

Abstract Abstract
The effects of the heptapeptide angiotensin-(1–7) (Ang-(1–7)), via its receptor Mas, oppose many of the effects of the classic angiotensin II signaling pathway, and pharmacological exploitation of this effect is currently actively pursued for a wide range of cardiovascular, neoplastic, or immunological disorders. On the basis of its vasodilatory and antiproliferative properties, Ang-(1–7) has consequentially also been proposed as a novel therapeutic strategy for the treatment of pulmonary arterial hypertension (PAH). In this study, we tested the effectiveness of Ang-(1–7) and its stable, cyclic analog cAng-(1–7) over a range of doses for their therapeutic potential in experimental PAH. In the monocrotaline (MCT) rat model of PAH, Ang-(1–7) or cAng-(1–7) were injected in doses of 30, 100, 300, or 900 μg kg−1 day−1, and effects on pulmonary hemodynamics and vascular remodeling were assessed. Five weeks after MCT injection, right ventricular systolic pressure (RVSP) was significantly reduced for 3 dose groups treated with Ang-(1–7) (100, 300, and 900 μg kg−1 day−1) and for all dose groups treated with cAng-(1–7), as compared to untreated controls, yet the total reduction of RVSP was <50% at best and thus markedly lower than that with a positive treatment control with ambrisentan. Medial-wall thickness in pulmonary arterioles was only slightly reduced, without reaching significance, for any of the tested Ang-(1–7) compounds and doses. The reported moderate attenuation of PAH does not confirm the previously postulated high promise of this strategy, and the therapeutic usefulness of Ang-(1–7) may be limited in PAH relative to that in other cardiovascular diseases.
Keywords: pulmonary hypertension, vascular remodeling, renin-angiotensin system
Pulmonary arterial hypertension (PAH) is a rare but ultimately fatal disease defined by an increase in mean pulmonary arterial pressure to >25 mmHg at rest or >30 mmHg during exercise.1 The pathogenesis of PAH is very diverse, in that PAH may be heritable or idiopathic or develop in association with a number of diseases, such as systemic sclerosis or HIV infection. The pathology of PAH is characterized by endothelial dysfunction, which becomes apparent as an imbalance in the release of vasoconstrictive and vasodilatory factors, such as endothelin and nitric oxide (NO), leading to pulmonary vasoconstriction and ultimately promoting structural remodeling of the pulmonary vasculature, resulting in a persistent elevation in pulmonary vascular resistance.2,3 A series of therapeutic drugs have been approved for the treatment of PAH that target three different pathways: prostacyclin analogs, endothelin receptor antagonists, and phosphodiesterase type 5 inhibitors.4 While these drugs have helped to improve life expectancy and quality of life in PAH patients in the past 2 decades, mortality remains high, as one-third of PAH patients die within 3 years of diagnosis; this emphasizes the need for further research and improved therapeutic options.5
The renin-angiotensin system (RAS) is a critical regulator of cardiovascular homeostasis, and cardiovascular diseases are accordingly frequently associated with an imbalance of the RAS.6 Its main effector, angiotensin II (AngII), is formed from AngI by the endopepdidase angiotensin-converting enzyme (ACE) and exerts its effects mostly by acting through the AngII receptor subtype 1 (AT1). Activation of the ACE/AngII/AT1 axis mediates vasoconstrictive, proliferative, fibrotic, and inflammatory effects and therefore is a key player in the onset of many cardiovascular diseases.7 In vivo, this pathway is endogenously opposed by the ACE2/Ang-(1–7)/Mas axis, which counteracts many of the effects of AngII.8 The heptapeptide Ang-(1–7) is predominantly formed by ACE2-mediated cleavage of the octapeptide AngII7 and exerts its effects through the G-protein-coupled receptor Mas.9 Ang-(1–7) contributes importantly to cardiovascular homeostasis by triggering a variety of downstream targets, most notably by activation of endothelial NO synthase (eNOS) and thus the release of NO,10,11 causing vasodilation and—in broadest terms—endothelial protection. Ang-(1–7) also blocks mitogen-activated protein kinase signaling, thereby limiting smooth muscle cell proliferation, and can in parallel attenuate cardiac myocyte hypertrophy by reducing transforming growth factor β levels.12-14 Taken together, these properties render Ang-(1–7) a promising drug candidate for the treatment of cardiovascular diseases in general and PAH in particular. Proof of concept for the efficacy of Ang-(1–7) as a novel therapeutic strategy in cardiovascular diseases has been described in a series of preclinical studies, including two studies on the effects of Ang-(1–7) in PAH.15,16
However, the translation of Ang-(1–7) as a novel drug therapy for PAH to the clinical setting is somewhat hampered by its rapid metabolism and degradation in plasma and tissue, predominantly, again, by ACE.17 To improve its pharmacokinetic properties, a cyclic form of Ang-(1–7), cAng-(1–7), which is linked with a thioether bridge, was developed with the intention of mitigating its rapid degradation and increasing its half-life. Indeed, during continuous infusion, cAng-1–7 displays a 34-fold higher presence than conventional Ang-(1–7) in the circulating blood of Sprague-Dawley (SD) rats,18 making it a promising candidate for clinical use in cardiovascular therapy.
