

Abstract Abstract
Increased proliferation and resistance to apoptosis of pulmonary arterial vascular smooth muscle cells (PAVSMCs), coupled with metabolic reprogramming, are key components of pulmonary vascular remodeling, a major and currently irreversible pathophysiological feature of pulmonary arterial hypertension (PAH). We recently reported that activation of mammalian target of rapamycin (mTOR) plays a key role in increased energy generation and maintenance of the proliferative, apoptosis-resistant PAVSMC phenotype in human PAH, but the downstream effects of mTOR activation on PAH PAVSMC metabolism are not clear. Using liquid and gas chromatography–based mass spectrometry, we performed pilot metabolomic profiling of human microvascular PAVSMCs from idiopathic-PAH subjects before and after treatment with the selective adenosine triphosphate–competitive mTOR inhibitor PP242 and from nondiseased lungs. We have shown that PAH PAVSMCs have a distinct metabolomic signature of altered metabolites—components of fatty acid synthesis, deficiency of sugars, amino sugars, and nucleotide sugars—intermediates of protein and lipid glycosylation, and downregulation of key biochemicals involved in glutathione and nicotinamide adenine dinucleotide (NAD) metabolism. We also report that mTOR inhibition attenuated or reversed the majority of the PAH-specific abnormalities in lipogenesis, glycosylation, glutathione, and NAD metabolism without affecting altered polyunsaturated fatty acid metabolism. Collectively, our data demonstrate a critical role of mTOR in major PAH PAVSMC metabolic abnormalities and suggest the existence of de novo lipid synthesis in PAVSMCs in human PAH that may represent a new, important component of disease pathogenesis worthy of future investigation.
Keywords: mammalian target of rapamycin, pulmonary arterial hypertension, pulmonary arterial vascular smooth muscle cell metabolome
Pulmonary arterial hypertension (PAH) is a progressive disease, manifested by vasoconstriction and remodeling of small muscular pulmonary arteries (PAs) and leading to increased PA pressure, elevated right ventricular afterload, right heart failure, and death.1 Increased proliferation and resistance to apoptosis of distal pulmonary arterial vascular smooth muscle cells (PAVSMCs) are major components of pulmonary vascular remodeling, the mechanisms of which are not completely understood.2
Several lines of evidence demonstrate a close connection between increased proliferation and survival of PAVSMCs in PAH and alterations in major metabolic pathways. PAVSMCs in human PAH and experimental pulmonary hypertension (PH) have reduced mitochondrial glucose oxidation and metabolic shift to glycolysis supported by inhibition of pyruvate dehydrogenase (PDH) and upregulation of PDH kinase (PDK) and hypoxia-inducible factor 1α.3,4 Recently, deregulation of malonyl–coenzyme A (CoA) decarboxylase, acetyl-CoA carboxylase (ACC), and peroxisome proliferator-activated receptor γ (PPARγ) has been linked with human PAH PAVSMC proliferation and survival, indicating the role of lipid metabolism in vascular remodeling.5,6 Alterations in the redox system and oxidative stress also play critical roles in imbalanced proliferation and apoptosis of PAVSMCs in PAH.7,8 Such multiple abnormalities in metabolic modulators strongly suggest that PAVSMCs in PAH undergo metabolic reprogramming to support an energy-consuming, proliferative, prosurvival cell phenotype.
We recently reported that increased adenosine triphosphate (ATP) generation, proliferation, and survival of PAVSMCs in human PAH require persistent activation of protein kinase mammalian target of rapamycin (mTOR),6 a catalytic core of two multiprotein complexes, mTOR complex 1 (mTORC1) and mTORC2. We found that both complexes are required for vascular smooth muscle remodeling, with mTORC2 playing a critical role in energy generation, survival, and mTORC1-dependent proliferation of PAH PAVSMCs.6 This complex (mTORC2) is an emerging key regulator of glycolytic shift in cancer, and the role of mTORC1 as a master regulator of protein and lipid synthesis is well confirmed by extensive basic and translational studies.9 Together, published studies from our group and others strongly suggest an important role of mTOR in PAVSMC metabolic reprogramming in PAH.
In this study, we aimed to dissect the metabolic alterations in PAVSMCs in human PAH, with a specific focus on mTOR as a potential driver of metabolic reprogramming. We performed exploratory metabolomic analysis of primary distal PAVSMCs derived from lungs of subjects with idiopathic PAH and compared them with distal PAVSMCs from nondiseased human lungs and with the same cultures of PAH PAVSMCs treated with the selective mTOR kinase inhibitor PP242, which blocks mTOR catalytic activity in both mTORC1 and mTORC2.6 We found that PAH PAVSMCs have a distinct metabolomic profile compared to cells from normal lungs, with marked changes in lipid, carbohydrate, and redox metabolism, the majority of which were mTOR dependent. We further report new, PAH-specific alterations in major biochemicals involved in glycosylation that are reversed by mTOR inhibition. Collectively, our data demonstrate a critical role of mTOR in major metabolic abnormalities in PAH PAVSMCs and suggest that PAH PAVSMCs have de novo lipid synthesis linked to mTOR, which may represent novel, potentially important pathological mechanisms contributing to disease pathogenesis.
Methods
Human-cell cultures
PAVSMCs from small (<1,500-μm outer diameter), muscular PAs from subjects with idiopathic PAH and unused donor lungs (control) were provided by the Pulmonary Hypertension Breakthrough Initiative (PHBI), under protocols approved by the PHBI and University of Pittsburgh institutional review boards. Cells were isolated and characterized by the presence of smooth muscle markers (smooth muscle actin [SMA], smooth muscle protein 22 [SM22], and smooth muscle–myosin heavy chain [SM-MHC]) to confirm smooth muscle origin, as described previously,6 and then cultured in smooth muscle growth medium (SmGM) supplemented with SmGM growth kit (Lonza, Walkersville, MD). Before experiments, cells were incubated in Lonza Sm basal media supplemented with 0.1% bovine serum albumin for 24 hours and then treated with 10 μM mTOR kinase inhibitor PP242 (Tocris Bioscience, Bristol, UK) or vehicle for 24 hours. Cells of the same passage5 were used through all experiments. Cells from 5 subjects per group were analyzed for each experimental condition.
Metabolomic profiling
Cell monolayers were washed with ice-cold phosphate-buffered saline, cells were harvested by trypsinization, and cell pellets were snap-frozen in liquid nitrogen. Metabolomic profiling was performed by Metabolon (Durham, NC), as previously described.10 Briefly, cell pellets were subjected to a series of organic and aqueous extractions, divided into two fractions, frozen, and dried under vacuum, and liquid chromatography– and gas chromatography–based mass spectrometry were performed (Fig. S1). A full description of the methodology is provided in the appendix.
Statistical analysis and data presentation
Cells from 5 control and 5 idiopathic-PAH subjects were analyzed for each metabolite in each experimental condition. Raw data were normalized to total protein concentration assessed by Bradford assay, and log transformation and imputation of missing values for each compound, if any, were performed with the minimum observed value for each compound. Statistical analysis was performed with the R GNU software (Free Software Foundation). Statistically significant differences among groups were assessed with the Welch 2-sample t test, with values of P ≤ 0.05 sufficient to reject the null hypothesis and values 0.05 < P < 0.1 approaching significance. Multiple comparisons were accounted for by estimating the false discovery rate (FDR) with q values from the permuted P values.11,12 Data were presented as box-and-whiskers plots (see Fig. S2 for details). Principal-component analysis was performed on log-transformed data. Hierarchical clustering and z-score plots were created for changed metabolites for PAH versus control cells and for PAH cells treated with an mTOR inhibitor versus those treated with vehicle. For z-score plots, the mean and standard deviation of the control samples were determined for each metabolite. Then, each sample was centered by the control mean and scaled by the control standard deviation. Hierarchical clustering was performed on the log-transformed normalized data. The log-transformed data were median centered, per metabolite, before clustering. Euclidean distance was used for the similarity metric. A yellow/blue color scheme was used in heat maps of the metabolites. The reaction schemes for carbohydrate, lipid, nicotinamide adenine dinucleotide (NAD), and glutathione metabolism are based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database pathway maps (references are provided in the appendix).
