

Abstract
Despite that Vibrio spp. have a significant impact on the health of humans and aquatic animals, the molecular basis of their pathogenesis is little known, mainly due to the limited genetic tools for the functional research of genes in Vibrio. In some cases, deletion of target DNAs in Vibrio can be achieved through the use of suicide vectors. However, these strategies are time-consuming and lack universality, and the widely used counterselectable gene sacB does not work well in Vibrio cells. In this study, we developed universal genetic tools for rapid and efficient deletion mutations in Vibrio species based on suicide T-Vectors carrying a novel counterselectable marker, vmi480. We explored two uncharacterized genes, vmi480 and vmi470, in a genomic island from Vibrio mimicus VM573 and confirmed that vmi480 and vmi470 constitute a two-component toxin-antitoxin system through deletion and expression of vmi480 and vmi470. The product of vmi480 exhibited strong toxicity to Escherichia coli cells. Based on vmi480 and the PBAD or PTAC promoter system, we constructed two suicide T-vectors, pLP11 and pLP12, and each of these vectors contained a multiple cloning region with two AhdI sites. Both vectors linearized by AhdI digestion could be stored and directly ligated with purified PCR products without a digestion step. By using pLP11 and pLP12 coupled with a highly efficient conjugation system provided by E. coli β2163, six genes from four representative Vibrio species were easily deleted. By using the counterselective marker vmi480, we obtained 3–12 positive colonies (deletion mutants) among no more than 20 colonies randomly selected on counterselection plates. The strategy does not require the digestion of PCR products and suicide vectors every time, and it avoids large-scale screening colonies on counterselective plates. These results demonstrate that we successfully developed universal genetic tools for rapid and efficient gene deletion in Vibrio species.
Introduction
Vibrio comprises at least 89 species with validly published names [1]. It is one of the most common bacterial groups in marine environments [1,2]. Although Vibrio spp. are of great importance for the remineralization of organic matter in the sea [3], the foremost attention brought to this genus is related to its many pathogenic strains. Until now, at least 12 Vibrio species have been reported to be pathogenic to human beings [4], among which V. cholerae, V. parahaemolyticus and V. vulnificus are the most notorious due to their significant threat to human health and seafood safety [4–6]. Some members of Vibrio are also causative pathogens for aquatic animals, which often cause enormous economic loss [7–9]. In contrast, some species such as V. alginolyticus have even been reported as probiotics for shrimp aquaculture [10,11].
Despite that Vibrio spp. has a significant impact on the health of humankind and aquatic animals, the molecular basis of their pathogenesis is little known, mainly due to the limited genetic tools available for the functional research of genes in most pathogenic Vibrio species [12]. Generally, gene deletion coupled with its complementation is one direct way to assess the function of a gene of interest. One of the most widely used methods is allelic exchange between a target gene and a mutation fragment carried by a suicide vector (plasmid) harboring an antibiotic-resistant cassette and a counterselectable gene [13]. In this way, a suicide vector is driven into recipient cells through conjugation. Under selective stress from an antibiotic environment, recipient cells and the plasmid cannot survive unless the plasmid integrates into a specific site through homologous recombination [14]. As allelic exchange mutants may represent only a small fraction of the transformants and may be difficult to isolate, a counterselectable marker is often instrumental for the acquisition of a deletion mutant [13,14]. In the presence of the counterselective compound, a counterselectable gene promotes the death of the microorganisms harboring this gene, and only the deletion mutant losing the integrated form of the vector through a second homologous recombination and the resultant wild-type clones can survive [14,15].
The Bacillus subtilis levansucrase gene sacB is now certainly the most commonly used of the different counterselectable markers due to its general efficiency in Gram-negative bacteria and for the simplicity of the counterselection protocol [12,16]. sacB-based suicide vectors have been occasionally used for allelic exchange in several Vibrio species, such as V. cholerae [16,17], V. anguillarum [18], and V. alginolyticus [19]. However, the use of this gene for allelic exchange in more Vibrio species or in other common bacterial species is seriously impeded by the necessary absence of NaCl in the counterselection medium [12]. Milton et al. reported that although they obtained a null flaA mutant of V. anguillarum through the use of a sacB-based suicide vector, pDM4, sacB did not work well, as both colonies lacking the flaA gene and colonies maintaining the integrated vector occurred on sucrose-containing plates [18]. In another case of a deletion mutation in V. vulnificus by using a sacB-based suicide vector, counterselection of sucrose sensitivity was not very efficient, as it was time-consuming and involved the screening of large numbers of colonies to find truly sucrose-resistant colonies [20]. In our laboratory, sacB-based pDM4 was also used for a deletion mutation in V. alginolyticus [19], but it is troublesome to arduously screen deletion mutants from numerous background false-positive colonies (internal communication). These cases strongly support the idea that sacB is not an ideal counterselectable marker for allelic exchange in halophilic Vibrio. Therefore, it is necessary to explore new counterselectable markers for genetic manipulation in Vibrio.
In 2005, Demarre et al. developed a series of pSW suicide plasmids and their cognate E. coli host strains. These plasmids have small sizes (without mob genes) and lack identity with any bacterial chromosome gene [21], which apparently enhances the transfer efficiency of the plasmids and largely reduces incorrect integration. ΔdapA and ΔthyA E. coli strains facilitate the counterselection if they are used in plasmid transfer experiments into markerless recipients [21]. Demarre et al. also verified the high conjugation–recombination frequency when this recombineering system was applied to allelic replacement of V. cholerae [21]. However, all of these plasmids lack the second necessary counterselectable markers, which limit their further application in complete gene knockout in Vibrio.
In the present study, we discovered that two uncharacterized genes, vmi480 (VMD_06480) and vmi470 (VMD_06470), in a V. mimicus strain VM573 (NZ_ACYV01000005) formed a new toxin-antitoxin (TA) system. The toxin gene vmi480 was introduced into a suicide plasmid constructed from pSW23T and pSW25T-ccdB. Finally, we obtained two novel suicide T-Vectors, pLP11 and pLP12, carrying the vmi480 gene as a counterselectable marker. Through the use of pLP11 and pLP12 coupled with a high-efficiency conjugation system provided by host strain E. coli β2163, we easily deleted six genes in V. alginolyticus, V. cholerae, V. parahaemolyticus, and V. vulnificus. Therefore, our results demonstrated that we successfully developed universal genetic tools for a rapid and efficient deletion mutation in Vibrio species.
Results
Functional Prediction of vmi480 and vmi470
Previously, we obtained an E. coli strain LP79 harboring a genomic island, MGIVmi1, from V. mimicus VM573 through conjugation [22]. MGIVmi1 includes two adjacent genes, vmi480 and vmi470, on the same strand. There is a 5-bp short intergenic region separating two genes, while the genes flanking vmi480 and vmi470 are located on the opposite strand and maintain 600-bp and 73-bp intervals from the vmi480-vmi470 locus. There is an intact promoter region upstream of vmi480 (Fig 1A). These features indicated that vmi480 and vmi470 belonged to the same transcriptional unit and were likely functionally related. Blastx searches revealed that vmi470 encoded a DNA-binding protein; however, conserved domain analysis showed that the product of vmi470 had a Zn-dependent peptidase domain at the C-terminal region besides a DNA-binding helix-turn-helix domain at the N-terminal region (Fig 1B). Blastx searches also revealed that vmi480 coded for a hypothetical protein with unknown function and that genes similar to them could only be found in V. mimicus SX-4, V. anguillarum 775, a strain from V. cholerae and a strain from V. ordalii. In spite of the lack of similarity between two genes and reported toxin-antitoxin genes, the genetic configuration in which vmi480 and vmi470 are tightly linked in an operon resemble the findings observed for most type II toxin-antitoxin (TA) systems [23,24]. Then web-based TA prediction was carried on vmi480 and vmi470. The result showed that vmi480 failed to match any identified toxin genes in database through conserved domain analysis; however the product of vmi470 contains a DNA-binding helix-turn-helix domain shared by several antitoxins.