In preparation for the translation of Ang-(1–7) therapy into first clinical PAH trials, we designed this study to (1) test a wide range of drug concentrations for their effectiveness in experimental PAH and (2) compare the efficacy of Ang-(1–7) with that of its stable, cyclic analog cAng-(1–7). To this end, we adopted a preventive drug administration approach, as the primary goal was to establish dose-response relationships for each individual compound before proceeding to therapeutic-intervention models.
Methods
Animals and experimental design
Male SD rats of 200–220 g body weight (bw) were obtained from Charles River Laboratories (St. Constant, QC, Canada). All animals received care in accordance with the Guide for the Care and Use of Laboratory Animals (published in 1996 by the Institute of Laboratory Animal Resources, National Research Council). The study was approved by the Animal Care Committee of St. Michael’s Hospital (protocol 397). All animals were allowed to acclimate for 5 days before start of the Ang-(1–7)/cAng-(1–7) injections. Ang-(1–7) and cAng-(1–7) were kindly provided by Tarix Pharmaceuticals (Cambridge, MA). At the inception of the study, rats were enrolled in a staggered manner, such that equal numbers of animals were enrolled per study group per day. PAH was induced by a single intraperitoneal injection of monocrotaline (MCT; 40 mg/kg), and 24 hours later once-daily subcutaneous administration of Ang-(1–7), cAng-(1–7) (30, 100, 300, or 900 μg kg−1 day−1 for both), or saline (vehicle) was started. Daily subcutaneous injections were maintained over 5 weeks. As a positive treatment control, an additional group of rats was treated with 90 μg kg−1 day−1 of ambrisentan with the drinking water. All rats were killed 5 weeks after MCT injection.
Assessment of pulmonary hemodynamics and right ventricular hypertrophy (RVH)
Hemodynamic and RVH measurements were performed as previously reported.19 In brief, 5 weeks after MCT treatment, rats were anesthetized, tracheotomized, and mechanically ventilated with a tidal volume of 6 mL/kg bw and a positive end-expiratory pressure of 3 mmHg at 80 breaths/min, as described elsewhere.20 A 2F microtip Millar catheter (Millar, Houston, TX) was introduced via the right jugular vein into the right ventricle for real-time monitoring of right ventricular systolic pressure (RVSP; DasyLab, Norton, MA). After recording of RVSP curves, animals were exsanguinated, and the left lung was excised and fixed for histology in 10% neutral buffered paraformaldehyde. The right lung was snap-frozen in liquid nitrogen for postmortem protein analyses. For assessment of RVH, the Fulton index was calculated as the ratio of the right ventricular weight to the left-ventricular-plus-septal weight (RV/[LV + S]).
Lung histology and analysis of vascular remodeling
In each experimental group, 5 lungs from 5 different rats were fixed by intratracheal instillation of 10% paraformaldehyde for 30 minutes, and tissue sections of 1 cm3 were excised and fixed for another 24 hours. Dehydrated tissues were embedded in paraffin, and 5-μm sections were cut and stained with hematoxylin and eosin. In each group, 100–200 pulmonary arterioles were analyzed, and these vessels were categorized according to their external diameters as small (20–50 μm in diameter) or medium-sized (50–100 μm in diameter). The medial-wall thickness was expressed as the difference between external vessel diameter and luminal diameter, as a percentage of the external vessel diameter.21
Western blot analysis
Snap-frozen lungs were homogenized in lysis buffer containing 100 mM NaCl, 30 mM HEPES, 1 mM EGTA (ethylene glycol tetraacetic acid), 1% Triton X-100, protease inhibitor mixture, and PhosSTOP (Roche, Mississauga, ON, Canada). Total protein concentration was determined with a bicinchoninic acid protein assay kit (Thermo Scientific, Waltham, MA). Equal amounts of protein (60 μg/slot) were loaded onto a sodium dodecyl sulfate (SDS) polyacrylamide gel, transferred to a nitrocellulose membrane (BioRad, Mississauga, ON, Canada), and blocked for 2 hours with 5% milk. Membranes were then incubated overnight with primary antibodies (mas receptor 1∶500 [AlomoneLab, Jerusalem], eNOS 1∶500, GAPDH 1∶1,000 [both Santa Cruz Biotechnology, San Diego, CA], and phospho-eNOS [p-eNOS] 1∶400 [CellSignaling, Danvers, MA]) in the appropriate dilution in 0.1% TBST (Tris-buffered saline and Tween 20) containing 1% milk. After 3 washes with 0.1% TBST, membranes were incubated with horseradish peroxidase–linked secondary goat antirabbit antibodies (Abcam, Cambridge, MA). After 3 more washes, membranes were developed by enhanced chemiluminescence (ECL plus, Thermo Scientific) and imaged by VersaDoc molecular imager (BioRad). Images were densitometrically quantified with ImageLab (ver. 3.0, BioRad).