Results
PAVSMCs from subjects with idiopathic PAH have a unique metabolomic signature
Comparison of metabolomic profiles of PAVSMCs from control and idiopathic-PAH lung (5 subjects/group) demonstrated that PAH PAVSMCs exhibit a distinct metabolic signature compared to control cells (Fig. 1A). Among 420 quantitatively detected compounds of known identity, 38 metabolites were found to be altered in PAH PAVSMCs compared to controls, 19 of which were significantly different (Fig. 1B, 1C). Nonsupervised hierarchical-clustering analysis of changed metabolites across the samples showed metabolic distinction between PAVSMCs from PAH and control subjects (Fig. 1D), demonstrating that distal PAVSMCs in PAH have a distinct metabolomic pattern compared to those from control lungs.
Figure 1.

Metabolomic profiling of microvascular pulmonary arterial vascular smooth muscle cells (PAVSMCs) from subjects with pulmonary arterial hypertension (PAH) and nondiseased donors. A, Principal-component analysis of the total metabolites detected in PAVSMCs. Red: nondiseased (control), blue: idiopathic PAH (5 subjects/group). B, Significantly different metabolites (P ≤ 0.05) and those approaching statistical significance (0.05 < P < 0.1) in PAH and control PAVSMCs (5 subjects/group), by the Welch 2-sample t test. Arrows indicate upregulated (red) and downregulated (green) metabolites. C, A z-score plot showing the levels of 38 differential metabolites normalized to the mean of the control samples. Red: cells from control subject; blue: cells from idiopathic PAH subject (5 subjects/group). D, Heat map representing the nonsupervised hierarchical clustering of 38 differential metabolites in PAH PAVSMCs (5 subjects/group) relative to normal sample data (5 subjects/group). Shades of yellow and blue represent the increase and decrease of a metabolite, respectively, relative to the median metabolite levels. The heat map scale (bottom) ranges from −4 to +4 on a log2 scale. UDP: uridine diphosphate.
mTOR is required for a majority of PAH-specific metabolomic changes
In order to evaluate the impact of mTOR on the metabolomic alterations in subjects with idiopathic PAH, we next performed metabolomic profiling of PAH PAVSMCs from the same subjects treated with the mTOR kinase inhibitor PP242 or vehicle. The mTOR inhibition had broad metabolic effects, as evident by Euclidean-distance shift, compared to vehicle-treated group (Fig. 2A) and resulted in significant alterations in 55 out of 420 metabolites (Fig. 2B, 2C). Hierarchical clustering revealed a clear distinction between treated and untreated groups (Fig. 2D), demonstrating the critical role of mTOR in PAH PAVSMC metabolism. Of note, PP242 improved the levels of 24 of the 38 metabolites that are differential between PAH and nondiseased cells (Table 1), showing the important role of mTOR in a majority of PAH-specific metabolomic changes.
Figure 2.

Effect of mTOR inhibition on metabolomic profile of pulmonary arterial hypertension (PAH) pulmonary arterial vascular smooth muscle cells (PAVSMCs). A, Principal-component analysis of the total metabolites detected in PAH PAVSMCs treated with the small adenosine triphosphate–competitive mTOR inhibitor PP242 (blue) compared to vehicle-treated PAH PAVSMCs (red); 5 subjects/group. B, Significantly changed metabolites (P ≤ 0.05) and those approaching statistical significance (0.05 < P < 0.1) after PP242 treatment, compared to nontreated cells, by the Welch 2-sample t test. Arrows indicate upregulated (red) and downregulated (green) metabolites. C, A z-score plot of the 55 named metabolites differential between PP242-treated (blue) and vehicle-treated (red) groups, normalized to the mean of the control samples (5 subjects/group). D, Heat map showing relative levels of 55 differential metabolites in vehicle-treated (left) and PP242-treated (right) PAH PAVSMCs, arranged by unsupervised hierarchical clustering. Shades of yellow and blue represent an increase and decrease of a metabolite, respectively, related to the median metabolite levels. The heat map scale (bottom) ranges from −4 to +4 on a log2 scale. UDP: uridine diphosphate. mTOR: mammalian target of rapamycin.
Table 1.
Effect of mTOR inhibitor PP242 on human PAH PAVSMC deregulated metabolites
| PAH PAVSMCs | ||||||
|---|---|---|---|---|---|---|
| Biochemical name | Control | PAH | PAH/control ratio | Vehicle | PP242 | PP242/vehicle ratio |
| Eicosapentaenoate (EPA; 20:5n3) | 0.479 | 1.2577 | 2.63↑* | 1.1575 | 1.3281 | 1.15 |
| Docosahexaenoate (DHA; 22:6n3) | 0.6719 | 1.1076 | 1.65↑* | 1.1328 | 1.0652 | 0.94 |
| Caprylate (8:0) | 0.629 | 1.9576 | 3.11↑* | 1.7127 | 0.7639 | 0.45↓** |
| Bisphenol A monosulfate | 0.606 | 1.4518 | 2.4↑* | 1.5235 | 0.3854 | 0.25↓** |
| Penicillin G | 0.2923 | 2.5761 | 8.81↑* | 2.5431 | 0.2688 | 0.11↓** |
| N-acetylglucosamine 6-phosphate | 0.5371 | 1.4396 | 2.68↑# | 1.3159 | 1.3697 | 1.04 |
| Citrate | 0.935 | 1.6895 | 1.81↑# | 1.0616 | 0.6472 | 0.61↓** |
| Caprate (10:0) | 0.9623 | 1.3458 | 1.4↑# | 1.2588 | 0.8663 | 0.69↓## |
| Arachidonate (20:4n6) | 0.6341 | 1.0516 | 1.66↑# | 1.0401 | 1.2266 | 1.18 |
| Choline | 0.9411 | 1.1652 | 1.24↑# | 1.3329 | 0.9087 | 0.68↓** |
| Glycerophosphorylcholine (GPC) | 0.5603 | 1.2177 | 2.17↑# | 1.2177 | 1.3078 | 1.07 |
| Riboflavin (vitamin B2) | 0.4164 | 1.1935 | 2.87↑# | 1.2907 | 0.698 | 0.54 |
| Pyridoxine (vitamin B6) | 0.9501 | 1.4166 | 1.49↑# | 1.4069 | 0.6516 | 0.46↓** |
| N-acetylneuraminate | 1.4949 | 0.9071 | 0.61↓* | 0.974 | 1.1743 | 1.21 |
| Gluconate | 1.4773 | 0.5806 | 0.39↓* | 0.5666 | 1.6347 | 2.89↑** |
| Xylitol | 2.0647 | 0.6786 | 0.33↓* | 0.7505 | 1.6655 | 2.22↑** |
| Urate | 2.3562 | 0.7913 | 0.34↓* | 1.3067 | 0.8703 | 0.67 |
| Cytidine-3′-monophosphate (3′-CMP) | 1.8755 | 0.7057 | 0.38↓* | 0.6412 | 1.4894 | 2.32 |
| Pseudouridine | 1.7391 | 0.6058 | 0.44↓* | 0.7452 | 1.3515 | 1.81↑** |
| Adenosine 5′ diphosphoribose | 4.661 | 0.5081 | 0.3↓* | 0.4202 | 1.1663 | 2.78↑** |
| Cysteinylglycine | 1.7283 | 0.7535 | 0.35↓* | 0.6759 | 1.8305 | 2.71↑** |
| Aspartyltryptophan | 1.7118 | 0.5208 | 0.49↓* | 0.3178 | 1.7057 | 5.37↑** |
| Aspartylphenylalanine | 1.6649 | 0.5908 | 0.38↓* | 0.4835 | 1.7727 | 3.67↑** |
| Aspartate-glutamate | 1.3535 | 0.6626 | 0.49↓* | 0.7953 | 1.7233 | 2.17↑** |
| Beta-alanine | 2.3868 | 0.9075 | 0.38↓* | 1.0082 | 1.4353 | 1.42 |
| C-glycosyltryptophan | 2.3443 | 0.8296 | 0.35↓* | 0.8774 | 1.2138 | 1.38↑** |
| Pyroglutamine | 2.0257 | 0.9363 | 0.46↓* | 1.1066 | 1.1858 | 1.07 |
| Ribulose | 2.2799 | 0.8186 | 0.36↓# | 0.9362 | 1.4586 | 1.56↑## |
| UDP-glucuronate | 2.2888 | 0.5078 | 0.22↓# | 0.4717 | 0.7584 | 1.61↑## |
| Fructose | 1.2145 | 0.4937 | 0.41↓# | 0.7146 | 1.6642 | 2.33↑** |
