as kinetic parameters used in the differential equations, allowing the fate of each object in the system to be tracked. In the context of cancer, the process of predicting a patient’s response to a drug or drug combination is a stepwise process involving: (i) adaptation of the existing ModCell™ generic cancer signaling network to incorporate additional pathway information relevant to the specific cancer being studied; (ii) validation of pathway integration; (iii) indi-vidualization of the network with omics data (eg, exome/transcriptome/proteome) generated from individual patient samples (germline and tumor). Inclusion of germline (control) data, eg, from a blood sample, allows identification of molecular data that are unique to the tumor; (iv) incorporation of relevant drug data, eg, mechanistic information and kinetic parameters; and (v) simulation of the effect of the drug(s) on a particular patient tumor using a molecular readout (eg, Myc levels, phosphorylation status of TP53, cleavage of PARP1, and GTP loading status of RAC1 and CDC42) as a proxy for phenotypic effects (eg, cell proliferation, senescence, apoptosis induction and cell migration; Fig. 1).
Figure 1.
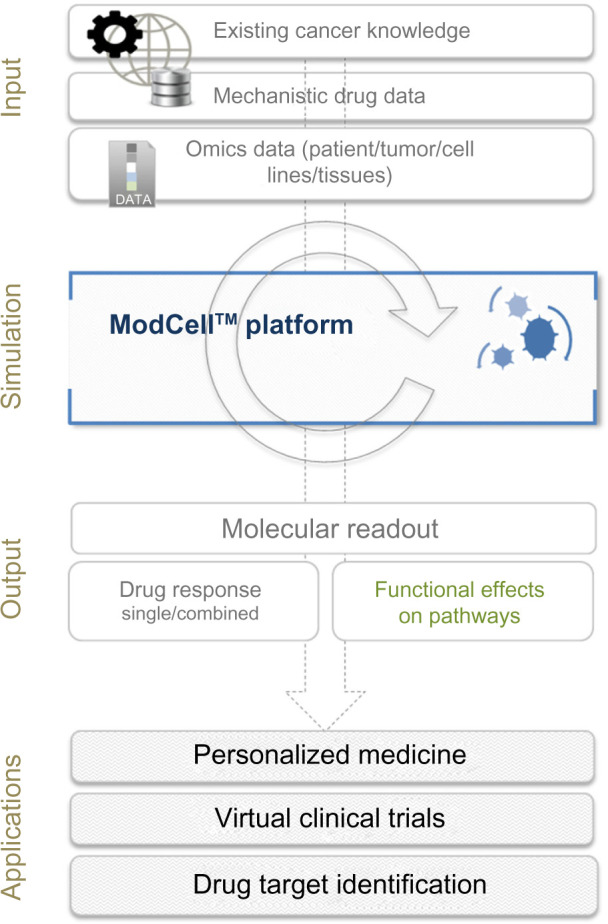
Overview of the ModCell™ predictive modeling approach in oncology. ModCell™ uses publicly available resources, representing the sum of knowledge on cancer, cell signaling, and drug action (eg, dissociation constants and molecular targets), to construct a large-scale mechanistic model of cellular signaling. A generic large-scale signaling network is established, which can be personalized with omics data (eg, transcriptome/exome/proteome) from individual patient tumors/cell lines/experimental tissues (public and/or private data resources). The effects of identified molecular alterations on pathway function and cross-talk can then be simulated using the mechanistic modeling approach implemented by ModCell™ and the underlying PyBioS modeling framework. Response to molecularly targeted drugs (single or in combination) can be predicted through establishment of a molecular readout (eg, MYC levels, phosphorylation status of TP53, cleavage of PARP1, and GTP loading status of RAC1 and CDC42) as a proxy for phenotypic effects (eg, cell proliferation, senescence, apoptosis induction and cell migration), allowing identification of the optimal treatment.