Fig 1. Features of vmi480 and vmi470.

(a) Schematic diagram of a potential toxin-antitoxin system comprising vmi480 and vmi470. vmi480 and vmi470 are located on the same strand and are separated from flanking genes, mobI and res2. There is a 5-bp intergenic region between vmi480 and vmi470. Upstream of vmi480, there is a promoter region for vmi480 and vmi470. These features suggest that vmi480 and vmi470 belong to one transcriptional unit and that they are functionally related. The bending arrow represents the position of the promoter, and the white letters on the black background represent the -10 and-35 regions of the promoter. (b) Conserved domain analysis of Vmi470. At the N-terminal of Vmi470, there is a DNA-binding helix-turn-helix domain, and at the C-terminal of Vmi470, there is a Zn-dependent peptidase domain.
vmi480 and vmi470 Constitute a Toxin-Antitoxin Module
To further explore the functions of vmi480 and vmi470, we attempted to delete them in E. coli through one-step inactivation based on the λ recombination system [25]. We easily deleted vmi480 or both genes but we failed to solely delete vmi470 in three attempts, which suggest that retaining vmi480 without vmi470 may be poisonous to the cells. It raised a concern that vmi480 and vmi470 may form a TA pair. vmi470, vmi480 or both genes were further cloned into the expression vector pBAD30. Transformants of E. coli NEB 5α hosting pBAD30-vmi480-470 (LP134) and pBAD30-vmi470 (LP135) were easily constructed. In the case of the strain LP134, co-expression of vmi480 and vmi470 under the control of PBAD promoter can be achieved as they locate on the same transcription unit. However, a transformant of E. coli NEB 5α hosting pBAD30-vmi480 could not be constructed in the initial attempts. This transformant (LP192) was finally constructed by using Luria-Bertani (LB) plates supplemented with D-glucose that blocked the basic expression of vmi480. The results showed that basic expression of vmi480 probably caused some stress on recovered cells, which prevented the formation of the transformant. When blocked by D-glucose or induced by L-arabinose, LP134 (pBAD30-vmi480-470) and LP135 (pBAD30-vmi470) grew well, and no differences were observed between block and induction treatment (Fig 2). When blocked by D-glucose, LP192 (pBAD30-vmi480) grew well, but when inactivated by L-arabinose, LP192 did not grow, as no clones were discovered (Fig 2).These results indicated that sole expression of vmi480 is lethal to the cells but the lethal effect can be eliminated when vmi480 and vmi470 are co-expressed in the same vector. It demonstrated that Vmi480 serves as a type of toxin that has a strong lethal effect, while Vmi470 is the antidote of Vmi480. Coupled with the above genetic analysis, the success of deleting vmi480 or vmi480-vmi470, and the failure of deleting vmi470, these results demonstrate that vmi480 and vmi470 compose a two component TA module. An outline of these evidences was shown in Table 1. The strong lethality of vmi480 product establishes a foundation for applying it as a counterselective marker.
Fig 2. The effect of the expression of vmi480 and vmi470 on the growth of E. coli cells.

a, b, c: expression of vmi480-470 (LP134), vmi470 (LP135), and vmi480 (LP192) were blocked by D-glucose (0.3%), respectively. d, e, f: expression of vmi480-470 (LP134), vmi470 (LP135), and vmi480 (LP192) were activated by L-arabinose (0.2%), respectively.
Table 1. An outline of evidences that vmi480 and vmi470 constitute a TA module.
| Aspects | Evidences |
|---|---|
| Genetic analysis | 1. vmi480 and vmi470 constitute an operon with their own promoter and their genetic configuration resemble those of most type II TA systems. |
| 2. vmi470 contains a DNA-binding helix-turn-helix domain shared by several antitoxins. | |
| Deletion mutants | 3. Deletion of vmi480 or vmi480-vmi470 can be achieved while deletion of sole vmi470 cannot fulfill. |
| Ectopic expression | 4. Sole expression of vmi480 is lethal to the cells. |
| 5. Sole expression of vmi470 has no effect on cell growth. | |
| 6. Lethality of Vmi480 can be eliminated when both genes are co-expressed in one vector. |
Suicide T-Vectors pLP11-T and pLP12-T for Gene Disruption in Vibrio
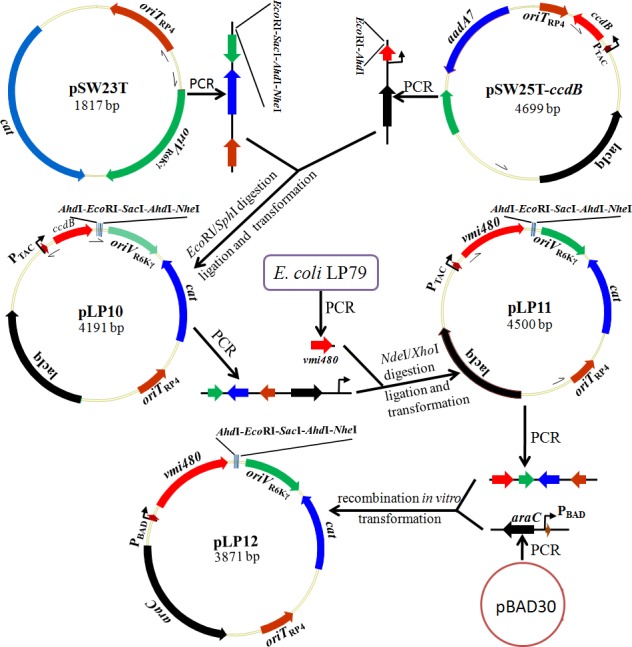
As shown in Fig 3, we first constructed a derivative plasmid pLP10. pLP10 consists of a fragment containing an R6K origin of replication (oriV R6Kγ), an RP4 origin of transfer (oriT RP4) and a chloramphenicol-resistant gene (cat) from pSW23T, and a fragment containing a lethal counterselectable gene ccdB and a PTAC promoter system from pSW25T-ccdB. A multiple cloning site (MCS, AhdI-EcoRI-SacI-AhdI-NheI) was introduced into pLP10 by primers pSW23T-F and pSW25T-F. The ccdB gene in pLP10 was replaced by vmi480 to generate a plasmid pLP11, and then, the PTAC promoter system in pLP11 was replaced by the PBAD promoter system to generate a new plasmid, pLP12. Therefore, the only difference between pLP11 and pLP12 is that the expression of vmi480 is controlled by the PTAC promoter (pLP11) or by the PBAD promoter (pLP12). Suicide T-vectors, pLP11-T and pLP12-T, were obtained by the amplification of pLP11 and pLP12 following AhdI digestion, which led to each T-vector having single base 3'-T overhangs on both ends. Thus, the overlap of the PCR products of target genes can be purified and directly ligated into linearized pLP11-T or pLP12-T through common TA cloning without digestion steps of overlap PCR products.