| Docosatrienoate (22:3n3) | 1.5111 | 1.0544 | 0.7↓# | 1.0975 | 1.0558 | 0.96 |
| Scyllo-inositol | 2.3482 | 0.392 | 0.17↓# | 0.3547 | 0.5779 | 1.63 |
| 1-palmitoleoylglycerophosphoethanolamine | 1.3694 | 0.6247 | 0.46↓# | 0.77 | 1.0805 | 1.4↑## |
| Nicotinamide | 2.7705 | 1.0521 | 0.38↓# | 1.2287 | 0.898 | 0.73 |
| Aspartylvaline | 1.2538 | 0.6864 | 0.55↓# | 0.9112 | 1.6156 | 1.77↑## |
| Aspartylleucine | 1.4226 | 0.6961 | 0.49↓# | 0.6184 | 1.5914 | 2.57↑** |
| Cysteine | 1.1987 | 0.6932 | 0.58↓# | 0.4638 | 4.3122 | 9.3↑** |
| Homocysteine | 1.3576 | 0.8643 | 0.64↓# | 0.8831 | 1.8607 | 2.11↑** |
Data are means from 5 subjects/group. Boldface indicates deregulated metabolites. Arrows indicate increased (↑) or decreased (↓) metabolite levels. Significance was based on the Welch 2-sample t test. mTOR: mammalian target of rapamycin; PAH: pulmonary arterial hypertension; PAVSMCs: pulmonary arterial vascular smooth muscle cells; UDP: uridine diphosphate.
P ≤ 0.05 for PAH versus control.
0.05 < P < 0.1 for PAH versus control.
P ≤ 0.05 for PP242-treated versus vehicle-treated.
0.05 < P < 0.1 for PP242-treated versus vehicle-treated.
mTOR and sugar metabolism
To dissect metabolic pathways dysregulated in PAH, we performed mapping of metabolites significantly deregulated in PAH PAVSMCs to their respective biochemical pathways based on the KEGG database. While metabolomic analysis detected subtle changes in intermediates of glycolysis, we found marked changes in the levels of sugars, amino sugars, and nucleotide sugars, which are the readouts of protein and lipid glycosylation capacity and polysaccharide biosynthesis between PAH and control cells (Fig. 3). Specifically, PAH samples displayed significantly lower levels of N-acetylneuraminate and a similar strong trend for uridine diphosphate (UDP)-glucuronate, key metabolites in synthesis of glycoproteins/glycolipids (gangliosides), and glucosaminoglycans, respectively, compared to control cells. A similar pattern was observed for UDP-acetylglucosamine-UDP-acetylgalactosamine, UDP-galactose, and mannose-6-phosphate (Fig. 3). In addition, PAH PAVSMCs showed a reduction in the levels of fructose and glucose-6-phosphate, universal metabolites that also serve as intermediates in carbohydrate pathways related to glycosylation. Interestingly, in addition to intermediates of N- and O-glycosylation pathways, PAH PAVSMCs also had a significant decrease in C-glycosyltryptophan, the key metabolite involved in C-mannosylation, a recently discovered protein modification playing a pathological role in metabolic syndrome complications.13 Inhibition of mTOR with PP242 attenuated or reversed glycosylation-associated sugar deficiency in PAH PAVSMCs, including significant elevation of UDP-acetylglucosamine-UDP-acetylgalactosamine, C-glycosyltryptophan, fructose, and sorbitol. This pattern was also evident for UDP-galactose, mannose-6-phosphate, and glucose-6-phosphate. Therefore, PAH PAVSMCs have a mTOR-dependent deficit of sugars, amino sugars, and nucleotide sugars, intermediates of protein and lipid glycosylation, strongly suggesting disturbed glycosylation processes in PAH PAVSMCs.
Figure 3.

Intermediates of carbohydrate metabolism and glycosylation pathways in control (CTRL), pulmonary arterial hypertension (PAH) pulmonary arterial vascular smooth muscle cells (PAVSMCs), and mTOR inhibitor PP242- and vehicle-treated PAH PAVSMCs. Box plots for the metabolites involved in glycosylation metabolism, combined with the reaction scheme including key components and enzymes, are shown. The metabolite data are presented as box-and-whiskers graphs as follows: light yellow: nondiseased (control) PAVSMCs; light blue: PAH PAVSMCs; dark blue: vehicle-treated PAH PAVSMCs (Veh); orange: PP242-treated PAH PAVSMCs (details in Fig. S2). Data are in arbitrary units normalized by protein concentration from 5 subjects/group; *P ≤ 0.05, #0.05 < P < 0.1 by the Welch 2-sample t test. Enzymes that catalyze particular steps in the pathway are shown in blue boxes labeled with their abbreviations according to the Kyoto Encyclopedia of Genes and Genomes database. AGX: UDP-N-acetylhexosamine pyrophosphorylase 1; CMAS: N-acylneuraminate cytidylyltransferase; GALE: UDP-glucose epimerase; GALT: UDP-glucose:alpha-D-galactose-1-phosphate uridylyltransferase; GNAT: glucosamine-6-phosphate-acetyl transferase; HK: hexokinase; MPI: mannose-6-phosphate isomerase; NAGD: N-acetyl-glucosamine 6-phosphate deacetylase; NAGK: N-acetyl-glucosamine kinase; NANP: N-acylneuraminate-9-phosphatase; OGTs: O-GLCNAc transferases; PGM: phosphoglucomutase; PGM-3: phosphoglucomutase 3; PPM1: phosphomannomutase 1; SODH: sorbitol dehydrogenase; UAGD: UDP-N-acetylgalactosamine diphosphorylase; UAGE: UDP-N-acetylglucosamine 4-epimerase; UGD: UDP-glucuronate decarboxylase; UGDH: UDP-glucose 6-dehydrogenase. Ac-CoA: acetyl coenzyme A; CMP: cytidine monophosphate; Gln: glutamine; Glu: glutamic acid; mTOR: mammalian target of rapamycin; UDP: uridine diphosphate.
mTOR and lipid metabolism
We found that intermediates of medium-chain fatty acid metabolism (caprylic and capric fatty acids, as well as citrate, the precursor of fatty acid biosynthesis; Fig. 4A) and the key intermediates of polyunsaturated fatty acid (PUFA) metabolism were increased in PAH PAVSMCs compared to controls. Specifically, we detected a significant increase in the major precursors of eicosanoids, eicosapentaenoic acid (EPA) and docosaexaenoic acid (DHA), and a trend to increase for arachidonic acid (AA; Fig. 4B). Inhibition of mTOR with PP242 reversed citrate, caprylate, and caprate levels and reduced the levels of palmitate, consistent with a role of mTOR in altered fatty acid biosynthesis in human PAH PAVSMCs. PP242, however, had no effect on PUFA levels in PAH PAVSMCs (Fig. 4B), indicating that mTOR acts in parallel or downstream of eicosanoid mediator formation. Interestingly, we also detected an imbalance of metabolites involved in choline-conjugated phospholipid biosynthesis and degradation, with changes that trended toward the opposite patterns (Fig. 4C). The effect of mTOR inhibition on choline-conjugated phospholipid metabolism in PAH PAVSMCs was also divergent. For instance, PP242 significantly abolished choline to the levels comparable with those in control cells and improved levels of cytidine diphosphate (CDP)-choline without evident effects on glycerophosphorylcholine (GPC; Fig. 4C). Taken together, these observed changes suggest that eicosanoid and medium-chain fatty acid metabolism are deregulated in PAH PAVSMCs and that mTOR likely controls medium-chain fatty acid synthesis. Trends in deregulation of choline-conjugated phospholipid metabolism are suggestive of potential alterations in membrane dynamics in PAH PAVSMCs.