Due to the vast complexity of the biological basis of a disease and our incomplete understanding of disease mechanisms, mechanistic modeling of large-scale systems generates substantial networks with many unknown parameters. In the absence of accurate knowledge on parameter values, a Monte Carlo approach can be used to sample the unknown para meters from appropriate probability distributions.46 The Monte Carlo approach can take into account all existing knowledge on reaction kinetics and their parameters as well as any existing experimental data that can be linked to model parameters, eg, RNAseq expression data describing protein synthesis rates. Unknown reaction kinetics can subsequently be modeled by simple kinetic laws, such as mass action kinetics, and their parameters can be sampled from appropriate probability distributions. The Monte Carlo approach enables prediction of the consequences (some) of the integrated molecular alterations related to cancer, including pathway cross-talk, on a patient-by-patient basis. Proof-of-principle data to support the effectiveness of this approach have been provided using the PyBioS modeling and simulation system to predict the effects of perturbations induced by anticancer drug target inhibitors.46 The effects of single drugs and drug combinations on cell proliferation were simulated using models of the epidermal growth factor signaling network, constructed with and without relevant mutations. The predictions generated clearly aligned with evidence from the literature; for example, simulation results showed that models with a specific KRAS mutation did not have a predicted response to the drugs gefitinib and erlotinib,49 while models with a PIK3CA mutation were resistant to cetuximab.50
In tandem, to improve model capabilities, approaches that either allow model reduction or enable parameter estimation in the context of large-scale ODE systems are applied (or improved).51 These include novel optimization techniques, such as deployment of a memetic algorithm based on local search chains (MA-SSW-Chain),52 which seeks to minimize the difference between predicted and experimental data, as well as logically derived predictions. Agreement between prediction and observation will differ depending on the parameter vectors used; therefore, improvements in the choice of parameter space, informed by which regions provide better predictions, will help to increase the accuracy of the model.
Given that most parameters of intermediate-to-large-sized models of biological systems are usually unknown and their values might not be identifiable unambiguously on the basis of experimental data, the analysis of uncertainty and sensitivity can help to judge the reliability of the predictions made by a given model. Uncertainty analysis qualifies the model’s output as generated from ambiguities in parameter inputs. This enables quantification of the degree of confidence in the estimated parameters as derived from the experimental data. Uncertainty analysis can be performed using Monte Carlo sampling, in particular, the Latin hypercube sampling method,53 for efficient implementation. In addition, sensitivity analysis enables measurement of the impact of each model parameter on its variables. Due to nonlinearity of a model, this usually depends on the individual parameter vector.
We anticipate that the large-scale and fine-grained cancer network biology reflected in ModCell™, together with improved parameter optimization approaches applied to individual molecular cancer profiles, will provide a competitive edge over statistical methods. Early evaluation of the model’s capabilities has proved that it is robust enough to assess the potential of miRNAs as a therapeutic target in colon cancer and can identify patient-specific responses to miRNA-based treatments.48 In the study conducted by Röhr et al, analysis of miRNA expression profiles within normal, tumor, and metastatic tissues from eight colorectal cancer patients revealed a number of miRNAs that were constantly over- or underexpressed in tumor and metastatic tissues. One of the identified miRNAs, miRNA-1, was selected as representative, and the effects of depletion as well as overexpression, as a potential treatment, were simulated using the generic cancer model (covering more than 42 signaling pathways, plus associated ligands and receptors; refer the study by Röhr et al,48 for further details of model components). RNA expression levels (in normal, tumor, and metastatic tissues) from four patients were used to initialize individual-specific models. Each patient model was treated with different miRNA-1 concentrations and gene expression levels were compared between the tissue types. Gene expression levels were graded (from beneficial to negative effects), with those comparable to the normal state being positive, and patients who were likely to benefit from miRNA-1 treatments were identified. Ongoing research as part of a number of international and national projects (eg, OncoTrack, SYBIL, and EPITREAT) is further establishing the validity and accuracy of the model’s predictive capacity, and the results generated will be made available to the wider scientific community in due course. Clinical validation of ModCell™ is also underway as part of a project involving 11 clinical centers with the aim of optimizing therapy choice for patients with metastatic melanoma (TREAT20plus).
ModCell™ Applications
In the context of predicting clinical efficacy of specific therapies for individual patients through computer simulations, the deployment of the large-scale mechanistic approach used by ModCell™ opens up a number of possible applications, including personalized medicine and virtual clinical trial scenarios, as well as streamlining the drug discovery and development processes (Fig. 2).
Figure 2.