Fig 3. Schematic diagram of the construction of suicide plasmids pLP11 and pLP12.
In-Frame Deletion of hem in V. alginolyticus
To test the feasibility of our strategy for gene knockout in Vibrio species, the hem gene (coding hemolysin) from V. alginolyticus E0601 was first selected as a target gene (Fig 4). Both suicide T-vectors pLP11-T and pLP12-T were used for hem knockout. The result showed that both pLP11-hem and pLP12-hem could integrate into a chromosomal site to form insertional mutants as PCR tests using an external primer hem-TF (anchoring upstream of integration site) and an internal primer pLP-UR (anchoring one vector-specific region) revealed that insertional mutants generated one predicted 946-bp band while wild-type E0601 could not as pLP-UR failed match any sites of genomic DNAs from E0601 (only the results from the pLP11-based knockout were shown in Fig 5, as both plasmids aimed at the same gene and led to an identical result). After counterselection driven by toxicity of Vmi480, both suicide vectors could lead to the generation of deletion mutants, as deletion mutants gave rise to one truncated PCR band (1141 bp) while wild-type E0601 gave rise to a normal PCR band (1699 bp) when using two external primers respectively targeting upstream and downstream of hem gene (Fig 4). The sequencing of PCR products also confirmed that insertion and deletion mutants were successfully obtained. In the counterselection step, 16 colonies resulting from strain LP204 (integrated pLP11-hem) and strain LP206 (integrated pLP12-hem) were randomly selected for a PCR test; 3 and 12 colonies were confirmed to be the deletion mutants of the hem gene. A higher occurrence of deletion mutants was observed when using the pLP12 plasmid.
Fig 4. Schematic diagram of wild type, insertional mutation and deletion mutation of targeted genes.

Targeted genes are shown with light gray arrows and their adjacent genes are shown with black arrows. Black triangles represent annealing sites of external or internal primers. The gene names prefixed with “T-” represent the names of the truncated genes.
Fig 5. PCR confirmation of insertional disruption and deletion of six genes in four Vibrio species.
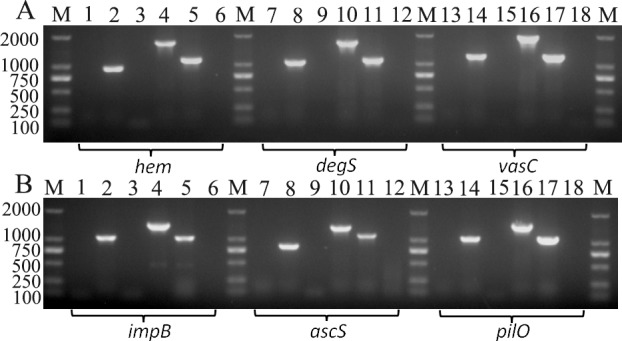
(A) Lane M: DNA marker DL2000; lanes 1–3: PCRs using primers hem-TF/pLP-UR to test wild type, insertional disruption and negative control (water as a template) of hem in V. alginolyticus E0601; lanes 4–6: PCRs using primers hem-TF/hem-TR to test wild type, deletion mutant and negative control (water as a template) of hem in V. alginolyticus E0601 (PCR tests for insertional disruption and deletion of hem using pLP11 and pLP12 had the same results, so only the results from pLP11 were shown); lanes 7–9: PCRs using primers degS-TF/pLP-UR to test wild type, insertional disruption and negative control (water as a template) of degS in V. cholerae HN375; lanes 10–12: PCRs using primers degS-TF/degS-TR to test wild type, deletion mutant and negative control (water as a template) of degS in V. cholerae HN375; lanes 13–15: PCRs using primers vasC-TF/pLP-UR to test wild type, insertional disruption and negative control (water as a template) of vasC in V. cholerae HN375; lanes 16–18: PCRs using primers vasC-TF/vasC-TR to test wild type, deletion mutant and negative control (water as a template) of vasC in V. cholerae HN375. (B) Lane M: DNA marker DL2000; lanes 1–3: PCRs using primers impB-TF/pLP-UR to test wild type, insertional disruption and negative control (water as a template) of impB in V. vulnificus ATCC 27562; lanes 4–6: PCRs using primers impB-TF/impB-TR to test wild type, deletion mutant and negative control (water as a template) of impB in V. vulnificus ATCC 27562; lanes 7–9: PCRs using primers ascS-TF/pLP-UR to test wild type, insertional disruption and negative control (water as a template) of ascS in V. parahaemolyticus E06135; lanes 10–12: PCRs using primers ascS-TF/ascS-TR to test wild type, deletion mutant and negative control (water as a template) of ascS in V. parahaemolyticus E06135; lanes 13–15: PCRs using primers pilO-TF/pLP-UR to test wild type, insertional disruption and negative control (water as a template) of pilO in V. parahaemolyticus E0680; lanes 16–18: PCRs using primers pilO-TF/pilO-TR to test wild type, deletion mutant and negative control (water as a template) of pilO in V. parahaemolyticus E0680.
In-Frame Deletion of degS and vasC in V. cholerae
To test the feasibility of the genetic tool for gene knockout in V. cholerae, pLP12-T was used for deletion mutation of degS (coding periplasmic serine peptidase) and vasC (coding uncharacterized protein in type VI secretion system) in V. cholerae HN375 (Fig 4). As shown in Fig 5, pLP12-degS was integrated into the chromosome, as it led to a 1047-bp PCR fragment when using an external primer degS-TF (anchoring upstream of integration site) and the internal vector-specific primer pLP-UR. However, wild-type HN375 could not produce any band when using the same primer pair due to the lack of annealing site by the primer pLP-UR. Under counterselection pressure, the integrated plasmid was removed through a second homologous recombination to form the deletion mutant, which produced a short 1126-bp PCR fragment in the deletion mutant while the wild-type strain HN375 only resulted in a normal band with a size of 1696 bp when using two external primers degS-TF/degS-TR. As expected, the insertional mutant of vasC generated by pLP12-vasC led to a 1224-bp PCR fragment but the wild-type strain HN375 could not. The deletion mutant of vasC led to a short 1226-bp PCR product while wild-type strain HN375 only generated a normal 1931-bp band. Insertional and deletion mutants of degS and vasC were also confirmed by subsequent sequencing of PCR products. In the counterselection step, 16 colonies resulting from integrated pLP12-degS or pLP12-vasC were randomly selected for a PCR test; 8 and 7 colonies were confirmed to be the deletion mutants for degS and vasC, respectively.