Figure 4.

Lipid metabolism in nondiseased (CTRL) and pulmonary arterial hypertension (PAH) pulmonary arterial vascular smooth muscle cells (PAVSMCs) in the presence or absence of mTOR inhibitor PP242. Box plots for changed metabolites/intermediates of lipid metabolism, combined with reaction schemes including key components and enzymes, are shown. Data for PAVSMCs (5 subjects/group) are presented as box-and-whiskers graphs as follows: light yellow: nondiseased (control); light blue: PAH PAVSMCs; dark blue: vehicle-treated PAH PAVSMCs (Veh); orange: PP242-treated PAH PAVSMCs (details in Fig. S2). Data are in arbitrary units specific to internal standards for each quantified metabolite normalized by total protein concentration. *P ≤ 0.05, #0.05 < P < 0.1 by the Welch 2-sample t test. Enzymes that catalyze particular steps in the pathway are shown in blue boxes labeled with their abbreviations according to the Kyoto Encyclopedia of Genes and Genomes database. A, Fatty acid biosynthesis. ACC: acetyl-coenzyme A (CoA) carboxylase; OLAH: dodecanoyl-acyl-carrier-protein hydrolase. B, Key metabolites of polyunsaturated fatty acid metabolism: intermediates of eicosanoid formation. COX: cyclo-oxygenase; LOX: lipoxygenase. CDP: cytidine diphosphate; CMP: cytidine monophosphate. C, Choline-conjugated phospholipid metabolism. CCT: phosphocholine cytidylyltransferase; CHK: choline kinase; CHPT: diacylglycerol cholinephosphotransferase; DAG: diacylglycerol; FA: fatty acid; GPC-PDE: glycerophosphocholine phosphodiesterase; LPLA: lysophospholipase. mTOR: mammalian target of rapamycin.
mTOR, redox homeostasis, and the γ-glutamyl cycle
Multiple pathways associated with thiol metabolism were found to be downregulated in PAH PAVSMCs, on the basis of its metabolite readouts (Fig. 5). Several key biochemicals along the biosynthetic pathway leading to glutathione, a key cellular antioxidant, were decreased in PAH, compared to control, PAVSMCs. Homocysteine, cysteine, both reduced and oxidized glutathione (GSH and GSSG, respectively), and 5-methyltetrahydrofolate (5MeTHF) showed a similar pattern of lower levels in PH compared to control cells. Also, cysteinylglycine, one of the major intermediates of γ-glutamyl cycle, and pyroglutamine, an amino modification of 5-oxoproline, were significantly lower in PAH compared to control group (Fig. 5). Importantly, inhibition of mTOR significantly increased 5MeTHF, homocysteine, cysteine, cysteinylglycine, and both GSH and GSSG content and decreased the level of 5-oxoproline (Fig. 5). Surprisingly, we found no effects of PP242 on pyroglutamine level. Taken together, these data indicate mTOR-dependent deficiency of the GSH-GSSG redox system in PAH PAVSMCs.
Figure 5.

Components of cysteine metabolism, redox homeostasis, and the γ-glutamyl cycle in control (CTRL), pulmonary arterial hypertension (PAH) pulmonary arterial vascular smooth muscle cells (PAVSMCs), and PAH PAVSMCs treated with mTOR kinase inhibitor PP242. Box plots for altered metabolites, combined with reaction schemes including key components and enzymes, are shown. Data for PAVSMCs (5 subjects/group) are presented as box-and-whiskers graphs as follows: light yellow: nondiseased (control); light blue PAH PAVSMCs; dark blue: vehicle-treated PAH PAVSMCs (Veh); orange: PP242-treated PAH PAVSMCs (details in Fig. S2). Data are in arbitrary units specific to internal standards for each quantified metabolite and normalized by total protein concentration. *P ≤ 0.05, #0.05 < P < 0.1 by the Welch 2-sample t test. Enzymes that catalyze particular steps in the pathway are shown in blue boxes labeled with their abbreviations according to the Kyoto Encyclopedia of Genes and Genomes database. CBS: cystathionine-β-synthase; CSE: cystathionine gamma-lyase (cystathionase); GCS: glutamylcysteine synthetase; GGT: gamma-glutamyltranspeptidase 1; GS: glutathione synthetase; GSR: glutathione reductase; MTR: methyltetrahydrofolate-homocysteine methyltransferase; OXRs: oxidoreductases. cys-gly: cysteinylglycine; GSH: reduced glutathione; GSSG: oxidized glutathione; SAH: S-adenosylhomocysteine; SAM: S-adenosylmethionine; THF: tetrahydrofolate. mTOR: mammalian target of rapamycin.
mTOR and nicotinamide cofactors
Comparative analysis revealed that PAH PAVSMCs have reduced levels of several cofactors, including the key energy metabolism redox pair NAD-NADH (reduced NAD); the products of NAD degradation, nicotinamide and adenosine 5′ diphosphoribose (ADPR), were also markedly reduced in PAH cells compared to controls (Fig. 6A). Inhibition of mTOR with PP242 significantly increased NAD, ADPR, and 1-methylnicotinamide levels without an evident effect on NADH and nicotinamide (Fig. 6A), suggesting a partial role for mTOR in nicotinamide cofactor biogenesis.
Figure 6.

Box plots for altered metabolites, combined in A with reaction schemes. Data from 5 subjects/group are presented as box-and-whiskers graphs as follows: light yellow: nondiseased (control [CTRL]); light blue: pulmonary arterial hypertension (PAH) pulmonary arterial vascular smooth muscle cells (PAVSMCs); dark blue: vehicle-treated PAH PAVSMCs (Veh); orange: PP242-treated PAH PAVSMCs (details in Fig. S2). Data are in arbitrary units specific to internal standards for each metabolite and normalized by total protein concentration. *P ≤ 0.05, #0.05 < P < 0.1 by the Welch 2-sample t test. A, Nicotinamide metabolism. Enzymes that catalyze particular steps in the pathway are shown in blue boxes labeled with their abbreviations according to the Kyoto Encyclopedia of Genes and Genomes database. CD157: ADP-ribosyl cyclase 2; CD38: cyclic ADP ribose hydrolase; NAD: nicotinamide adenine dinucleotide; NNMT: nicotinamide N-methyltransferase; SAM: S-adenosylmethionine. ADP: adenosine diphosphate. B, Aspartyl-containing dipeptides are significantly reduced in PAH PAVSMCs, in an mTOR-dependent manner. mTOR: mammalian target of rapamycin.
mTOR and aspartyl-containing dipeptides
Interestingly, we found that all detected aspartyl-containing dipeptides were significantly decreased or showed a trend to decreased levels (Fig. 6B). This decrease appeared to be controlled by mTOR and reversed by mTOR inhibition by PP242 (Fig. 6B).