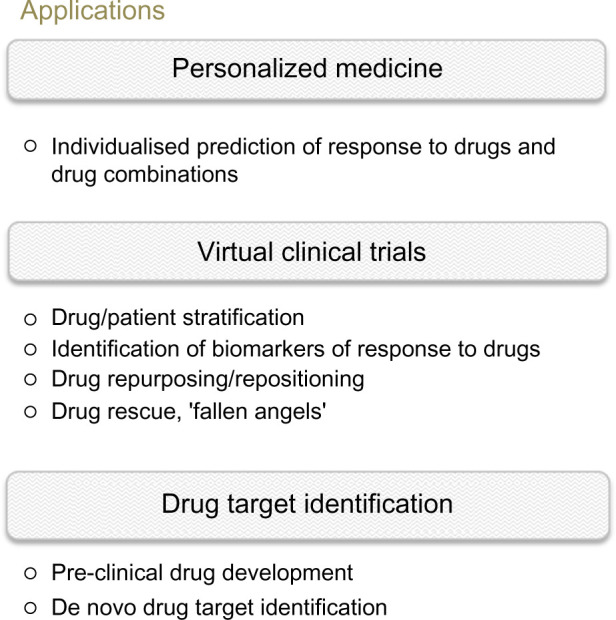
Applications of virtual patient modeling in oncology. The ability to predict the effects of drugs in silico opens up numerous avenues of application, from personalized medicine in the clinic to virtual clinical trial scenarios, enabling in silico testing of drug effects (single or combination) and potential side effects on individual or large patient (or preclinical model) cohorts. In virtual clinical trial scenarios, the patients who are most likely to benefit from a particular drug/drug combination can be selected, based on biomarkers identified, for inclusion in smaller, less risky, and less-expensive real-life clinical trials. A test bed is also created for assessing the efficacy of existing (drug repurposing) or failed drugs (‘fallen angels’), again providing a low risk and cost-effective route for further development. For early drug development, in silico models can be deployed for selecting the most relevant drugs/models for further development.
Personalized medicine
Given that the majority of cancer patients do not respond to first-line treatment,8 increasing the likelihood that treatments administered actually have beneficial outcomes would not only be a significant step toward improving patient welfare but also help to reduce the spiraling healthcare costs. In particular, due to the growing appreciation of the complexity of cancer, and the realization that multiple drugs with different mechanisms may provide more effective treatment options, there has been a shift from the “one-target-one-drug” approach to the application of multiple drugs.54 Determination of the right combination is, however, extremely cost and time intensive, even in preclinical settings. Implementation of ModCell™ for simulating patient responses to drug combinations provides a cost-effective and risk-attenuated approach for determining the optimal multi-targeted treatment strategy for individual patients. Due to the ongoing optimization of modeling algorithms, treatment decisions within the clinic could be supported by a tool that can operate essentially in real time, ie, within minutes once analyzed genome and/or transcriptome data are available.
Virtual clinical trials
Through upscaling of the application, virtual clinical trial scenarios are feasible. Virtual clinical trials can be conducted with virtual patient (or preclinical model) cohorts, each comprising multiple in silico models individualized with patient-generated and/or publicly available omics data. In such a scenario, predictions for a 1000-patient cohort with two drugs in combination would take days to model in silico, compared to months or even years required for a “real-life” clinical trial. Predictions generated by these models, plus potential biomarkers of responders/nonresponders derived in the analysis process, provide a robust starting point to tailor smaller, more rapid, and cost-effective targeted clinical trials. Such a stratification of the patient cohort prior to treatment, according to defined cancer bio-markers, allows the identification of responders and the most beneficial targeted treatment. As part of the validation process, ModCell™ is currently being deployed within a clinical trial scenario as part of the TREAT20plus project, aimed at optimizing therapy choices for patients with metastatic melanoma. Further clinical validation studies are currently in the planning phase.
Virtual patient technology and virtual clinical trials also provide many opportunities for streamlining the drug development and approval process, offering avenues for improving the current approval rates for newly discovered and already existing drugs. Prediction of responder groups from available omics data (public or private) can help to focus early drug development stages, guiding experimental validation in animal and cell line experiments. The technology can also be deployed in drug discovery projects to determine the effects of specific chemical inhibitors in vitro. In silico modeling based on whole genome and transcriptome data in a virtual (pre) clinical trial scenario can provide suggestions as to which cancer cell lines and cancers are most likely to respond to the chosen compounds.