In-Frame Deletion of pilO and ascS in V. parahaemolyticus
pLP12-T was also used for deletion mutation of pilO (coding type IV pilus biogenesis protein) and ascS (coding preprotein translocase S) in V. parahaemolyticus (Fig 4). To verify that the method can be used in different strains of the same Vibrio species, we aimed to delete pilO in V. parahaemolyticus E0680 and ascS in V. parahaemolyticus E06135. As shown in Fig 5, the result revealed that pLP12-pilO integrated into the chromosomal site of E0680 to generate a 1062-bp PCR fragment as expected, while the wild-type strain didn’t result in an amplicon when using primers pilO-TF and pLP-UR. After counterselection, the integrated plasmid was removed through a second homologous recombination, and it generated a short 835-bp PCR product representing in-frame deletion. Compared with deletion mutants, wild-type E0680 gave rise to a normal PCR product of 1491 bp when using the same primer pair. Knockout of ascS in E06135 had the same outcome. An insertional mutant of ascS resulted in one 835-bp PCR fragment and an deletion mutant of the gene resulted in truncated PCR fragment of 1108 bp compared with a normal PCR fragment of 1357 bp from the wild-type strain. PCR results were confirmed by the following sequencing. In the counterselection step, 16 colonies resulting from LP246 (integrated pLP12-pilO) or LP248 (integrated pLP12-ascS) were randomly selected for a PCR test; 7 and 3 colonies were confirmed to be the deletion mutants of pilO and ascS, respectively.
In-Frame Deletion of impB in V. vulnificus
To test the feasibility of the genetic tool for gene knockout in V. vulnificus, impB (coding uncharacterized protein in type VI secretion system) in V. vulnificus ATCC 27562 was selected as target gene (Fig 4). As shown in Fig 5, pLP12-impB integrated into the desired chromosomal site to generate a 1061-bp PCR fragment, while the wild-type strain didn’t. After counterselection, PCR for the clone representing the deletion mutant generated a short 1045-bp DNA fragment as expected, while the wild-type strain generated a normal 1426-bp DNA fragment when using the same external primer pair. The PCR results were confirmed by the following sequencing. In the counterselection step, 20 colonies were randomly selected for a PCR test; 6 colonies resulting from integrated pLP12-impB were confirmed to be the deletion mutants. Thus, the results demonstrated that deletion of impB was successfully achieved.
Discussion
Though there are several successful cases of using sacB-based suicide vectors for gene knockout in Vibrio [e.g., 16–19], SacB toxicity is susceptible to the presence of NaCl in the selective medium [26], while the addition of NaCl to the media is absolutely necessary for the growth of nearly all Vibrio species [27].The inadequate toxicity of SacB must largely decrease the occurrence ratio of the correct deletion mutants on counterselective plates, which inevitably increases the difficulty of screening. Therefore, the lack of a suitable counterselective marker has actually become a primary obstacle to genetic manipulation in Vibrio. In 2007, Roux et al. constructed a deletion mutant of the vsm gene in V. splendidus by using a ccdB-based suicide vector [12]; that study shed some light on the application of a new counterselective marker in the genetic manipulation of Vibrio species. However, there is still concern about whether CcdB originated from the F plasmid of E. coli, which has extensive and strong toxicity in many other Vibrio species because CcdB poisons GyrA (the subunit of DNA gyrase) by forming a binding complex [28] while GyrA proteins from Vibrio are only distantly related with those from E. coli strains (comparison by Blastp). In the discrepant intracelluar environment of Vibrio (halophilic bacteria), the affinity of CcdB to GyrA in Vibrio is likely not as strong as it is in E. coli. In light of these facts and considerations, exploring new alternative marker genes from rare Vibrio strains could help develop genetic tools for gene knockout in Vibrio.
First, we aimed to explore the function of vmi480 and vmi470 because a Blastx search suggested that vmi470 codes a DNA-binding protein and its orthologous proteins includes several transcription factors. Thus, we doubted whether vmi470 plays an important role in the genomic island, MGIVmi1. In another aspect, vmi480 and vmi470 constitute an operon with their own promoter and their genetic configuration resemble those of most type II TA systems [23, 24], and thus it also raised a question whether they represent a TA module. In most cases for identification of new TA pairs, gene deletion mutation in vivo and ectopic expression of TA genes are key procedures besides theoretical prediction [24,29]. In our case, the attempt to construct a null mutant of a single vmi470 failed, while deletion of vmi480 and deletion of vmi470 and adjacent vmi480 together were easily achieved. The features of the conserved domains of vmi470 are similar to antitoxins CcdA and ParD because both of them have a DNA-binding domain at their N-terminal region and have a catalytic domain at their C-terminal region that reacts with their toxins, CcdB and ParE, respectively [30–32]. This suggests that vmi480 and vmi470 probably serve as a TA system. Sole expression of vmi480 was lethal to the cells but the lethal effect could be eliminated when vmi480 and vmi470 were co-expressed in the same vector. It clearly showed that the product of vmi470 serves as an antidote of Vmi480. Generally, co-expression of toxin and antitoxin gene is achieved through adopting two compatible vectors and using different inducers [33–35]. But in our case, we take the full advantages of the genetic structure that not only vmi480 and vmi470 are tightly linked (only a 5-bp intergenic region) but also candidate toxin gene vmi480 locates upstream of candidate antitoxin vmi470. It ensures that both genes can be expressed in one expression vector. Suppose that the downstream vmi470 in the same transcription unit cannot be expressed, Vmi480 will kill cells without the participation of Vmi470. Compared with most of type IITA system, vmi480-vmi470 exhibits two distinct characteristics. One is that candidate toxin consisting of 198 amino acids is encoded by the first gene; this organization is opposite to the widespread TA gene order but occurs in several TA systems such as higBA [29,35], hicAB [36] and mqsR-ygiT [24]. Another is that vmi470 code a candidate antitoxin with a big size of 362 amino acids. Generally, toxin and antitoxin have the small sizes (31–204 amino acids for antitoxins and 41–206 amino acids for toxins) [23]. However, vmi470 is not the only exception: gene VC0815 from a TA system of V. cholerae even codes a toxin with much bigger size of 453 amino acids [34]. All these results and analysis well support the idea that vmi480 and vmi470 compose a new TA system. These findings actually diversify the TA families and expand our knowledge on TA systems.
Vmi480 exhibited a strong lethal effect to E. coli cells, and it suggests that although this toxin originates from Vibrio, it may have toxicity to bacterial cells from extensive sources. In another aspect, vmi480 and vmi470 only exist in rare Vibrio strains, which guarantee that vmi480 is suitable as a counterselection marker for gene deletion in Vibrio in most cases. These features of vmi480 and its product Vmi480 suggest that the vmi480 gene can be used as an ideal counterselection marker, especially in the construction of suicide vectors. Gaining insight into the mechanism of vmi480 and vmi470 is currently ongoing.
Consequently, we constructed suicide vectors based on the plasmid pSW23T and the toxin gene vmi480 and attempted to use them in the deletion mutation in Vibrio strains. V. cholerae, V. parahaemolyticus and V. vulnificus were selected because they are the most harmful Vibrio pathogens to humans [4]; thus, there is a more urgent need to develop convenient tools for their genetic manipulation. On the other hand, V. alginolyticus widely distributes in estuary and marine environments [37,38], and this bacterium has garnered increased concern, as some strains have been reported as pathogenetic to aquatic animals and have caused huge economic losses [4,37,39]. Therefore, V. alginolyticus was also selected as a desired target for attempted gene knockout. We easily obtained six deletion mutants in five strains from these Vibrio species by using new suicide plasmids, pLP11 and pLP12, and the highly efficient conjugation system developed by Demarre et al. [21]. In this conjugation system, donor strain E. coli β2163 not only contains the transfer apparatus to drive suicide plasmids but also cannot grow on LB plates without thymidine and diaminopimelic acid (DAP) [21]. Therefore, it can minimize the sizes of suicide plasmids and does not require selective markers from the recipient cells [21]. By using this conjugation system coupled with pSW27, V. cholerae suicide transconjugants were obtained with an overall conjugation–recombination frequency of approximately 2× 10−7 transconjugants per recipient [21]. In our experiments, we could obtain 30–70 colonies (suicide transconjugants) on each plate (100 dilution) every time in the first selection (data not shown), which was sufficient to perform the following counterselection. Furthermore, in the counterselection step, we could obtain 3–12 positive colonies (deletion mutants) by randomly screening no more than 20 colonies. The high occurrence of deletion mutants is primarily due to the expression of vmi480 under induction conditions. As a result, the application of vmi480 actually saves time for the screening of deletion mutants.