Statistical analysis
Data are given as means ± SEM. Different groups were compared by ANOVA followed by the Dunnett multiple-comparison test (Prism, ver. 5.0, GraphPad Software, La Jolla, CA). Statistical significance was assumed at P < 0.05.
Results
To probe for the effectiveness and potential dose-response relationships of Ang-(1–7)-based therapies in experimental PAH, we first focused on the effects of linear Ang-(1–7) on pulmonary hemodynamics and lung vascular remodeling. RVSP values showed a significant increase in MCT-treated animals compared to nontreated controls, consistent with the induction of PAH. In groups treated with 100, 300, or 900 μg kg−1 day−1 of Ang-(1–7), RVSP was significantly reduced compared to that in the untreated MCT group, yet even in the most effective treatment groups (100 or 300 μg kg−1 day−1), Ang-(1–7) reversed less than 50% of the MCT-induced RVSP increase. No significant reduction of RVSP was detectable for rats treated with the lowest (30 μg kg−1 day−1) dose of Ang-(1–7) (Fig. 1A). In the right ventricle, MCT treatment induced the marked hypertrophic response that is characteristic of the development of PAH, as demonstrated by a significant increase in the Fulton index compared to healthy control rats. RVH showed a trend to reduced values in all Ang-(1–7) groups, yet this trend reached significance only in the group treated with 300 μg kg−1 day−1 (Fig. 1B). As compared to healthy controls, MCT induced marked vascular remodeling that was characteristically more prominent in the small, as compared to medium-sized, pulmonary arterioles. There was a trend to reduced medial-wall thickness in small arterioles in virtually all Ang-(1–7)-treated groups, yet without reaching statistical significance (Fig. 1C). No marked attenuation of vascular remodeling by any of the applied Ang-(1–7) doses was detectable in medium-sized arterioles.
Figure 1.

Dose-dependent effects of angiotensin-(1–7) (Ang-(1–7)) on pulmonary arterial hypertension, lung vascular remodeling, and right ventricular hypertrophy in the monocrotaline (MCT) model. Rats were injected with either vehicle (Contr), MCT only (MCT 0), or MCT plus Ang-(1–7) in doses of 30, 100, 300, or 900 μg kg−1 day−1. Group data show right ventricular systolic pressure (RVSP; A), the ratio of right ventricular to left-ventricular-plus-septal weight (RV/(LV + S); B), and medial-wall thickness for pulmonary arterioles 20–50 μm (C) or 50–100 μm (D) in diameter. Each bar represents mean ± SEM of 4–9 experiments; *P < 0.05 versus MCT-only treatment.
Despite moderate reductions in RVSP, the overall therapeutic effectiveness of Ang-(1–7) was thus rather limited throughout all groups. In a second set of experiments, we therefore tested its chemically modified, more stable cyclic analog cAng-(1–7) in the identical model and dosing regime. For all groups treated with cAng-(1–7) (30, 100, 300, or 900 μg kg−1 day−1), RVSP showed a significant reduction compared to the untreated MCT group, yet again MCT-induced PAH was at best reversible by <50%, and RVSP still markedly exceeded hemodynamic values in healthy control rats (Fig. 2A). RVH was significantly reduced in groups treated with either the lowest (30 μg kg−1 day−1) or the highest (900 μg kg−1 day−1) dose of cAng-(1–7), compared to the untreated MCT group (Fig. 2B). Vascular remodeling showed a trend to reduced medial-wall thickness in small arterioles in most cAng-(1–7)-treated groups (with the notable exception of the 100-μg kg−1 day−1 group), but, yet again, this trend did not reach statistical significance (Fig. 2C). Vascular remodeling was again virtually unchanged in medium-sized arterioles (Fig. 2D). Overall, treatment with cAng-(1–7), as compared to Ang-(1–7), caused a shift in the dose-response curve, in that the lowest dose of cAng-(1–7) already showed moderate therapeutic benefits on RVSP and vascular remodeling. However, cAng-(1–7) treatment failed in general to yield striking therapeutic benefits in any of the studied dose groups.
Figure 2.

Dose-dependent effects of cyclic angiotensin-(1–7) (cAng-(1–7)) on pulmonary arterial hypertension, lung vascular remodeling, and right ventricular hypertrophy in the monocrotaline (MCT) model. Rats were injected with either vehicle (Contr), MCT only (MCT 0), or MCT plus cAng-(1–7) in doses of 30, 100, 300, or 900 μg kg−1 day−1. Group data show right ventricular systolic pressure (RVSP; A), the ratio of right ventricular to left-ventricular-plus-septal weight (RV/(LV + S); B), and medial-wall thickness for pulmonary arterioles 20–50 μm (C) or 50–100 μm (D) in diameter. Each bar represents mean ± SEM of 4–9 experiments; *P < 0.05 versus MCT-only treatment.