Drug repositioning (or repurposing) and rescue (‘fallen angels’), a process of recycling with the aim of finding new therapeutic uses for existing drugs, is another promising area of application. Due to the fact that drugs can perturb multiple biological targets, each potentially being involved in multiple biological processes, there is scope to discover new cancer treatments within the large pool of drugs that already have approval for different diseases or that have failed clinical trials due to lack of efficacy rather than safety issues. Drug repositioning efforts span a continuum from purely serendipitous observations to targeted knowledge-based efforts that rely on more complex computational strategies.55,56 Recent efforts have highlighted the usefulness of network-based computational approaches for prediction of drug–target interactions, as well as network visualization of drug–target, target–disease, and disease–gene associations, to provide information that could facilitate discovery of new therapeutic indications or adverse effects associated with old drugs.57,58 In the context of ModCell™, information generated by such resources can be used to extend the model with further molecular pathway information, helping to define the topology of the model and reflecting additional kinase activities of interest.
An integrated approach to drug repositioning and rescue that incorporates the heterogeneity of patients and their disease and provides options for elucidating unknown disease mechanisms can be taken. By capitalizing on the knowledge already available on well-characterized drugs, including mechanisms of action, as well as the growing molecular information base on individual cancer patients generated by initiatives such as the TCGA and ICGC, new drug candidates can be selected for individual patients or patient cohorts. In this way, virtual patient models in oncology provide an ideal testing ground for drug repurposing, accelerating timelines, reducing risks to patients, and improving cost-effectiveness of the drug discovery and development pipelines.
Drug target identification
An enhanced understanding of the functional effects of disease-related molecular alterations on cellular signaling and subsequent gene regulation also provides opportunities for de novo drug target identification. Based on omics data and/or disease-specific mechanistic changes as well as disease-specific molecular readouts, the generic model of cellular pathways in ModCell™ can be adapted to a specific disease by defining appropriate molecular alterations. A subsequent systematic alteration of each model component or a comprehensive sensitivity analysis could provide a list of candidates to which the disease-specific readouts are most sensitive. These candidates could potentially be the most promising targets for subsequent drug development. Such an in silico prescreening process can help focus the drug target identification process and hence make the development of new drugs (eg, for orphan diseases) feasible and cost-efficient.
ModCell™ also holds promise for predicting off- target effects of selective drugs. Many cancer drugs target the selective expression of mutations within tumor tissues; for example, the activating BRAF V600E mutation that is often expressed in melanoma and colon cancer.59 Vemurafenib, a selective inhibitor of the BRAF V600E mutant protein, is effective in treating V600E-positive melanoma; however, it can also cause severe side effects due to off-target activation of the wild-type RAF-dependent signaling cascade. The combination of knowledge on the molecular mechanisms underlying the effectiveness of such inhibitors with in-depth analysis of an individual’s genetic background offers opportunities for ModCell™ to provide a more comprehensive analysis of drug effectiveness.
Future Perspectives
The application of virtual patient models such as ModCell™ opens up a number of novel avenues of application, from the development of companion tools to inform clinical decision making to positively impacting the drug development and approval pipeline for the pharmaceutical industry. In the medium to the long term, application of the virtual patient mechanistic modeling approach would provide the opportunity to implement a more systematic approach to the treatment of patients. Clinical decision making would be informed by a clinical companion tool that would categorize patients through an iterative and standardized decision-making pipeline, identifying patients likely to respond to a specific drug or drug combination and those at risk of developing side effects. In the first instance, this would be applicable within oncology, with the potential to expand the area of application to a broader disease (and drug type) panel, in particular, diseases with primarily genetic causes, eg, diabetes and vascular diseases.
Identification of more effective treatment options using single drugs repurposed for a new disease type or even using combinations of targeted drugs approved for the same disease could clearly benefit the patient in terms of effectiveness of the initial treatment regime and help to avoid multiple rounds of stressful and costly treatment cycles. In their present form, clinical trials assess the safety of a single drug with respect to concentration tolerance, side effects on healthy individuals, etc., whereas combinations of drugs are not rigorously assessed in the same way. Due to toxicity concerns, application of targeted drug combinations is often hindered by regulatory obstacles. ModCell™ can predict drug combinations that would make most sense with respect to therapeutic outcomes, but that may prove impractical as a treatment approach due to other issues, such as unforeseen toxic side effects. An expansion of phase 1 clinical trial design may be required to allow combinatory drug treatments with a broader scope than the current, “off label”/“compassionate use” of drugs within individual patients.