In the conventional strategies of gene deletion through suicide vectors, PCR products and suicide vectors are generally digested by the same restriction enzymes and are then purified before ligation. Occasionally, altering the cloning sites must be performed due to the difference in sequences of inserted DNA fragments. Therefore, the operations are somewhat laborious. To overcome these flaws, we introduced double AhdI sites in MCS and digested pLP11 and pLP12 with AhdI to generate suicide T-vectors. This strategy does not require consideration of restriction sites and evades the laborious steps of digestion and the following purification. In this strategy, purified PCR products are directly ligated with linearized suicide T-vectors. Once linearized suicide T-vectors are prepared, they can be stored and used repeatedly. Therefore, this accelerates the process of gene knockout based on suicide vectors. We do not need to consider the insertional orientation of allelic DNA fragments in suicide vectors because no matter how they are inserted, the suicide vectors carrying allelic DNA fragments will integrate into the desired chromosomal sites in the correct direction due to the homologous recombination mechanism, which will not influence the function of suicide vectors. Of course, we still retain MCS sites in two vectors to meet the preference of different users.
The expression of target genes under the tight control of the PBAD promoter from E. coli is induced by L-arabinose [40]. To date, the PBAD promoter expression system has been used in many Gram-negative bacteria, such as E. coli, Salmonella typhimurium and Xanthomonas sp. [41]. PBAD-based suicide vectors or expression vectors were also successfully used in some Vibrio species [12,42–44], which suggested that the permeability of L-arabinose may not be a problem in Vibrio species. However, there are few reports that PTAC-based suicide vectors or expression vectors are used in Vibrio cells. Furthermore, the low permeability of isopropyl β-D-1-thiogalactopyranoside (IPTG) has been observed in Corynebacterium glutamicum strains, which hampers the application of a PTAC-based induction system in this bacterium [41]. It is well known that IPTG acts as an analogue of lactose in expression systems based on the lac operon; lactose is mainly transported through lactose permease on the cell membrane of E. coli [45,46]. However, no lactose permease has been discovered in Vibrio to date. This suggests that induction from IPTG in Vibrio may not be as efficient as induction in E. coli. In our case, the loss of integrated pLP11 was not observed in V. alginolyticus when using 1 mM of IPTG (usual dosage for induction) to induce the expression of vmi480 (data not shown) until we increased the dosage of IPTG to 5 mM. Given these considerations, although we successfully developed two suicide plasmids carrying vmi480 and different promoter systems, we prefer to use the suicide plasmid pLP12 containing the PBAD promoter activated by L-arabinose.
Finally, we must note that although our genetic tools were designed to be used in gene disruption in Vibrio, they likely have the potential to be applied in other Gram-negative bacteria because the toxicity of Vmi480 may be broad-spectrum to various bacteria and donor strain E. coli β2163 can conjugate with a wide range of Gram-negative bacteria [21].
Conclusions
In this study, we confirmed that vmi480 and vmi470 in a genomic island, MGIVmi1, from V. mimicus VM573 belong to a type of two-component toxin-antitoxin system through their knockout and expression. vmi480 is an uncharacterized toxin gene. We constructed two suicide T-vectors featuring the toxin gene vmi480, a PBAD or PTAC promoter system, and a MCS region including two AhdI sites. Linearized vectors by AhdI digestion can be stored and directly ligated with purified PCR products without digestion steps. Using two suicide T-vectors coupled with a highly efficient conjugation system from E. coli β2163, we easily deleted six genes from four representative Vibrio species. Through the use of counterselective marker vmi480, we could obtain 3–12 positive colonies (deletion mutants) among no more than 20 randomly selected colonies on counterselection plates. These results demonstrated that we successfully developed universal genetic tools for rapid and efficient gene deletion in Vibrio species.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Conditions
The bacterial strains and plasmids used in this study are listed in Table 2. E. coli and Vibrio strains were cultured in LB medium. DAP were supplemented to a final concentration of 0.3 mM when necessary. Antibiotics were used at the following concentrations: nalidixic acid (Nx), 40 μg/ml; ampicillin (Ap), 100 μg/ml; spectinomycin (Sp), 50 μg/ml; kanamycin (Kn), 50μg/ml; chloramphenicol (Cm), 20 μg/ml for the propagation of suicide plasmids in host strains, and 10 μg/ml for integrated plasmids in Vibrio genomes. Induction of gene expression under the control of the PBAD promoter (from pBAD30 or pLP12) was achieved by the addition of 0.2% L-arabinose to the growth media, and the expression was repressed by the addition of 0.3% D-glucose. Induction of vmi480 expression under control of the PTAC promoter was carried out by the addition of IPTG to the LB plates at a final concentration of 5 mM.
Table 2. Strains and plasmids used in this study.