Ambrisentan is a specific endothelin receptor A antagonist that is clinically approved for the treatment of PAH.22 Since the effectiveness of both Ang-(1–7) and cAng(1–7) to attenuate PAH and lung vascular remodeling in MCT rats was modest overall, we probed for a potential systematic error by using ambrisentan as a positive treatment control. MCT-treated animals receiving ambrisentan at a dose of 90 μg kg−1 day−1 with the drinking water showed a distinct improvement in PAH and vascular remodeling, evident as a significant reduction in RVSP, Fulton index, and arteriolar-wall thickness, compared to the untreated MCT group, confirming the ability of the applied model to detect beneficial treatment effects with sufficient sensitivity (Fig. 3A–3D).
Figure 3.

Effects of ambrisentan (AMB) on pulmonary arterial hypertension, lung vascular remodeling, and right ventricular hypertrophy in the monocrotaline (MCT) model. Rats were injected with either vehicle (Contr), MCT only (MCT 0), or MCT with subsequent administration of 90 μg kg−1 day−1 AMB with the drinking water. Group data show right ventricular systolic pressure (RVSP; A), the ratio of right ventricular to left-ventricular-plus-septal weight (RV/(LV + S); B), and medial-wall thickness for pulmonary arterioles 20–50 μm (C) or 50–100 μm (D) in diameter. Each bar represents mean ± SEM of 4–9 experiments; *P < 0.05 versus MCT-only treatment.
In order to probe whether the limited effectiveness of Ang-(1–7) and cAng-(1–7) in the MCT model may have been the result of a loss of target, we next probed for expression levels of the Ang-(1–7) receptor Mas by Western blot. Mas receptor expression was comparable between lungs collected from control and MCT animals. Moreover, in all lungs from animals treated with Ang-(1–7), Mas expression tended to be higher, as compared to the control groups, especially in groups treated with doses of 30, 100, or 300 μg kg−1 day−1 (Fig. 4A). A similar trend to increased Mas expression was detectable in animals treated with lower cAng-(1–7) doses of 30 or 100 μg kg−1 day−1, while Mas expression was lower in rats treated with cAng-(1–7) doses of 300 or 900 μg kg−1 day−1 (Fig. 4B). Notably, none of these changes reached statistical significance.
Figure 4.

Dose-dependent effect of angiotensin-(1–7) (Ang-(1–7); A) or cyclic Ang-(1–7) (cAng-(1–7); B) on relative Mas receptor expression in the monocrotaline (MCT) pulmonary arterial hypertension model. Representative immunoblots are shown, and quantitative densitometric data were normalized to GAPDH and expressed as percentage of control (Contr); each bar represents mean ± SEM of 3–6 experiments.
Endothelial NO synthase (eNOS), which becomes activated when it is phosphorylated at its Ser1177 residue (p-eNOS), represents a potential downstream target of the ACE2/Ang-(1–7)/Mas axis and serves at the same time as a marker of intact endothelial function in PAH.3,10 Expression of eNOS was similar between control and MCT-treated animals. A trend to a gradual increase in eNOS levels was evident with increasing doses of Ang-(1–7) (Fig. 5A) or cAng-(1–7) (Fig. 5D), with the notable exception of the highest cAng-(1–7) dose. The p-eNOS levels were distinctly lower in lungs from MCT animals than in controls, in line with reduced eNOS activity, as previously reported in this model.23 In contrast, p-eNOS levels were reversed almost to control levels in rats receiving higher Ang-(1–7) doses (100, 300, or 900 μg kg−1 day−1; Fig. 5B). Treatment with either the lowest (30 μg kg−1 day−1) or the highest (900 μg kg−1 day−1) dose of cAng-(1–7) did not result in a detectable change in p-eNOS levels, while groups treated with medium doses, 100 or 300 μg kg−1 day−1, showed a trend to increased p-eNOS levels (Fig. 5E). Finally, as a surrogate measure of eNOS activation, we calculated the ratio of p-eNOS to eNOS, which was again markedly reduced in the MCT-only group. A distinct, though not significant, increase in eNOS activity was measured in lungs from animals treated with 100 μg kg−1 day−1 Ang-(1–7), compared to the MCT-only group, whereas all other doses failed to show marked changes, and the group receiving the lowest dose (30 μg kg−1 day−1) showed a trend to reduced activation compared to the MCT-only group (Fig. 5C). Likewise, animals treated with the lowest dose of cAng-(1–7) (30 μg kg−1 day−1) showed a reduction in eNOS activation, which remained virtually unchanged in groups receiving higher doses, compared to the MCT-only group (Fig. 5F).
Figure 5.

Dose-dependent effect of angiotensin-(1–7) (Ang-(1–7); A–C) or cyclic Ang-(1–7) (cAng-(1–7); D–F) on endothelial NO synthase (eNOS) expression and phosphorylation in the monocrotaline (MCT) pulmonary arterial hypertension model: A, D, Relative eNOS expression; B, E, relative phospho-eNOS (p-eNOS) expression; C, F, ratio of p-eNOS to eNOS expression. Representative immunoblots are shown, and quantitative densitometric data were normalized to GAPDH and expressed as percentage of control (Contr). Each bar represents mean ± SEM of 3–6 experiments.