Virtual patient models in oncology – and beyond – are set to become an integral part of the normal clinical diagnosis and treatment process, increasing the likelihood of beneficial outcomes for patients and cost-effectiveness of already cash-strapped healthcare systems. Such technology not only holds promise for improving patient outcomes by focusing diagnoses and treatment decisions but through virtual clinical trials will also help to ensure that patients most likely to respond positively to a drug will be enrolled in clinical trials, with the associated impact for drug development pipelines. Virtual clinical trials also present opportunities to reduce or even abolish animal testing in preclinical drug developmental stages. In the future, virtual clinical trials will be an obligatory ethical responsibility, helping to avoid unnecessary suffering of both animals and patients.
However, concerted effort is still required to reap the benefits of the ongoing progress in computational modeling technologies and the accumulating knowledge base on disease processes at the level of the individual. Personalized, precision medicine, based on the systematic molecular characterization of patients and their diseases, requires development of the relevant technological infrastructure, comprising an improved environment for data handling, data processing, and data exchange, as well as the accompanying legal, regulatory, and educational framework.
Acknowledgments
The authors would like to thank their colleagues at Alacris Theranostics GmbH and the Dahlem Centre for Genome Research and Medical Systems Biology (DCGMS) for fruitful discussions and constructive criticism.
Footnotes
ACADEMIC EDITOR: J. T. Efird, Editor in Chief
PEER REVIEW: Nine peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1701 words, excluding any confidential comments to the academic editor.
FUNDING: The authors receive support from OncoTrack, an Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115234, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013) and EFPIA companies; SYBIL, funded by FP7/2007–2013 under grant agreement no. 602300; TREAT20plus (031 A512C) and EPITREAT (031619B), funded by the German Federal Ministry for Research (BMBF). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: LAO, CW, TK, and BML are employees, and HL is the founder and scientific director of Alacris Theranostics GmbH, Berlin, Germany. The company holds an exclusive commercial license for ModCell™.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Wrote the first draft of the manuscript: LAO. Contributed to the writing of the manuscript: LAO, CW, TK. Jointly developed the structure and arguments for the paper: LAO, CW, TK, HL, BMHL. Made critical revisions and approved the final version: LAO, CW, TK, HL, BMHL. All the authors reviewed and approved the final manuscript.
REFERENCES
- 1.Stewart Bernard W, Wild Christopher P., editors. World Cancer Report 2014. [Google Scholar]
- 2.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schulze K, Imbeaud S, Letouzé E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–11. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hovestadt V, Jones DTW, Picelli S, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510(7506):537–41. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- 7.Weischenfeldt J, Simon R, Feuerbach L, et al. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer Cell. 2013;23(2):159–70. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Chute CG, Ullman-Cullere M, Wood GM, Lin SM, He M, Pathak J. Some experiences and opportunities for big data in translational research. Genet Med. 2013;15(10):802–9. doi: 10.1038/gim.2013.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spear BB, Heath-Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med. 2001;7(5):201–4. doi: 10.1016/s1471-4914(01)01986-4. [DOI] [PubMed] [Google Scholar]
- 10.Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35(1):41–50. doi: 10.1016/j.tips.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 11.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 12.Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–83. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paplomata E, Nahta R, O’Regan RM. Systemic therapy for early-stage HER2-positive breast cancers: time for a less-is-more approach? Cancer. 2015;121(4):517–26. doi: 10.1002/cncr.29060. [DOI] [PubMed] [Google Scholar]
- 14.Gullo G, Zuradelli M, Sclafani F, Santoro A, Crown J. Durable complete response following chemotherapy and trastuzumab for metastatic HER2-positive breast cancer. Ann Oncol. 2012;23(8):2204–5. doi: 10.1093/annonc/mds221. [DOI] [PubMed] [Google Scholar]