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| E. coli | ||
| DH5α λpir | F− φ80 lacZ ΔM15 recA1 end A1 hsdR17 supE44 thi-1 gyrA96 relA1 (lacZYA-argF) U169 λpir lysogen | Laboratory collection |
| NEB5α | NEB | |
| β2163 | (F-) RP4-2-Tc::Mu ΔdapA::(erm-pir) | [21] |
| LP79 | MG1655 MGIVmi1 res2::pSW23T (NxR, CmR) | [22] |
| LP86 | MG1655 MGIVmi1 res2::pSW23T pKD46 (NxR, CmR, ApR) | This study |
| LP116 | MG1655 MGIVmi1 res2::pSW23T Δvmi480-470 (NxR, CmR) | This study |
| LP128 | MG1655 MGIVmi1 res2::pSW23T Δvmi480 (NxR, CmR) | This study |
| LP134 | NEB5α pBAD30-vmi480-470 (ApR) | This study |
| LP135 | NEB5α pBAD30-vmi470 (ApR) | This study |
| LP192 | NEB5α pBAD30-vmi480 (ApR) | This study |
| LP194 | DH5α λpir pLP10 (CmR) | This study |
| LP196 | DH5α λpir pLP11 (CmR) | This study |
| LP197 | DH5α λpir pLP12 (CmR) | This study |
| V. alginolyticus | ||
| E0601 | Isolated from seawater in Yangjiang, China | This study |
| LP204 | E0601 hem::pLP11 (CmR) | This study |
| LP206 | E0601 hem::pLP12 (CmR) | This study |
| LP207 | E0601 Δhem (resulted from LP204) | This study |
| LP208 | E0601 Δhem (resulted from LP206) | This study |
| V. cholerae | ||
| HN375 | Isolated from seawater in Zhanjiang, China | This study |
| LP228 | HN375 degS::pLP12 (CmR) | This study |
| LP233 | HN375 ΔdegS | This study |
| LP230 | HN375 vasC::pLP12 (CmR) | This study |
| LP235 | HN375 ΔvasC | This study |
| V. parahaemolyticus | This study | |
| E0680 | Isolated from an oyster in Yangjiang, China | This study |
| E06135 | Isolated from Litopenaeus vannamei in Yangjiang, China | This study |
| LP246 | E0680 pilO:: pLP12 (CmR) | This study |
| LP250 | E0680 ΔpilO | This study |
| LP248 | E06135 ascS:: pLP12 (CmR) | This study |
| LP252 | E06135 ΔascS | This study |
| V. vulnificus | This study | |
| ATCC 27562 | ATCC | |
| LP239 | ATCC 27562 impB:: pLP12 (CmR) | This study |
| LP244 | ATCC 27562 ΔimpB | This study |
| Plasmids | ||
| pSW23T | oriT RP4 oriV R6K (CmR) | [21] |
| pSW25T-ccdB | oriT RP4 oriV R6K ccdB (SpR) | Laboratory collection |
| pBAD30 | ori p15A araC PBAD (ApR) | [40] |
| pKD4 | PCR template for one-step gene inactivation (KnR) | [25] |
| pKD13 | PCR template for one-step gene inactivation (KnR) | [25] |
| pKD46 | λ- recombination plasmid (ApR) | [25] |
| pCP20 | Helper plasmid to delete resistant gene with FRT sites (ApR) | [25] |
| pLP10 | oriT RP4 oriV R6K ccdB PTAC promoter (CmR) | This study |
| pLP11 | oriT RP4 oriV R6K vmi480 PTAC promoter (CmR) | This study |
| pLP12 | oriT RP4 oriV R6K vmi480 PBAD (CmR) | This study |
| pLP11-hem | pLP11 containing homologous arms of hem gene of E0601 | This study |
| pLP12-hem | pLP12 containing homologous arms of hem gene of E0601 | This study |
| pLP12-degS | pLP12 containing homologous arms of degS gene of HN375 | This study |
| pLP12-vasC | pLP12 containing homologous arms of vasC gene of HN375 | This study |
| pLP12-pilO | pLP12 containing homologous arms of pilO gene of E0680 | This study |
| pLP12-ascS | pLP12 containing homologous arms of ascS gene of E06135 | This study |
| pLP12-impB | pLP12 containing homologous arms of impB gene of ATCC 27562 | This study |
PCR, Sequencing and Other Molecular Methods
PCR assays were performed using the primers described in S1 Table. When PrimSTAR Max DNA Polymerase (Takara, China) was used, PCR conditions were as follows: 2 min at 98°C; 30 cycles of 10 sec at 98°C, 10 sec at the appropriate annealing temperature, 30 sec/kb at 72°C; and 5 min at 72°C. When rTaq DNA Polymerase (Takara) was used, PCR conditions were as follows: 4 min at 94°C; 30 cycles of 20 sec at 94°C, 30 sec at the appropriate annealing temperature, 1 min/kb at 72°C; and 7 min at 72°C. When necessary, PCR products were purified using a DNA Purification and Concentration Kit (ZhongDing, China). When necessary, PCR products were also sent to the company (BGI, China) for direct sequencing. Plasmids were extracted using the PureYield Plasmid Miniprep System (Promega, USA) according to the manufacturer’s instruction.
Construction of Null Mutants for vmi480, vmi470 and vmi480-470 in E. coli
E. coli LP79 was first transformed with the recombination plasmid pKD46 by electroporation according to Dower et al. [47]. Electroporation was carried out in a BioRad Gene Pulser Xcell apparatus set at 25 μF and 1.8 kV using 1-mm gap electroporation cuvettes. One-step inactivations of vmi480, vmi470, and vmi480-470 in the transformant LP86 (carrying pKD46) were carried out with the protocol from Datsenko et al. [25]. pKD4 and pKD13 were used to generate PCR fragments containing homologous arms of the abovementioned genes and a kanamycin-resistant cassette.
Construction of Expression Vectors of vmi480, vmi470 and vmi480-470
Complete genes of vmi480, vmi470 and vmi480-470 were amplified by primer pairs, 480-exF/480-exR, 470-exF/470-exR and 480-470-exF/480-470-exR, respectively. PCR products of vmi480, vmi470 and vmi480-470 were purified and digested with EcoR I and Xba I, and they were ligated with vector pBAD30 digested with the same restriction enzymes. The ligation products were transformed into competent E. coli NEB5a cells (NEB) according to the manufacturer’s instructions. During the construction of the transformant hosting the plasmid pBAD30-vmi480, D-glucose was added into the medium to repress expression of vmi480. Transformants were screened on LB plates supplemented with Ap. Transformants were identified by PCR using the primer pair pBAD30-TF/pBAD30-TR and were confirmed by subsequent sequencing.
Assay of the Lethal Effect of Toxin Vmi480 and Its Antidote Vmi470
To test the lethal effect of Vmi480 and the antagonism of Vmi470 against Vmi480, transformants of E. coli LP134 (pBAD30-vmi480-470), LP135 (pBAD30-vmi470) and LP192 (pBAD30-vmi480) were grown in LB broth supplemented with D-glucose at 37°C for 6 hr and serially diluted. Samples (10−4 dilution) were spread on LB plates with D-glucose or with L-arabinose, and they were then incubated at 37 C overnight.
Construction of Suicide T-Vectors carrying the Lethal Gene vmi480
In the process of constructing suicide T-vectors, pSW23T and pSW25T-ccdB were used as the initiator plasmids. PCR of pSW23T was carried out using the primer pair pSW23T-F/pSW23T-R to generate a fragment containing oriV R6Kγ, oriT RP4 and cat. PCR of pSW25T-ccdB was carried out using the primer pair pSW25T-F/pSW25T-R to generate a fragment containing ccdB, lacIq and PTAC. A multiple cloning site (AhdI-EcoRI-SacI-AhdI-NheI) was introduced by primers pSW23T-F and pSW25T-F. Two fragments were purified, digested by EcoRI and SphI, and ligated together. The ligation product was transformed into E. coli DH5α λ pir cells to obtain a strain LP194 hosting a resultant plasmid, pLP10. Chemical transformations were performed according to the method by Swords [48]. The construction of pLP10 was tested by PCR with two primer pairs, pLP10L-TF1/ pLP10L-TR1 and pLP10L-TF2/ pLP10L-TR2, and confirmed by sequencing.
A fragment containing all the parts of pLP10 except ccdB was acquired by reverse PCR amplification from pLP10 using primers pLP10-F and pLP10-R. vmi480 was amplified from E. coli LP79 using primers vmi480-F and vmi480-R. They were purified, digested by NdeI and XhoI, and ligated together, and then the ligation product was transformed into E. coli DH5α λ pir cells to obtain a strain LP196 hosting a resultant plasmid, pLP11. The construction of pLP11 was confirmed by the primer pair pLP11L-TF/pLP11L-TR followed by sequencing.
pLP11 was amplified with primers pLP11-F and pLP11-R to generate a fragment retaining all the parts of the plasmid except the PTAC promoter and lacIq gene. The PBAD promoter system was amplified from plasmid pBAD30 using primers pBAD30-PF and pBAD30-PR. Two fragments were recombined together through in vitro recombination using ClonExpress® II One Step Cloning Kit (Vazyme, China), where recombinase Exnase was used to avoid the excess introduction of restriction sites into the resulting plasmid. The recombination product was transformed into E. coli DH5α λ pir cells to obtain a strain LP197 hosting the resultant plasmid, pLP12. The construction of pLP12 was confirmed by PCR testing with the primer pair pLP12L-TF/pLP12L-TR followed by sequencing.