Discussion
In this study, we aimed to identify the optimal dosing strategy for Ang-(1–7), which has recently evolved as a promising therapeutic strategy for PAH, and to compare the effectiveness of the linear Ang-(1–7) peptide to that of its more stable cyclic derivative, cAng-(1–7). While medium doses of Ang-(1–7) (100 or 300 μg kg−1 day−1) and a wide range of doses of cAng-(1–7) given from day 1 after PAH induction by MCT showed therapeutic benefit in terms of reduced RVSP, the overall hemodynamic improvement achieved was moderate and was not equally reflected by benefits in terms of lung vascular remodeling or RVH. Ambrisentan, as a positive control, showed excellent efficacy, thus ruling out model-immanent systematic effects. Mas expression was found to have increased rather than decreased in Ang-(1–7)- or cAng-(1–7)-treated rats, precluding loss of target as a potential cause for limited drug effectiveness. Furthermore, expression analyses of eNOS and p-eNOS are overall in line with effective drug delivery and action in the pulmonary vasculature, which, however, failed to have a decisive impact on disease severity.
The results from this study can therefore not unequivocally confirm the exciting promise for the therapeutic potential of Ang-(1–7) in experimental PAH suggested in two previous studies. Both Shenoy and colleagues16 and Chen and coworkers15 had reported significant, striking reductions in RVSP, medial-wall thickness, and RVH in an MCT rat model after, respectively, overexpression (by lenti-Ang-(1–7) viral particles) or continuous infusion (24 μg kg−1 h−1 via an osmotic minipump) of Ang-(1–7). In contrast, in our study, only moderate beneficial effects for both Ang-(1–7) and cAng-(1–7) were detectable at the applied concentrations. Frequently, these effects were too small to reach the level of significance and were far less pronounced than what could be expected from those prior studies.
On the side of therapeutic benefit, RVSP was found to be significantly reduced in three clinical-dose-treated Ang-(1–7) groups and in all cAng-(1–7)-treated groups, attesting to the general effectiveness of the treatment and possibly to the higher potency/effectiveness of the more stable cAng-(1–7) compound, as compared to Ang-(1–7). A similar beneficial effect, albeit less pronounced, was detectable at the level of lung vascular remodeling. The fact that these changes were less pronounced than the effects on RVSP and missed the level of significance may indicate that the hemodynamic effects of Ang-(1–7) and cAng-(1–7) were, at least in part, attributable to the vasodilatory potential of Ang-(1–7) rather than to anti- or reverse-remodeling effects of the drug. Yet, notably, the dose-response pattern for the hemodynamic effects of Ang-(1–7) and cAng-(1–7) was closely reflected in the vascular-remodeling response, in that the most prominent effects were detectable for animals treated with 100 μg kg−1 day−1 of Ang-(1–7) and with 30, 300, or 900 μg kg−1 day−1 of cAng-(1–7). Overall, the therapeutic effects of both Ang-(1–7) and cAng-(1–7) were more evident in small than in medium-sized arterioles, which is in line with the finding that MCT-induced vascular remodeling was most pronounced in the 20–50-μm pulmonary blood vessels. RVH was also reduced in all treatment groups, as compared to the MCT-only group; however, this effect was significant only for the group treated with 300 μg kg−1 day−1 of Ang-(1–7) and groups treated with 30 or 900 μg kg−1 day−1 of cAng-(1–7). Notably, these groups also showed a marked decline in RVSP yet only a modest vascular-remodeling response, which may again indicate that Ang-(1–7) acted more as a pulmonary vasodilator than as an antiremodeling drug.
To specifically examine the effects of Ang-(1–7) and cAng-(1–7) on endothelial function, we probed for eNOS and p-eNOS expression in lung tissue. Interestingly, for groups treated with Ang-(1–7), the combined ratio of p-eNOS to eNOS was inversely related to the effects on RVSP, in that the two treatment groups with the most prominent decrease in RVSP (100 and 300 μg kg−1 day−1) were at the same time the two groups with the most prominent increase in eNOS activity. Accordingly, the results for RVSP and eNOS activation for both the lowest and the highest Ang-(1–7) dose groups were virtually unchanged, compared to the MCT control. A similar inverse correlation between RVSP and eNOS activation was, however, not evident for groups treated with cAng-(1–7). Most treatment groups showed no discernible increase in eNOS activity or—in case of the lowest-dose group—even showed values below those for the MCT control, raising doubts about the efficacy of cAng-(1–7) on lung endothelial function in this model.
To test whether loss of the Ang-(1–7) receptor Mas may have limited the effectiveness of Ang-(1–7) or cAng-(1–7) treatment in our study, we probed lung samples for Mas receptor expression. Except for the two highest-dose groups of cAng-(1–7), all treatment groups showed strong Mas receptor expression above the level of both the control and the untreated MCT groups, indicating that the modest effectiveness of Ang-(1–7) or cAng-(1–7) was not attributable to a loss of target.