Discussion
Increased proliferation and resistance to apoptosis of microvascular PAVSMCs are major components of pulmonary vascular remodeling, a key and currently irreversible feature of PAH.14,15 PAVSMCs in PAH undergo reprogramming of major metabolic and synthetic pathways, as well as the redox system, to support an energy-consuming, proliferative, apoptosis-resistant diseased phenotype.3,5-7,16-18 Metabolomic analysis of whole-lung samples from PAH subjects showed disrupted glycolysis, de novo synthesis of bile salts, increased tricarboxylic acid cycle, and fatty acid metabolites with altered oxidation pathways compared to nondiseased lungs,19,20 but targeted metabolomic analysis of PAVSMCs in PAH has never been performed, and the mechanisms regulating PAVSMC metabolic reprogramming are not fully understood.
In this study, we aimed to explore the role of mTOR in the PAH PAVSMC metabolome, using unbiased metabolomic profiling. Studies by our group and others demonstrate that early-passage PAVSMCs retain molecular and functional abnormalities found in PAH lungs in vivo (i.e., increased proliferation, survival, and persistent mTOR activation),6,21 which makes these cells an ideal model for exploratory research. We report that PAH PAVSMCs have a unique metabolomic signature distinct from that of nondiseased cells and that mTOR inhibition attenuates or reverses the levels of ∼60% of altered metabolites, showing a broad effect of mTOR on PAH-specific metabolomic changes. Mapping the metabolites to their respective pathways revealed marked differences in lipid, sugar, redox, and NAD metabolism in human PAH PAVSMCs, most of which appeared to be mTOR dependent.
Sugar metabolism
We found that human PAH PAVSMCs have a deficiency of multiple amino sugars and nucleotide sugars—essential components of protein and lipid glycosylation, including key components of glycoproteins, glycolipids (gangliosides), and glucosaminoglycans, as well as C-glycosyltryptophan, the central metabolite involved in C-mannosylation.13 These alterations appeared to be highly mTOR dependent and reversible by mTOR inhibition. While understudied compared to other posttranslational modifications, cellular glycans play important roles in key biological processes, including cell adhesion, receptor activation, molecular trafficking, clearance, and signal transduction, and they modulate Ca2+ release and metabolic and proliferative responses.22,23 Thus, mammalian glycans in the Golgi modulate the endocytosis of cell-surface glycoproteins that controls receptor expression and, in turn, respective signaling. As an example, N-acetylglucosamine GlcNAcT-V linkage retards epidermal growth factor and transforming growth factor (TGF)-β receptor endocytosis, and O-fucose linkage regulates Notch receptor trafficking, ligand binding, and activation,23 suggesting potential links between disturbed glycosylation, growth factors, and Notch-signaling abnormalities detected in PAH PAVSMCs. Dysregulated glycosylation, caused mostly by failure of distinct steps in glycan formation, leads to severe morphogenic and metabolic disorders and is linked to a number of genetic diseases and cancer,23,24 and a recent study from the Dweik group provides a strong mechanistic link between dysregulated glycosylation and PAVSMC proliferation in PAH.25 Such a broadly shared deficit of sugars, amino sugars, and nucleotide sugars in the PAH cohort and its high dependence on mTOR activity strongly suggest that PAVSMCs in PAH have mTOR-dependent alterations of protein and lipid glycosylation that may dramatically affect multiple signaling pathways involved in disease pathogenesis.
Lipid metabolism
Analysis of the biochemicals involved in fatty acid metabolism showed that human PAH PAVSMCs have increased levels of the key components of medium-chain fatty acid synthesis pathways, including citrate, caprylate, and caprate. Together with our previous report showing that PAH PAVSMCs have increased levels of active dephosphorylated ACC,6 a key enzyme catalyzing a committed step in fatty acid biosynthesis, these data suggest existence of de novo lipid synthesis in PAVSMCs in human PAH. Of note, de novo fatty acid synthesis is one of the hallmarks of proliferative cancer cells,26 and chemical inhibition of ACC suppresses the self-renewal growth of cancer stem cells27 and selectively induces cancer cell growth arrest and apoptosis.28 Importantly, mTOR inhibition dramatically reduced the levels of fatty acid synthesis intermediates, which is in good agreement with our previous findings showing the requirement of mTORC2 for ACC activation in PAH PAVSMCs6 and with the reports from the Sheng and Kim groups demonstrating the critical role of mTOR in fatty acid synthase expression, ACC activation, and increased growth and survival of breast cancer cells.29,30 Given that targeting de novo fatty acid synthesis is considered a promising therapeutic option to develop anticancer therapies,31 mTOR-dependent lipogenesis might be considered a new, potentially attractive target pathway for therapeutic intervention in PAH.
PAH PAVSMCs also showed changed levels of biochemicals involved in choline-conjugated phospholipid metabolism, also proving the link between proliferative human PAH PAVSMCs and cancer. Increased choline metabolism is an emerging metabolic hallmark of oncogenesis. Elevated levels of choline-containing precursors and breakdown products of membrane phospholipids are strongly associated with cancer cell proliferation and are suggested as noninvasive biomarkers of tumor transformation and response to therapy,32 and choline kinase (CHK), the key enzyme of choline metabolism, is under consideration as a novel cancer therapeutic target.32 Our data show an mTOR-dependent increase in the levels of choline, a CHK substrate, in PAH PAVSMCs. Our new data and findings from the cancer field suggest a potential link between mTOR, CHK, and choline metabolism in PAVSMCs in human PAH, further exploration of which could lead to establishing a new target pathway.
In addition to mTOR-dependent changes in lipid metabolites, we detected an increase in the intermediates of the PUFA pathway— omega-6 AA, omega-3 eicosapentaenoate (EPA), and docosahexaenoate (DHA)—that was insensitive to mTOR inhibition. Given that AA and EPA control biosynthesis of eicosanoids (reviewed by Serhan et al.33), the observed significant increase in key PUFAs likely contributes to altered eicosanoid metabolism, a known pathological manifestation of idiopathic PAH.34 AA and its metabolites are also confirmed stimulators of the mammalian transient receptor potential 6 (TRPC6) channel that regulates intracellular Ca2+ release and is upregulated in PAH PAVSMCs,35 suggesting a strong link between increased AA levels and altered Ca2+ release detected in PAH. Unesterified AA is liberated from membrane phospholipids by class A2 phospholipases (PLA2), suggesting potential PLA2 upregulation in PAVSMCs from idiopathic-PAH lungs. While there is no direct evidence linking PLA2 and human PAH, endothelin 1 and reactive oxygen species (ROS) activate cPLA2 and induce AA release in human and rabbit PAVSMCs,36,37 and PLA2 activation was reported as a regulator of acute PA pressure in rats exposed to hypoxia.38 Interestingly, increased PLA2 and AA levels in human breast cancer are required for mTORC1 and mTORC2 activation and cell proliferation,39 suggesting that increased levels of PUFAs in PAH PAVSMCs might contribute to PAVSMC-specific mTOR activation in human PAH and experimental PH that was recently reported by our group.6
Redox homeostasis and the γ-glutamyl cycle
Oxidative stress is one of the features of PAH pathogenesis made evident by increased levels of DNA, protein, and lipid oxidation in the lungs of PH subjects.7 A major cellular endogenous soluble antioxidant is cysteine-containing tripeptide glutathione, a pair of reduced (GSH) and oxidized (GSSG) forms of which, controlled by the γ-glutamyl cycle, maintains redox homeostasis in mammalian cells.40 We found that PAH PAVSMCs have reduced levels of major intermediates of the γ-glutamyl cycle, associated with a decrease in both reduced and oxidized forms of glutathione. These data suggest a deficiency in the GSH-GSSG redox system that might contribute to the alterations in ROS levels reported in PAH PAVSMCs.7,17 Interestingly, significant downregulation of expression and activity of cystathionine gamma-lyase (CSE), an enzyme of the γ-glutamyl cycle converting cystathionine to cysteine (and cysteine to pyruvate and H2S), is detected in the lung of rats with hypoxia-induced PH,41 also indicating that glutathione metabolism is defective in PH. Inhibition of mTOR led to a marked increase in key intermediates of glutathione synthesis as well as significant upregulation of both reduced and oxidized forms of glutathione, suggesting that mTOR inhibition may restore PAH-specific defects in the GSH-GSSG redox system, which will likely improve oxidant/antioxidant imbalance and increase PAVSMC antioxidant capacity. Of note, mTOR also controls mitochondrial oxidative function42 and contributes to ROS production,43 supporting its role in regulating redox signaling.
