

Abstract Abstract
Pulmonary fibrosis is often complicated by pulmonary hypertension (PH), and previous studies have shown a potential link between bone morphogenetic protein receptor II (BMPR2) and PH secondary to pulmonary fibrosis. We exposed transgenic mice expressing mutant BMPR2 and control mice to repetitive intraperitoneal injections of bleomycin for 4 weeks. The duration of transgene activation was too short for mutant BMPR2 mice to develop spontaneous PH. Mutant BMPR2 mice had increased right ventricular systolic pressure compared to control mice, without differences in pulmonary fibrosis. We found increased hypoxia-inducible factor (HIF)1-α stabilization in lungs of mutant-BMPR2-expressing mice compared to controls following bleomycin treatment. In addition, expression of the hypoxia response element protein connective tissue growth factor was increased in transgenic mice as well as in a human pulmonary microvascular endothelial cell line expressing mutant BMPR2. In mouse pulmonary vascular endothelial cells, mutant BMPR2 expression resulted in increased HIF1-α and reactive oxygen species production following exposure to hypoxia, both of which were attenuated with the antioxidant TEMPOL. These data suggest that expression of mutant BMPR2 worsens secondary PH through increased HIF activity in vascular endothelium. This pathway could be therapeutically targeted in patients with PH secondary to pulmonary fibrosis.
Keywords: idiopathic pulmonary fibrosis (IPF), pulmonary hypertension (PH), bone morphogenetic protein receptor II (BMPR2), hypoxia-inducible factor (HIF)
Idiopathic pulmonary fibrosis (IPF) is a diffuse lung disease marked by an insidious onset and a poor overall prognosis. Pulmonary hypertension (PH) complicating IPF, also known as secondary PH (World Health Organization Group III), affects a significant number of patients, with some studies suggesting anywhere from one-third to one-half of all patients with IPF having concurrent elevated pulmonary pressures and right ventricle (RV) failure. Secondary PH is defined by excessive vascular remodeling beyond the degree expected for chronic hypoxic vasoconstriction and is associated with both a decreased exercise capacity and increased mortality.1,2 Unfortunately, therapies approved for the treatment of idiopathic pulmonary arterial hypertension (PAH) have not proven effective in the treatment of PH associated with IPF, including ambrisentan3 and sildenafil.4 This patient population also fares worse with respect to lung transplantation, having increased instances of primary graft dysfunction.5
There is an urgent need to identify pathways involved in the development of secondary PH in order to develop novel therapeutics. One study examining genome-wide RNA expression profiling in lung samples from three populations (normal controls, PAH, and IPF with associated PH) identified a number of characteristic differences between these groups at the level of gene expression, among which was a significant change in the level of bone morphogenetic protein receptor II (BMPR2) in the secondary-PH group versus controls.6 BMPR2, a member of the transforming growth factor-β family, has previously been shown to play a major role in the development of PAH, though its exact mechanism is unknown.7 The importance of BMPR2 within vascular cells is known, with loss-of-function BMPR2 mutation resulting in increased endothelial cell apoptosis and development of PAH.8,9 In addition, a recent case report details development of IPF in a BMPR2-mutation-carrying patient with PAH, suggesting at least some shared mechanistic pathways between the diseases.10
Another pathway that is potentially relevant to the development and progression of PH associated with IPF involves hypoxic signaling, which is known to cause worsened vasoconstriction and elevated pulmonary artery pressure in BMPR2 mutation carriers compared to non-BMPR2-mutation-carrying controls.11 In addition, targets of hypoxia-inducible factor (HIF) signaling have been implicated in the development and progression of PH, including vascular endothelial growth factor.12 HIF is an oxygen-sensing transcription factor that is stabilized in response to low-oxygen tension, resulting in increased expression of hypoxia response element (HRE)–associated genes.13 Normoxic stabilization of HIF has previously been described in patients with PH,14 possibly mediated through a BMPR2-facilitated increase in reactive oxygen species (ROS) production.15 In addition, HIF has previously been implicated in the development of secondary PH in a chronic hypoxia murine model.16-19
Because of these previous reports of interaction between BMPR2 and HIF responsive signaling and because both pathways have been reported as important in the development of PH secondary to fibrosis, we investigated HIF and HIF target activity in the context of BMPR2 mutation in bleomycin-treated mice.