As a positive control, we treated rats with the endothelin receptor antagonist ambrisentan, an established drug for the treatment of PAH, which received approval by the Food and Drug Administration in 200724 and has been proven effective in PAH treatment in various studies.22,25,26 In line with these reports, treatment with ambrisentan significantly reduced RVSP, RVH, and vascular remodeling in our MCT rat model, therefore ruling out systematic errors in the experimental model.
It should be considered that this study tested the effects of Ang-(1–7) and cAng-(1–7) in only a single animal model of PAH. While the classic MCT model remains the most frequently utilized PAH model, other approaches, such as the Sugen-hypoxia model, have been proposed to yield a more accurate reflection of human PAH disease because of a lesser initial inflammatory component and the development of plexiform lesions.27 While we cannot exclude the possibility that Ang-(1–7) therapy may prove more beneficial in such models (and, ultimately, in patients), as compared to the MCT model, this notion seems unlikely, because (1) we are, at present, not aware of any treatment approach that has provided striking therapeutic benefit in the Sugen-hypoxia model yet not in the MCT model of PAH, and (2) Ang-(1–7) would, on the basis of its well-documented anti-inflammatory effects,28,29 be expected to yield false-positive rather than false-negative results in the MCT model.
Finally, we considered whether the moderate therapeutic effects in our study may have been attributable to the dosing regimen or route of delivery. Importantly, the bioavailability of Ang-(1–7) after subcutaneous injection is reported as 98%, suggesting that almost all injected Ang-(1–7) enters the bloodstream, which makes this route of delivery highly effective.30 However, the half-life of Ang-(1–7) in plasma is relatively short17 and may therefore have limited the therapeutic effectiveness in our study, as the drug was administered only once daily. As a consequence, effective Ang-(1–7) therapy may require more frequent administration of the drug, which, however, limits the feasibility of such an approach in the clinical scenario. In line with this view, ambrisentan, which was presumably taken up in a more continuous fashion with the drinking water, showed clear beneficial effects in our MCT model. Recently, studies by Magalhães and colleagues31 in which Ang-(1–7) was delivered continuously via implanted osmotic minipumps showed beneficial effects on lung vascular remodeling and RVH in a murine model of ovalbumin-induced chronic allergic lung disease. While we cannot exclude that continuous delivery via minipumps might have yielded more advantageous effects of Ang-(1–7) or cAng-(1–7) in our study, it is important to note that Magalhães and coworkers31 studied the effects of Ang-(1–7) in a model of chronic lung inflammation. In view of the potent anti-inflammatory action of Ang-(1–7),28,29 it may not be surprising that Ang-(1–7) was more potent in alleviating pathological changes in these chronically inflamed lungs independent of the actual mode of drug delivery.
In a series of studies, Ang-(1–7) has been applied successfully as a single daily injection with distinct beneficial effects in various disease models other than PAH.32,33 Consistently, measurements of Ang-(1–7) levels in lung homogenate (Fig. S1) show a good dose-response relation, demonstrating effective delivery of the drug to the lung and its accumulation in the pulmonary parenchyma. Nevertheless, the argument that the limited therapeutic effect in our study may have resulted from the short half-life of Ang-(1–7) cannot be completely refuted. However, the same argument cannot be equally applied for cAng-(1–7), which is largely resistant to degradation by ACE34 and, as a result, has a 34-fold longer half-life in vivo than Ang-(1–7).18 However, the improved bioavailability of cAng-(1–7) did not increase its therapeutic effectiveness over Ang-(1–7) in our study. Finally, a wide range of Ang-(1–7) concentrations, from 30 to 900 μg kg−1 day−1, were applied in our study, including concentrations below as well as above those commonly used in previous studies,33,35 again effectively arguing against insufficiency of the dosing regimen.
In summary, both Ang-(1–7) and cAng-(1–7), when administered immediately after induction of PAH, showed moderate beneficial effects in the MCT rat model. Because of the overall limited therapeutic effectiveness of the approach, we refrained from further pursuing this strategy in a therapeutic regime where Ang-(1–7) or cAng-(1–7) therapy would be initiated 3 weeks after MCT treatment, at a time when PAH and vascular remodeling were already established. The optimal dose for Ang-(1–7) in our study seems to be around 100–300 μg kg−1 day−1, whereas cAng-(1–7) already showed a moderate effect at the lowest dose (30 μg kg−1 day−1), in line with its longer half-life. Yet the extent of the therapeutic benefit was overall far less pronounced than what could be expected from previous reports. The results of our study therefore dampen enthusiasm for the therapeutic use of Ang-(1–7) and its derivatives in the treatment of PAH, which had been spurred by reports of a high therapeutic efficiency. While Ang-(1–7) clearly remains a promising strategy for a variety of hematological, immunological, and cardiovascular diseases, our results cannot encourage the pursuit of this strategy for the treatment of PAH in clinical trials.