- 15.Lièvre A Bachet J-B, D Le Corre, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 16.Wilson PM, Labonte MJ, Lenz HJ. Molecular markers in the treatment of metastatic colorectal cancer. Cancer J. 2010;16(3):262–72. doi: 10.1097/PPO.0b013e3181e07738. [DOI] [PubMed] [Google Scholar]
- 17.Wilson PM, Lenz HJ. Integrating biomarkers into clinical decision making for colorectal cancer. Clin Colorectal Cancer. 2010;9(suppl 1):S16–27. doi: 10.3816/CCC.2010.s.003. [DOI] [PubMed] [Google Scholar]
- 18.Seymour MT, Brown SR, Middleton G, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14(8):749–59. doi: 10.1016/S1470-2045(13)70163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 20.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Barretina J, Caponigro G, Stransky N, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubio-Perez C, Tamborero D, Schroeder MP, et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015;27(3):382–96. doi: 10.1016/j.ccell.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Ghahramani Z. Probabilistic machine learning and artificial intelligence. Nature. 2015;521(7553):452–9. doi: 10.1038/nature14541. [DOI] [PubMed] [Google Scholar]
- 25.Le Novère N. Quantitative and logic modelling of molecular and gene networks. Nat Rev Genet. 2015;16(3):146–58. doi: 10.1038/nrg3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelander S, Wang W, Nilsson B, et al. Models from experiments: combinatorial drug perturbations of cancer cells. Mol Syst Biol. 2008;4:216. doi: 10.1038/msb.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Jong H. Modeling and simulation of genetic regulatory systems: a literature review. J Comput Biol. 2002;9(1):67–103. doi: 10.1089/10665270252833208. [DOI] [PubMed] [Google Scholar]
- 28.Xu H, Ang Y-S, Sevilla A, Lemischka IR, Ma’ayan A. Construction and validation of a regulatory network for pluripotency and self-renewal of mouse embryonic stem cells. PLoS Comput Biol. 2014;10(8):e1003777. doi: 10.1371/journal.pcbi.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saez-Rodriguez J, Alexopoulos LG, Epperlein J, et al. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol Syst Biol. 2009;5:331. doi: 10.1038/msb.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, Kholodenko BN. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J Biol Chem. 2006;281(29):19925–38. doi: 10.1074/jbc.M600482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barua D, Hlavacek WS. Modeling the effect of APC truncation on destruction complex function in colorectal cancer cells. PLoS Comput Biol. 2013;9(9):e1003217. doi: 10.1371/journal.pcbi.1003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klinger B, Sieber A, Fritsche-Guenther R, et al. Network quantification of EGFR signaling unveils potential for targeted combination therapy. Mol Syst Biol. 2013;9:673. doi: 10.1038/msb.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirouac DC, Du JY, Lahdenranta J, et al. Computational modeling of ERBB2-amplified breast cancer identifies combined ErbB2/3 blockade as superior to the combination of MEK and AKT inhibitors. Sci Signal. 2013;6(288):ra68. doi: 10.1126/scisignal.2004008. [DOI] [PubMed] [Google Scholar]
- 34.Bader GD, Cary MP, Sander C. Pathguide: a pathway resource list. Nucleic Acids Res. 2006;34(Database issue):D504–6. doi: 10.1093/nar/gkj126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42(Database issue):D199–205. doi: 10.1093/nar/gkt1076. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Croft D, Mundo AF, Haw R, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):D472–7. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. Con-sensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39(Database issue):D712–7. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chelliah V, Juty N, Ajmera I, et al. BioModels: ten-year anniversary. Nucleic Acids Res. 2015;43(Database issue):D542–8. doi: 10.1093/nar/gku1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olivier BG, Snoep JL. Web-based kinetic modelling using JWS Online. Bioinformatics. 2004;20(13):2143–4. doi: 10.1093/bioinformatics/bth200. [DOI] [PubMed] [Google Scholar]