To avoid repetitive extraction of plasmids for generating suicide T-vectors and to decrease the false clones in the subsequent construction of recombinant T-vectors, pLP11 and pLP12 were amplified using PrimSTAR Max DNA Polymerase and the primer pair STVU-F/STVU-R. PCR products were digested by AhdI and purified to finally obtain the linearized suicide T-vectors pLP11-T and pLP12-T.
Construction of Deletion Mutants of Six Genes from Four Vibrio Species
Six genes from four representative Vibrio species were targeted for deletion mutation. In-frame deletion fragments consisting of two flanking regions of each target locus were made by overlap extension PCR [19]. PrimSTAR Max DNA Polymerase was adopted in the first PCR, and rTaq DNA polymerase was used in the second PCR to conveniently add the single base A to the 3' end of the PCR products. The final PCR products were purified and ligated with pLP11-T or pLP12-T. The ligation products were transformed into competent E. coli DH5α λ pir cells to generate recombinant suicide plasmids carrying these homologous fragments for allelic exchange of targeted genes. Recombinant plasmids were extracted and transformed into E. coli β2163 by electroporation. Then, the recombinant plasmids were transferred into Vibrio strains through conjugation.
Conjugations were performed by mixing equal volumes of recombinant E. coli β2163 and each Vibrio strain grown overnight at 37°C. The cells were harvested by centrifugation for 2 min at 8000 g, washed in 400 μL of LB broth and resuspended in 10 μL of LB broth. Mating mixtures were then deposited on LB plates supplemented with DAP and D-glucose and incubated at 37°C for 8 hr. The cells were recovered from the plates in 1 ml of LB broth. Each of the 100-μL of mixed cells was spread on LB plates supplemented with Cm and D-glucose for screening of single-crossover cells with integrated plasmids into specific chromosomal loci. The clones were purified on the same LB plates to make sure that stable and correct insertional mutants were obtained. Then, these insertional mutants were checked by PCR with external primers targeting upstream of integration sites and an internal primer targeting vector-specific region. In this condition, wild-type strains will not result in any predicted PCR bands. Insertional mutants were grown at 37°C for 6 hr, serially diluted, and spread on LB plates supplemented with IPTG or L-arabinose for counterselection of deletion mutants (double-crossover recombination). The clones on counterselection plates were randomly selected and purified before PCR assays. Two external primers respectively anchoring upstream and downstream of targeted genes were adopted for the detection of deletion mutants. All of the insertional mutants and deletion mutants were confirmed by PCR using the same primer pair and subsequent sequencing.
Bioinformatics Analysis and Nucleotide Sequence Accession Numbers
Blastx searches against vmi480 and its adjacent vmi470 were performed to predict their function. The promoter was predicted by online tools (http://molbiol-tools.ca/Promoters.htm). A web-based TA prediction tool was adopted to identify vmi480 and vmi470 (http://genoweb1.irisa.fr/duals/RASTA-Bacteria/index.php?page=form). The sequences of the suicide plasmids pLP11 and pLP12 have been deposited at GenBank under accession numbers KT326152 and KT326153, respectively.
Supporting Information
(DOCX)
Acknowledgments
We would like to thank Dr. Vincent Burrus for kindly providing some E. coli strains and the plasmids pSW23T and pSW25T-ccdB.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funded by Natural Science Foundation of China (31370149), http://www.nsfc.gov.cn/, Comprehensive Strategic Cooperation Project of Guangdong Province and Chinese Academy of Sciences (2012B091100269), http://gdcas.gdstc.gov.cn/, and Knowledge Innovation Program of Chinese Academy of Sciences (KSCX2-EW-G-12B), http://www.cas.cn/.
References
- 1. Jin C, Luo P, Zuo H, Chen J, Chen M, Wang W. Vibrio Zhuhaiensis sp. nov., isolated from Japanese prawn (Marsupenaeus japonicus). Antonie van Leeuwenhoek. 2013; 103:989–96. 10.1007/s10482-013-9878-4 [DOI] [PubMed] [Google Scholar]
- 2. Howard RJ, Bennett NT. Infections caused by halophilic marine Vibrio bacteria. Ann Surg. 1993; 217:525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fukami K, Simidu U, Taga N. Microbial decomposition of phyto- and zooplankton in seawater. II. Changes in the bacterial community. Mar Ecol Prog Ser. 1985; 21:7–23. [Google Scholar]
- 4. Daniels NA, Shafaie A. A review of pathogenic Vibrio infections for clinicians. Infect Med. 2000; 17:665–85. [Google Scholar]
- 5. Bhunia AK. Vibrio cholerae, V. parahaemolyticus, V. vulnificus In: Bhunia AK, editor. Foodborne microbial pathogens. Springer; 2008. p. 241–52. [Google Scholar]
- 6. Turner JW, Malayil L, Guadagnoli D, Cole D, Lipp EK. Detection of Vibrio parahaemolyticus, Vibrio vulnificus and Vibrio cholerae with respect to seasonal fluctuations in temperature and plankton abundance. Environ Microbiol. 2014; 16:1019–28. 10.1111/1462-2920.12246 [DOI] [PubMed] [Google Scholar]
- 7. Lafferty KD, Harvell CD, Conrad JM, Friedman CS, Kent ML, Kuris AM, et al. Infectious diseases affect marine fisheries and aquaculture economics. Ann Rev Mar Sci. 2015; 7:471–96. 10.1146/annurev-marine-010814-015646 [DOI] [PubMed] [Google Scholar]
- 8. Mahbub KR, Paul KP, Ahmed MM. Prevalence of Vibrio spp. and antibiogram of isolates from shrimp rearing ponds in Bangladesh. J Adv Sci Res. 2011; 2:74–80. [Google Scholar]
- 9. Shruti C, Soumya H. Vibrio related diseases in aquaculture and development of rapid and accurate methods. J Mar Sci Res Dev. 2012; S1:002. [Google Scholar]
- 10. Vandenberghe J, Thompson FL, Gomez-Gill B, Swings J. Phenotypic diversity amongst Vibrio isolates from marine aquaculture systems. Aquaculture. 2003; 219:9–20. [Google Scholar]
- 11. Direkbusaram S, Yoshimizu M, Ezura Y, Ruangpan L, Danayadol Y. Vibrio spp., the dominant flora in shrimp hatchery against some fish pathogenic viruses. J Mar Biotech. 1998; l6:266–67. [PubMed] [Google Scholar]
- 12. Roux FL, Binesse J, Soulnier D, Mazel D. Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl Environ Microbiol. 2007; 73:777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakashima N, Miyazaki K. Bacterial cellular engineering by genome editing and gene silencing. Int J Mol Sci. 2014; 15:2773–93. 10.3390/ijms15022773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reyrat J, Pelicic V, Gicquel B, Rappuoli R. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect Immun. 1998; 66:4011–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stibitz S. Use of conditionally counterselectable suicide vectors for allelic exchange. Methods Enzymol. 1994; 235:458–65. [DOI] [PubMed] [Google Scholar]