Appendix. Supplementary Methods
Ang-(1–7) levels in rat lung homogenates were determined in duplicate with a rat Ang-(1–7) enzyme-linked immunosorbent assay kit (NeoScientific, Cambridge, MA) according to the manufacturer’s instructions.
Figure S1.
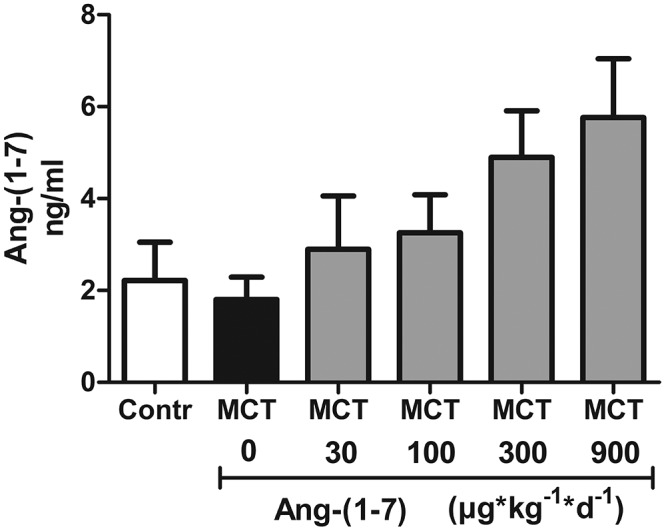
Angiotensin-(1–7) (Ang-(1–7)) levels in lung homogenate. Ang-(1–7) concentration was determined by enzyme-linked immunosorbent assay in lung homogenates of rats who had been injected with either vehicle (Contr), monocrotaline (MCT) only (MCT 0), or MCT plus Ang-(1–7) in doses of 30, 100, 300, or 900 μg kg−1 day−1. Each bar represents mean ± SEM of 6 experiments.
Source of Support: This work was supported by Tarix Pharmaceuticals. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest: None declared.
References
- 1.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S43–S54. [DOI] [PubMed]
- 2.Galiè N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res 2004;61(2):227–337. [DOI] [PubMed]
- 3.Hampl V, Herget J. Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev 2000;80(4):1337–1372. [DOI] [PubMed]
- 4.Rhodes CJ, Davidson A, Gibbs JSR, Wharton J, Wilkins MR. Therapeutic targets in pulmonary arterial hypertension. Pharmacol Ther 2009;121(1):69–88. [DOI] [PubMed]
- 5.Thenappan T, Shah SJ, Rich S, Tian L, Archer SL, Gomberg-Maitland M. Survival in pulmonary arterial hypertension: a reappraisal of the NIH risk stratification equation. Eur Respir J 2010;35(5):1079–1087. [DOI] [PMC free article] [PubMed]
- 6.Bader M, Santos RA, Unger T, Steckelings UM. New therapeutic pathways in the RAS. J Renin Angiotensin Aldosterone Syst 2012;13(4):505–508. [DOI] [PubMed]
- 7.Farag E, Maheshwari K, Morgan J, Esa WAS, Doyle DJ. An update of the role of renin angiotensin in cardiovascular homeostasis. Anesth Analg 2015;120(2):275–292. [DOI] [PubMed]
- 8.Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, Castellano RK, et al. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med 2009;179(11):1048–1054. [DOI] [PMC free article] [PubMed]
- 9.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 2003;100(14):8258–8263. [DOI] [PMC free article] [PubMed]
- 10.Sampaio WO, Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1–7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007;49(1):185–192. [DOI] [PubMed]
- 11.Heitsch H, Brovkovych S, Malinski T, Wiemer G. Angiotensin-(1–7)-stimulated nitric oxide and superoxide release from endothelial cells. Hypertension 2001;37(1):72–76. [DOI] [PubMed]
- 12.Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1–7). Am J Physiol Heart Circ Physiol 2007;292(2):H736–H742. [DOI] [PubMed]
- 13.Zhang F, Hu Y, Xu Q, Ye S. Different effects of angiotensin II and angiotensin-(1–7) on vascular smooth muscle cell proliferation and migration. PLoS ONE 2010;5(8):e12323. doi:10.1371/journal.pone.0012323. [DOI] [PMC free article] [PubMed]
- 14.Tallant EA, Diz DI, Ferrario CM. Antiproliferative actions of angiotensin-(1–7) in vascular smooth muscle. Hypertension 1999; 34(4):950–957. [DOI] [PubMed]
- 15.Chen L, Xiao J, Li Y, Ma H. Ang-(1–7) might prevent the development of monocrotaline induced pulmonary arterial hypertension in rats. Eur Rev Med Pharmacol Sci 2011;15(1):1–7. [PubMed]
- 16.Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva RA, Díez-Freire C, Dooies A, Jun JY, et al. The angiotensin-converting enzyme 2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 2010;182(8):1065–1072. [DOI] [PMC free article] [PubMed]
- 17.Yamada K, Iyer SN, Chappell MC, Ganten D, Ferrario CM. Converting enzyme determines plasma clearance of angiotensin-(1–7). Hypertension 1998;32(3):496–502. [DOI] [PubMed]
- 18.Durik M, van Veghel R, Kuipers A, Rink R, Haas Jimoh Akanbi M, Moll G, Danser AH, Roks AJ. The effect of the thioether-bridged, stabilized angiotensin-(1–7) analogue cyclic Ang-(1–7) on cardiac remodeling and endothelial function in rats with myocardial infarction. Int J Hypertens 2012;2012:536426. doi:10.1155/2012/536426. [DOI] [PMC free article] [PubMed]