NAD metabolism
We found that intermediates of NAD metabolism (ADPR and nicotinamide) are significantly reduced in PAH PAVSMCs compared to the control group. The NAD-NADH pair acts as a cofactor in multiple processes, and reduced NAD levels may have a broad impact on other synthetic pathways. NAD is a substrate for CD38 (cyclic ADPR hydrolase) or CD157 (ADP-ribosyl cyclase) bifunctional enzymes, which convert NAD to cyclic ADPR, a well-known Ca2+-releasing second messenger, and then hydrolyze it to ADPR. Of note, hypoxia, one of known triggers of experimental PH, inhibits hydrolase activities of both CD38 and cyclic ADPR in rabbit PAVSMCs,44 suggesting a potential mTOR-dependent disruption of NAD-ADPR conversion in pulmonary hypertensive conditions that may affect both proliferation and vasomotor tone in PAH pulmonary vasculature.
The major limitation of this work is a small sample size that limits power of analysis and may be associated with the inability to detect potential differences in certain metabolites. Idiopathic PAH is a rare disease that limits availability of well-characterized primary microvascular PAVSMCs from the PAH cohort and prevents us from expanding the quantity of subjects to the 40–50 per group required to achieve low FDR, indicative of strong statistical performance as calculated by power analysis. Current FDRs are 36% for the PP242-vehicle PAH PAVSMC cohort and 98% for the control-PAH PAVSMC cohort. While corroborating previous mechanistic findings on human PAH and experimental PH, because of such limitations the data generated in this pilot study cannot be generalized to the broader human PAH population and must be integrated with prior knowledge to identify specific metabolites for targeted analysis in future studies and to validate/confirm findings.
In conclusion, we report that mTOR is a significant positive regulator of multiple, but not all, metabolic abnormalities found in PAH PAVSMCs, including altered fatty acid synthesis, a deficiency of glycosylation donors, alterations in choline-conjugated phospholipid metabolism, and a defective glutathione metabolism.
Recognizing the complexity of metabolic regulation and the important role of other signaling pathways in the development and maintenance of the proliferative, apoptosis-resistant PAVSMC phenotype in PAH, we anticipate that multiple cross talks might exist between mTOR and other known pathways/modulators of PAH pathogenesis. Indeed, mTORC2 contributes to upregulation of hypoxia-inducible factor 1α and Akt, known triggers of metabolic glycolytic shift in PH and cancer and positive regulators of PAVSMC proliferation and survival in human PAH and experimental PH.6,9,45-49 Recently reported mTORC2-dependent activation of ACC,6 one of the key enzymes/regulators of lipogenesis, provides a mechanistic link between mTOR and signaling pathways regulating lipid metabolism in PAH. Further, mTOR is regulated by NADPH oxidase Nox46 and, in turn, controls mitochondrial oxidative function42 and contributes to ROS production,43 strongly suggesting its multiple-level involvement in redox signaling. Although there is no direct evidence linking mTOR with bone morphogenetic protein receptor 2 (BMPR2) deficiency, the BMPR2 downstream effector peroxisome PPARγ inhibits mTORC1 in cancer cells,50,51 and Nox4 modulates expression of PPARγ under chronic hypoxia,52 suggesting potential cross talk between mTOR, BMPR2, redox signaling, and lipid metabolism in human PAH. While more studies should be done to dissect the exact role of mTOR in the PAH signaling network, it is now becoming clear that mTOR may act as an important component of several other signaling pathways, further supporting its potential attractiveness as a molecular target for therapeutic intervention.
Given recent advances in the pharmacological use of PP242 and other ATP-competitive mTOR inhibitors in animal models of PH and cancer,6,9,15 targeting mTOR may represent an attractive potential strategy to improve PAH-specific metabolic abnormalities supporting a proliferative, apoptosis-resistant PAVSMC phenotype. Our data also suggest that PAVSMCs in PAH have de novo lipid synthesis, which may represent an important component of disease pathogenesis that has never been studied in PAH. Taken together, metabolomic profiling highlights the important role of mTOR in PAH-specific metabolic alterations in human PAVSMCs and suggests new areas that are worthy of further investigation.
Acknowledgments
We thank the Pulmonary Hypertension Breakthrough Initiative for human lung specimens, Drs. Robert Mohney and Elisabeth Morin-Kensicki from Metabolon for their help with data analysis and presentation, and Rachel Farrell from the University of Pittsburgh Vascular Medicine Institute for critical reading of the manuscript.
Appendix. Supplemental methods
Sample preparation for metabolomic profiling analysis
Sample preparation from frozen cell pellets used the automated MicroLab STAR (Hamilton, Reno, NV). Recovery standards were added before the first step in the extraction process for quality control (QC) purposes. Sample preparation was conducted with a proprietary series of organic and aqueous extractions to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into two fractions: one for analysis by liquid chromatography (LC) and one for analysis by gas chromatography (GC). Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. Each sample was then frozen and dried under vacuum. Samples were then prepared for the appropriate instrument, either LC mass spectrometry (LC/MS) or GC/MS.
Quality assurance (QA) and QC
For QA/QC purposes, a number of additional samples were included with each analysis. A selection of QC compounds were added to every sample, including those under test. These compounds were carefully chosen so as not to interfere with the measurement of the endogenous compounds. These QC samples were primarily used to evaluate the process control for each study as well as aid in data curation.
LC/MS, LC/MS2 (tandem MS)
The LC/MS portion of the platform was based on a Waters ACQUITY ultraperformance liquid chromatography (UPLC) system and a Thermo-Finnigan LTQ mass spectrometer, which consisted of an electrospray ionization (ESI) source and linear ion trap (LIT) mass analyzer. The sample extract was split into two aliquots, dried, and then reconstituted in acidic or basic LC-compatible solvents, each of which contained 11 or more injection standards at fixed concentrations. One aliquot was analyzed with acidic positive ion–optimized conditions and the other with basic negative ion–optimized conditions in two independent injections using separate dedicated columns. Extracts reconstituted in acidic conditions were gradient eluted with water and methanol, both containing 0.1% formic acid, while the basic extracts, which also used water/methanol, contained 6.5 mM ammonium bicarbonate. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion.
GC/MS
The samples destined for GC/MS analysis were redried under vacuum desiccation for a minimum of 24 hours before being derivatized under dried nitrogen using bistrimethyl-silyl-triflouroacetamide (BSTFA). The GC column was 5% phenyl, and the temperature ramp was from 40° to 300°C in a 16-minute period. Samples were analyzed on a Thermo-Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization. The instrument was tuned and calibrated daily for mass resolution and mass accuracy. The information output from the raw data files was automatically extracted as discussed below.