- 40.Chang A, Schomburg I, Placzek S, et al. BRENDA in 2015: exciting developments in its 25th year of existence. Nucleic Acids Res. 2015;43(Database issue):D439–46. doi: 10.1093/nar/gku1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wittig U, Kania R, Golebiewski M, et al. SABIO-RK-database for biochemical reaction kinetics. Nucleic Acids Res. 2012;40(Database issue):D790–6. doi: 10.1093/nar/gkr1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bento AP, Gaulton A, Hersey A, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42(Database issue):D1083–90. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Funahashi A, Matsuoka Y, Jouraku A, Morohashi M, Kikuchi N, Kitano H. CellDesigner 3.5: a versatile modeling tool for biochemical networks. Proc IEEE. 2008;96:1254–65. [Google Scholar]
- 44.Hoops S, Sahle S, Gauges R, et al. COPASI – a complex pathway simulator. Bioinformatics. 2006;22(24):3067–74. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
- 45.Wierling C, Herwig R, Lehrach H. Resources, standards and tools for systems biology. Brief Funct Genomic Proteomic. 2007;6(3):240–51. doi: 10.1093/bfgp/elm027. [DOI] [PubMed] [Google Scholar]
- 46.Wierling C, Kühn A, Hache H, et al. Prediction in the face of uncertainty: a Monte Carlo-based approach for systems biology of cancer treatment. Mutat Res. 2012;746(2):163–70. doi: 10.1016/j.mrgentox.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Henderson D, Ogilvie LA, Hoyle N, Keilholz U, Lange B, Lehrach H. Personalized medicine approaches for colon cancer driven by genomics and systems biology: OncoTrack. Biotechnol J. 2014;9(9):1104–14. doi: 10.1002/biot.201400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Röhr C, Kerick M, Fischer A, et al. High-throughput miRNA and mRNA sequencing of paired colorectal normal, tumor and metastasis tissues and bioinformatic modeling of miRNA-1 therapeutic applications. PLoS One. 2013;8(7):e67461. doi: 10.1371/journal.pone.0067461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jhawer M, Goel S, Wilson AJ, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68(6):1953–61. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK. Physicochemical modelling of cell signalling pathways. Nat Cell Biol. 2006;8(11):1195–203. doi: 10.1038/ncb1497. [DOI] [PubMed] [Google Scholar]
- 52.Molina D, Lozano M, García-Martínez C, Herrera F. Memetic algorithms for continuous optimisation based on local search chains. Evol Comput. 2010;18(1):27–63. doi: 10.1162/evco.2010.18.1.18102. [DOI] [PubMed] [Google Scholar]
- 53.Helton JC, Davis FJ. Latin hypercube sampling and the propagation of uncertainty in analyses of complex systems. Reliab Eng Syst Saf. 2003;81(1):23–69. [Google Scholar]
- 54.Al-Lazikani B, Banerji U, Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679–92. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 55.Jin G, Wong STC. Toward better drug repositioning: prioritizing and integrating existing methods into efficient pipelines. Drug Discov Today. 2014;19(5):637–44. doi: 10.1016/j.drudis.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J, Zheng S, Chen B, Butte AJ, Swamidass SJ, Lu Z. A survey of current trends in computational drug repositioning. Brief Bioinform. 2015 doi: 10.1093/bib/bbv020. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng F, Liu C, Jiang J, et al. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput Biol. 2012;8(5):e1002503. doi: 10.1371/journal.pcbi.1002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheng F, Jia P, Wang Q, Zhao Z. Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget. 2014;5(11):3697–710. doi: 10.18632/oncotarget.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–3. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 60.Jensen LJ, Kuhn M, Stark M, et al. STRING 8 – a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–6. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fernández JM, Hoffmann R, Valencia A. iHOP web services. Nucleic Acids Res. 2007;35(Web Server issue):W21–6. doi: 10.1093/nar/gkm298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaefer CF, Anthony K, Krupa S, et al. PID: the pathway interaction database. Nucleic Acids Res. 2009;37(Database issue):D674–9. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Matos P, Adams N, Hastings J, Moreno P, Steinbeck C. A database for chemical proteomics: ChEBI. Methods Mol Biol. 2012;803:273–96. doi: 10.1007/978-1-61779-364-6_19. [DOI] [PubMed] [Google Scholar]
- 65.Wang Y, Suzek T, Zhang J, et al. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D1075–82. doi: 10.1093/nar/gkt978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pawson AJ, Sharman JL, Benson HE, et al. The IUPHAR/BPS guide to pharmacology: an expert-driven knowledgebase of drug targets and their ligands. Nucleic Acids Res. 2014;42(Database issue):D1098–106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]