- 19.Yin N, Kaestle S, Yin J, Hentschel T, Pries AR, Kuppe H, Kuebler WM. Inhaled nitric oxide versus aerosolized iloprost for the treatment of pulmonary hypertension with left heart disease. Crit Care Med 2009;37(3):980–986. [DOI] [PubMed]
- 20.Bueltmann M, Kong X, Mertens M, Yin N, Yin J, Liu Z, Koster A, Kuppe H, Kuebler WM. Inhaled milrinone attenuates experimental acute lung injury. Intensive Care Med 2009;35(1):171–178. [DOI] [PubMed]
- 21.Hoffmann J, Yin J, Kukucka M, Yin N, Saarikko I, Sterner-Kock A, Fujii H, et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur Respir J 2011;37(6):1400–1410. [DOI] [PubMed]
- 22.Galiè N, Badesch D, Oudiz R, Simonneau G, McGoon MD, Keogh AM, Frost AE, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2005;46(3):529–535. [DOI] [PubMed]
- 23.Haga S, Tsuchiya H, Hirai T, Hamano T, Mimori A. A novel ACE2 activator reduces monocrotaline-induced pulmonary hypertension by suppressing the JAK/STAT and TGF-β cascades with restored caveolin-1 expression. Exp Lung Res 2015;41(1):21–31. [DOI] [PubMed]
- 24.US Food and Drug Administration. FDA approves new orphan drug for treatment of pulmonary arterial hypertension. http://web.archive.org/web/20070623055608/http://www.fda.gov/bbs/topics/NEWS/2007/NEW01653.html. Published June 15, 2007. Accessed January 29, 2015.
- 25.Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, Badesch DB, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008;117(23):3010–3019. [DOI] [PubMed]
- 26.Nishida M, Okada Y, Akiyoshi K, Eshiro K, Takoaka M, Gariepy CE, Yanagisawa M, Matsumura Y. Role of endothelin ETB receptor in the pathogenesis of monocrotaline-induced pulmonary hypertension in rats. Eur J Pharmacol 2004; 496 (1–3):159–165. [DOI] [PubMed]
- 27.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010;121(25):2747–2754. [DOI] [PubMed]
- 28.Gaddam RR, Chambers S, Bhatia M. ACE and ACE2 in inflammation: a tale of two enzymes. Inflam Allergy Drug Targets 2014;13(4):224–234. [DOI] [PubMed]
- 29.Simões E, Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1–7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol 2013;169(3):477–492. [DOI] [PMC free article] [PubMed]
- 30.de Vries L, Reitzema-Klein CE, Meter-Arkema A, van Dam A, Rink R, Moll GN, Haas Jimoh Akanbi M. Oral and pulmonary delivery of thioether-bridged angiotensin-(1–7). Peptides 2010;31(5):893–898. [DOI] [PubMed]
- 31.Magalhães GS, Rodrigues-Machado MG, Motta-Santos D, Silva AR, Caliari MV, Prata LO, Abreu SC, et al. Angiotensin-(1–7) attenuates airway remodeling and hyperresponsiveness in a model of chronic allergic lung inflammation. Br J Pharmacol 2015;172(9):2330–2342. [DOI] [PMC free article] [PubMed]
- 32.El-Hashim AZ, Renno WM, Raghupathy R, Abduo HT, Akhtar S, Benter IF. Angiotensin-(1–7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways. Br J Pharmacol 2012;166(6):1964–1976. [DOI] [PMC free article] [PubMed]
- 33.Benter IF, Yousif MHM, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1–7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol 2007;292(1):H666–H672. [DOI] [PubMed]
- 34.Kluskens LD, Nelemans SA, Rink R, de Vries L, Meter-Arkema A, Wang Y, Walther T, Kuipers A, Moll GN, Haas M. Angiotensin-(1–7) with thioether bridge: an angiotensin-converting enzyme-resistant, potent angiotensin-(1–7) analog. J Pharmacol Exp Ther 2009;328(3):849–854. [DOI] [PubMed]
- 35.Yousif MH, Makki B, El-Hashim AZ, Akhtar S, Benter IF. Chronic treatment with Ang-(1–7) reverses abnormal reactivity in the corpus cavernosum and normalizes diabetes-induced changes in the protein levels of ACE, ACE2, ROCK1, ROCK2 and omega-hydroxylase in a rat model of type 1 diabetes. J Diabetes Res 2014;2014:142154. doi:10.1155/2014/142154. [DOI] [PMC free article] [PubMed]