- 16. Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991; 59:4310–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karlsson SL, Ax E, Nygren E, Källgård S, Blomquist M, Ekman A, et al. Development of stable Vibrio cholerae O1 Hikojima type vaccine strains co-expressing the Inaba and Ogawa lipopolysaccharide antigens. PLoS One. 2014; 9:e108521 10.1371/journal.pone.0108521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milton DL, OToole R, Hörstedt P, Wolf-Watz H. Flagellin A is essential for the virulence of Vibrio anguillarum . J Bacteriol. 1996; 178:1310–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen C, Zhao JJ, Ren CH, Hu CQ. Deletion of valR, a homolog of Vibrio harveyi luxR generates an intermediate colony phenotype between opaque/rugose and translucent/smooth in Vibrio alginolyticus . Biofouling. 2010; 26:595–601. 10.1080/08927014.2010.499511 [DOI] [PubMed] [Google Scholar]
- 20. Gulig PA, Tucker MS, Thiaville PC, Joseph JL, Brown RN. USER friendly cloning coupled with chitin-based natural transformation enables rapid mutagenesis of Vibrio vulnificus . Appl Environ Microbiol. 2009; 75:4936–49. 10.1128/AEM.02564-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Demarre G, Guérout AM, Matsumoto-Mashimo C, Rowe-Magnus DA, Marlière P, Mazel D. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPα) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol. 2005; 156:245–55. [DOI] [PubMed] [Google Scholar]
- 22. Carraro N, Matteau D, Luo P, Rodrigue S, Burrus V. The master activator of IncA/C conjugative plasmids stimulates genomic islands and multidrug resistance dissemination. PLoS Genet. 2014; 10:e1004714 10.1371/journal.pgen.1004714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005; 33:966–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kasari V, Kurg K, margus T, Tenson T, Kaldalu N. The Escherichia coli mqsR and ygiT genes encode a new toxin-antitoxin pair. J Bacteriol. 2010; 192:2908–19. 10.1128/JB.01266-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000; 97: 6640–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blomfield IC, Vaughn V, Rest RF, Eisenstein BI. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol. 1991; 5:1447–57. [DOI] [PubMed] [Google Scholar]
- 27. Baumann P, Schubert RHW. Genus II. Vibrionaceae Veron 1965 In: Krieg NR, Holt JG, editors. Bergey’s manual of systematic bacteriology, vol 1 Baltimore: Williams & Wilkins; 1984; p. 516–17. [Google Scholar]
- 28. Kampranis SC, Howells AJ, Maxwell A. The interaction of DNA gyrase with the bacterial toxin CcdB: evidence for the existence of two gyrase-CcdB complexes. J Mol Biol. 1999; 293:733–44. [DOI] [PubMed] [Google Scholar]
- 29. Budde PP, Davisn BM, Yuan J, Waldor MK. Characterization of a higBA toxin-antitoxin locus in Vibrio cholerae . J Bacteriol. 2007; 189: 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bernard P, Couturier M. The 41 carboxy-terminal residues of the mini F plasmid CcdA protein are sufficient to antagonize the killer activity of the CcdB protein. Mol Gen Genet. 1991; 226:297–304. [DOI] [PubMed] [Google Scholar]
- 31. Madl T, Van Melderen L, Mine N, Respondek M, Oberer M, Keller W, et al. Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J Mol Biol. 2006; 364:170–85. [DOI] [PubMed] [Google Scholar]
- 32. Oberer M, Zangger K, Gruber K, Keller W. The solution structure of ParD, the antidote of the ParDE toxin–antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci. 2007; 16:1676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leplae R, Geeraerts D, Hallez R, Guglielmini J, Drèze P. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011; 39:5513–25. 10.1093/nar/gkr131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iqbal N, Guérout A, Krin E, Roux FL, Mazel D. Comprehensive functional analysis of the 18 Vibrio cholerae N16961 toxin-antitoxin systems substantiates their role in stabilizing the superintegron. J Bacteriol. 2015; 197:2150–59. 10.1128/JB.00108-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Christensen-Dalsgaard M, Gerdes K. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol Microbiol. 2006; 62:397–411. [DOI] [PubMed] [Google Scholar]
- 36. Jorgensen MG, Pandey DP, Jaskolska M, Gerdes K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J Bacteriol. 2009; 191:1191–99. 10.1128/JB.01013-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luo P, Hu CQ. Analysis of gyrB sequences of Vibrio alginolyticus and gyrB-targeted rapid PCR identification of the bacterium from environmental isolates. Dis Aquat Org. 2008; 82:209–16. 10.3354/dao01984 [DOI] [PubMed] [Google Scholar]
- 38. Reilly GD, Reilly CA, Smith EG, Baker-Austin C. Vibrio alginolyticus-associated wound infection acquired in British waters, Guernsey, July 2011. Euro Surveill. 2011; 16:19994 [PubMed] [Google Scholar]
- 39. Liu CH, Cheng W, Hsu JP, Chen JC. Vibrio alginolyticus infection in the white shrimp Litopenaeus vannamei confirmed by polymerase chain reaction and 16S rDNA sequencing. Dis Aquat Org. 2004; 61:169–74. [DOI] [PubMed] [Google Scholar]
- 40. Guzman LM, Belin D, Carson MJ, Beckwith J. Tight 342 regulation, modulation, and high-level expression by vectors containing the 343 arabinose PBAD promoter. J Bacteriol. 1995; 177:4121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y, Shang X, Lai SJ, Zhang GQ, Liang Y, Wen TY. Development and application of an arabinose-inducible expression system by facilitating inducer uptake in Corynebacterium glutamicum . Appl Environ Microbiol. 2012; 78:5831–38. 10.1128/AEM.01147-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamaichi Y, Fogel MA, Waldor MK. Par genes and the pathology of chromosome loss in Vibrio cholerae . Proc Natl Acad Sci USA. 2007; 104:630–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bordeleau E, Brouillette E, Robichaud N, Burrus V. Beyond antibiotic resistance: integrating conjugative elements of the SXT/R391 family that encode novel diguanylate cyclases participate to c-di-GMP signaling in Vibrio cholerae . Environ Microbiol. 2010; 12:510–23. 10.1111/j.1462-2920.2009.02094.x [DOI] [PubMed] [Google Scholar]
- 44. Lo Scrudato M, Blokesch M. A transcriptional regulator linking quorum sensing and chitin induction to render Vibrio cholerae naturally transformable. Nucleic Acids Res. 2013; 41:3644–58. 10.1093/nar/gkt041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hansen LH, Knudsen S, Sørensen SJ. The effect of the lacY gene on the induction of IPTG inducible promoters, studied in Escherichia coli and Pseudomonas fluorescens . Curr Microbiol. 1998; 36:341–47. [DOI] [PubMed] [Google Scholar]
- 46. Marbach A, Bettenbrock K. Lac operon induction in Escherichia coli: systematic comparison of IPTG and TMG induction and influence of the transacetylase LacA. J Biotech. 2012; 1578:82–8. [DOI] [PubMed] [Google Scholar]
- 47. Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988; 16:6127–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Swords WE. Chemical transformation of E. coli . Methods Mol Biol. 2003; 235:49–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.