Accurate mass determination and MS/MS fragmentation (LC/MS, LC/MS/MS)
The LC/MS portion of the platform was based on a Waters ACQUITY UPLC and a Thermo-Finnigan LTQ-FT mass spectrometer, which had an LIT front end and a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer back end. For ions with counts greater than 2 million, an accurate mass measurement could be performed. Accurate mass measurements could be made on the parent ion as well as fragments. The typical mass error was less than 5 ppm. Ions with less than 2 million counts required a greater amount of effort to characterize. Fragmentation spectra (MS/MS) were typically generated in a data-dependent manner, but if necessary targeted MS/MS could be employed, as in the case of lower-level signals.
Data extraction and QA
The data extraction of the raw MS data files yielded information that could be loaded into a relational database and manipulated without resorting to BLOB manipulation. Once in the database, the information was examined and appropriate QC limits were imposed. Peaks were identified with Metabolon’s proprietary peak integration software, and component parts were stored in a separate and specifically designed complex data structure.
Compound identification
Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Identification of known chemical entities was based on comparison to metabolomic library entries of purified standards. More than 1,000 commercially available purified standard compounds have been acquired and registered into the Laboratory Information Management System (LIMS) for distribution to both the LC and GC platforms for determination of their analytical characteristics. The combination of chromatographic properties and mass spectra gave an indication of a match to the specific compound or an isobaric entity.
Reaction schemes
The reaction schemes depicted in Figures 3–6 are based on pathways from KEGG databases as follows:
-
1
Amino sugar and nucleotide sugar metabolism: http://www.genome.jp/kegg-bin/show_pathway?map00520+C00043 (Fig. 3).
-
2
Fructose and mannose metabolism: http://www.genome.jp/kegg-bin/show_pathway?map00051.
-
3
Biosynthesis of unsaturated fatty acids: http://www.genome.jp/kegg-bin/show_pathway?map01040 (Fig. 4).
-
4
Fatty acid biosynthesis: http://www.genome.jp/kegg/pathway/map/map00061.html.
-
5
Glycerophospholipid metabolism: http://www.genome.jp/kegg/pathway/map/map00564.html.
-
6
Glutathione metabolism: http://www.genome.jp/kegg/pathway/map/map00480.html (Fig. 5).
-
7
Cysteine and methionine metabolism: http://www.genome.jp/kegg/pathway/map/map00270.html.
-
8
Nicotinate and nicotinamide metabolism: http://www.genome.jp/kegg/pathway/map/map00760.html (Fig. 6).
Supplementary figures
Figure S1.
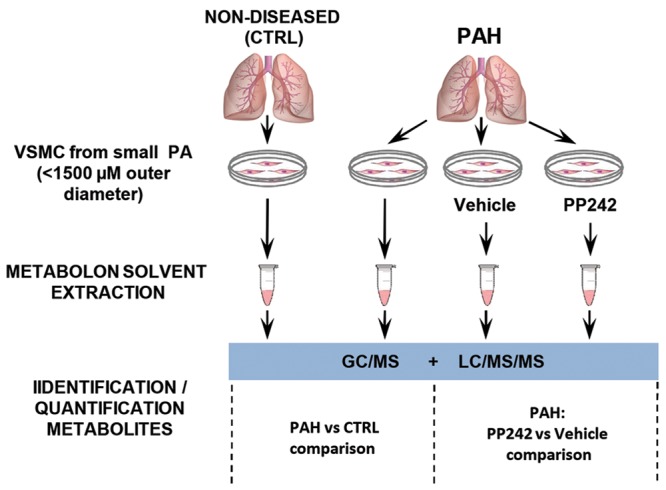
Experimental scheme of metabolomic profiling. Distal pulmonary artery (PA) vascular smooth muscle cells (VSMCs) from nondiseased (control) and idiopathic pulmonary arterial hypertension (PAH) subjects were snap-frozen, and cell pellets were subjected to liquid chromatography– and gas chromatography–based mass spectrometry analysis (LC/MS/MS and GC/MS, respectively). Comparisons between control and PAH groups and between PAH PAVSMCs treated with vehicle and those treated with PP242 were performed for each indicated metabolite.
Figure S2.
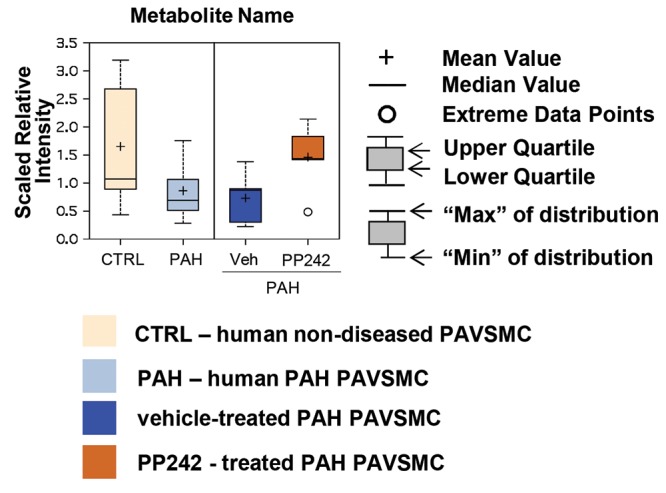
Metabolite box plots. Data for each metabolite obtained from pulmonary arterial vascular smooth muscle cells (PAVSMCs) from 5 subjects/group are presented as box-and-whiskers graphs. Data are quantity of metabolite in PAVSMCs, presented in arbitrary units specific to internal standards for each quantified metabolite and normalized by protein concentration. Light yellow: control (CTRL); light blue: pulmonary arterial hypertension (PAH) PAVSMCs; dark blue: vehicle-treated PAH PAVSMCs (Veh); orange: PP242-treated PAH PAVSMCs.
Source of Support: This work is supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants R01HL113178 (EAG) and K24 HL103844 (SMK). The Pulmonary Hypertension Breakthrough Initiative is supported by the Cardiovascular Medical Research and Educational Fund and National Institutes of Health/National Heart, Lung, and Blood Institute grant R24HL123767.
Conflict of Interest: None declared.
Supplements
Appendix: Data supplementPulmCirc-005-667.s001.pdf (920.5KB, pdf)
References
- 1.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, Elliott CG, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S43–S54. [DOI] [PubMed]
- 2.Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, et al. Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186(3):261–272. [DOI] [PMC free article] [PubMed]
- 3.Sutendra G, Dromparis P, Bonnet S, Haromy A, McMurtry MS, Bleackley RC, Michelakis ED. Pyruvate dehydrogenase inhibition by the inflammatory cytokine TNFα contributes to the pathogenesis of pulmonary arterial hypertension. J Mol Med 2011;89(8):771–783. [DOI] [PubMed]
- 4.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 2012;185(3):260–266. [DOI] [PMC free article] [PubMed]
- 5.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2010;2(44):44ra58. doi:10.1126/scitranslmed.3001327. [DOI] [PubMed]
- 6.Goncharov DA, Kudryashova TV, Ziai H, Ihida-Stansbury K, DeLisser H, Krymskaya VP, Tuder RM, Kawut SM, Goncharova EA. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014;129(8):864–874. [DOI] [PMC free article] [PubMed]
- 7.Wong CM, Bansal G, Pavlickova L, Marcocci L, Suzuki YJ. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal 2013;18(14):1789–1796. [DOI] [PMC free article] [PubMed]
- 8.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 2010;121(24):2661–2671. [DOI] [PMC free article] [PubMed]
- 9.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011;12(1):21–35. [DOI] [PMC free article] [PubMed]
- 10.Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci USA 2011;108(8):3270–3275. [DOI] [PMC free article] [PubMed]
- 11.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 2003;100(16):9440–9445. [DOI] [PMC free article] [PubMed]
- 12.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009;457(7231):910–914. [DOI] [PMC free article] [PubMed] [Research Misconduct Found]
- 13.Ihara Y, Manabe S, Kanda M, Kawano H, Nakayama T, Sekine I, Kondo T, Ito Y. Increased expression of protein C-mannosylation in the aortic vessels of diabetic Zucker rats. Glycobiology 2005;15(4):383–392. [DOI] [PubMed]
- 14.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 2009;54(1 suppl.):S20–S31. [DOI] [PMC free article] [PubMed]
- 15.Goncharova EA. mTOR and vascular remodeling in lung diseases: current challenges and therapeutic prospects. FASEB J 2013;27(5):1796–1807. [DOI] [PMC free article] [PubMed]
- 16.Law RE, Goetze S, Xi XP, Jackson S, Kawano Y, Demer L, Fishbein MC, Meehan WP, Hsueh WA. Expression and function of PPARγ in rat and human vascular smooth muscle cells. Circulation 2000;101(11):1311–1318. [DOI] [PubMed]
- 17.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 2008;294(2):H570–H578. [DOI] [PubMed]
- 18.Gong K, Xing D, Li P, Aksut B, Ambalavanan N, Yang Q, Nozell SE, Oparil S, Chen YF. Hypoxia induces downregulation of PPAR-γ in isolated pulmonary arterial smooth muscle cells and in rat lung via transforming growth factor-β signaling. Am J Physiol Lung Cell Mol Physiol 2011;301(6):L899–L907. [DOI] [PMC free article] [PubMed]
- 19.Zhao Y, Peng J, Lu C, Hsin M, Mura M, Wu L, Chu L, et al. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS ONE 2014;9(2):e88727. doi:10.1371/journal.pone.0088727. [DOI] [PMC free article] [PubMed]
- 20.Zhao YD, Yun HZ, Peng J, Yin L, Chu L, Wu L, Michalek R, et al. De novo synthesize of bile acids in pulmonary arterial hypertension lung. Metabolomics 2014;10(6):1169–1175. [DOI] [PMC free article] [PubMed]
- 21.Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014;129(7):786–797. [DOI] [PubMed]
- 22.Dietrich A, Mederos y Schnitzler M, Emmel J, Kalwa H, Hofmann T, Gudermann T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J Biol Chem 2003;278(48):47842–47852. [DOI] [PubMed]
- 23.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell;126(5):855–867. [DOI] [PubMed]
- 24.Varki A, Kannagi R, Toole BP. Glycosylation changes in cancer. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, eds. Essentials of glycobiology. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2009:617–632. [PubMed]
- 25.Barnes JW, Tian L, Heresi GA, Farver CF, Asosingh K, Comhair SAA, Aulak KS, Dweik RA. O-linked β-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015;131(14):1260–1268. [DOI] [PMC free article] [PubMed]
- 26.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J 2012;279(15):2610–2623. [DOI] [PubMed]
- 27.Corominas-Faja B, Cuyàs E, Gumuzio J, Bosch-Barrera J, Leis O, Martin ÁG, Menendez JA. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014;5(18):8306–8316. [DOI] [PMC free article] [PubMed]
- 28.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res 2007;67(17):8180–8187. [DOI] [PubMed]
- 29.Yan C, Wei H, Minjuan Z, Yan X, Jingyue Y, Wenchao L, Sheng H. The mTOR inhibitor rapamycin synergizes with a fatty acid synthase inhibitor to induce cytotoxicity in ER/HER2-positive breast cancer cells. PLoS ONE 2014;9(5):e97697. doi:10.1371/journal.pone.0097697. [DOI] [PMC free article] [PubMed]
- 30.Yoon S, Lee M-Y, Park SW, Moon J-S, Koh Y-K, Ahn Y-H, Park B-W, Kim K-S. Up-regulation of acetyl-CoA carboxylase α and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J Biol Chem 2007;282(36):26122–26131. [DOI] [PubMed]
- 31.Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer 2009;100(9):1369–1372. [DOI] [PMC free article] [PubMed]
- 32.Glunde K, Bhujwalla ZM, Ronen SM. Choline metabolism in malignant transformation. Nat Rev Cancer 2011;11(12):835–848. [DOI] [PMC free article] [PubMed]
- 33.Serhan CN, Haeggström JZ, Leslie CC. Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J 1996;10(10):1147–1158. [DOI] [PubMed]
- 34.Christman BW. Lipid mediator dysregulation in primary pulmonary hypertension. Chest 1998;114(3_suppl.):205S–207S. [DOI] [PubMed]
- 35.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 2004;101(38):13861–13866. [DOI] [PMC free article] [PubMed]
- 36.Chakraborti S, Michael JR. Role of protein kinase C in oxidant-mediated activation of phospholipase A2 in rabbit pulmonary arterial smooth muscle cells. Mol Cell Biochem 1993;122(1):9–15. [DOI] [PubMed]
- 37.Meves H. Arachidonic acid and ion channels: an update. Br J Pharmacol 2008;155(1):4–16. [DOI] [PMC free article] [PubMed]
- 38.Li H, He J, Lee S, Quan C, Ding J. The study of the relationship between the activity of phospholipase A2 and acute hypoxic pulmonary arterial pressure. Ann Ist Super Sanita 1997;33(2):273–277. [PubMed]
- 39.Wen ZH, Su YC, Lai PL, Zhang Y, Xu YF, Zhao A, Yao GY, et al. Critical role of arachidonic acid-activated mTOR signaling in breast carcinogenesis and angiogenesis. Oncogene 2013;32(2):160–170. [DOI] [PubMed]
- 40.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct 2004;22(6):343–352. [DOI] [PubMed]
- 41.Zhang C, Du J, Bu D, Yan H, Tang X, Tang C. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun 2003;302(4):810–816. [DOI] [PubMed]
- 42.Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 2007;450(7170):736–740. [DOI] [PubMed]
- 43.Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, Salgia R, Podar K, Griffin JD, Sattler M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood 2005;105(4):1717–1723. [DOI] [PubMed]
- 44.Wilson HL, Dipp M, Thomas JM, Lad C, Galione A, Evans AM. ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase act as a redox sensor: a primary role for cyclic ADP-ribose in hypoxic pulmonary vasoconstriction. J Biol Chem 2001;276(14):11180–11188. [DOI] [PubMed]
- 45.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, et al. Hypoxia inducible-factor1α regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 2010;176(3):1130–1138. [DOI] [PMC free article] [PubMed]
- 46.Guignabert C, Tu L, Le Hiress M, Ricard N, Sattler C, Seferian A, Huertas A, Humbert M, Montani D. Pathogenesis of pulmonary arterial hypertension: lessons from cancer. Eur Respir Rev 2013;22(130):543–551. [DOI] [PMC free article] [PubMed]
- 47.BelAiba RS, Bonello S, Zähringer C, Schmidt S, Hess J, Kietzmann T, Görlach A. Hypoxia up-regulates hypoxia-inducible factor-1α transcription by involving phosphatidylinositol 3-kinase and nuclear factor κB in pulmonary artery smooth muscle cells. Mol Biol Cell 2007;18(12):4691–4697. [DOI] [PMC free article] [PubMed]
- 48.Shimoda LA, Laurie SS. HIF and pulmonary vascular responses to hypoxia. J Appl Physiol 2014;116(7):867–874. [DOI] [PMC free article] [PubMed]
- 49.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2015;308(2):L208–L220. [DOI] [PMC free article] [PubMed]
- 50.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, et al. An antiproliferative BMP-2/PPARγ/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest 2008;118(5):1846–1857. [DOI] [PMC free article] [PubMed]
- 51.Han S, Zheng Y, Roman J. Rosiglitazone, an agonist of PPARγ, inhibits non-small cell carcinoma cell proliferation in part through activation of tumor sclerosis complex-2. PPAR Res 2007;2007:29632. doi:10.1155/2007/29632. [DOI] [PMC free article] [PubMed]
- 52.Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol 2012;47(5):718–726. [DOI] [PMC free article] [PubMed]