Methods
Transgenic mice
TetO7CMV-BMPR2Mut transgenic mice were generated as previously described.20 These mice have a dominantly acting BMPR2 mutation driven by a doxycycline-sensitive promoter. Transgenic mice were on the FVB/N background. Rosa26-rtTA2 and TetO7CMV-BMPR2Mut mice were crossed to produce double-transgenic mice with doxycycline-inducible expression of transgene in all tissue types.21 Mice were housed in animal care facilities at Vanderbilt University (Nashville, TN) with food/water ad lib. and provision of standard recommended room temperature (20°–26°C) and day∶night cycle (12∶12 hour cycle). The animal care facility is certified by the Association for Assessment and Accreditation of Laboratory Animal Care International, and all procedures were performed in compliance with the National Research Council for ethical handling of laboratory animals. The experimental protocol was approved by Vanderbilt’s Institutional Animal Care and Utilization Committee.
Primers and antibodies
Primer sequences used are itemized in Table 1. Antibodies used were as follows: α-smooth muscle actin (α-SMA), rabbit polyclonal (Abcam, Cambridge, MA); von Willebrand factor, rabbit polyclonal (DakoCytomation, Glostrup, Denmark); HIF1-α and connective tissue growth factor (CTGF), both rabbit polyclonal (Novus Biologicals, Littleton, CO); and BMPR2, goat polyclonal (Abcam).
Table 1.
List of primer sequences
| Species and gene | Sequence |
|---|---|
| Mouse: | |
| 18S, forward | ACCTGGTTGATCCTGCCAGTAG |
| 18S, reverse | TTAATGAGCCATTCGCAGTTTC |
| Bmpr2, forward | TGGGCGCACCAGCCGATTTC |
| Bmpr2, reverse | CAGCTGGCCAGGCAGCCAAC |
| Id-1, forward | GTGAGCAAGGTGGAGATCCTG |
| Id-1, reverse | GGTGGTCCCGACTTCAGACTC |
| Hif1-α, forward | TGGAGATGCTGGCTCCCTAT |
| Hif1-α, reverse | TGGAGGGCTTGGAGAATTGC |
| Ctgf, forward | GGGAGAACTGTGTACGGAGC |
| Ctgf, reverse | AGTGCACACTCCGATCTTGC |
| Human: | |
| 18S, forward | GTAACCCGTTGAACCCCATT |
| 18S, reverse | CCATCCAATCGGTAGTAGCG |
| CTGF, forward | GTGAGCCTCGTGCTGGAC |
| CTGF, reverse | GAAGAGGCCCTTGTGCGG |
Experimental design
Twelve-week-old 20–30-g Rosa26-rtTA2 mice (controls) and Rosa26-rtTA2 × TetO7CMV-BMPR2Mut mice had transgene expression induced by addition of doxycycline (Sigma-Aldrich, St. Louis, MO) in chow (1 g/kg) for 1 week. Mice then underwent intraperitoneal injection with 0.018 U/g bleomycin (reconstituted under sterile conditions with sterile phosphate-buffered saline and stored in 4°C for no more than 1 week after reconstitution; obtained from Bedford Laboratories, Bedford, OH) or vehicle twice weekly for 4 weeks before undergoing euthanasia.22 The mode of delivery of bleomycin and time point of pulmonary hemodynamic measurement were chosen based on previous work that detailed more consistent fibrosis with this method versus intratracheal bleomycin delivery. The time point chosen was too early for the mutant BMPR2 mice to spontaneously develop PAH.23 On induction with doxycycline, mice do not develop spontaneous PH until at least 6 weeks. One week after the last injection (approximately 40 days after doxycycline administration), mice were euthanized.
Hemodynamic measurements: right ventricular systolic pressure (RVSP), RV hypertrophy, and transthoracic echocardiogram
Transthoracic echocardiograms were collected 1 day before euthanasia using the Vivo 770 high-resolution image system (VisualSonics, Toronto, Canada).24 Invasive hemodynamic measurement was conducted as described in previous studies.20 In brief, mice were given 0.75 mg/g of 2.5% avertin (a mixture of tert-amyl alcohol and 2,2,2-tribromoethanol; Sigma-Aldrich) to induce anesthesia. Mice were then placed on a heating pad, and systemic blood pressure and pulse were measured via a tail cuff and transducer. The jugular vein was dissected and catheterized with a 1-French pressure catheter (SPR-1000; Millar Instruments, Houston, TX). The RV pressure tracing was then recorded utilizing Chart 5.3 software (AD Instruments). After completion of the measurements, blood was collected. The heart was then excised with removal of the atria, and the RV and left ventricle (LV) plus septum were isolated for measurement of the Fulton index (RV∶(LV + S)) as previously described.25
Histology, immunostaining, Western blot, semiquantitative scoring, and collagen content
On harvest, the left lung was inflated and placed in 10% formalin for histological processing, and the right lung was divided and snap frozen in liquid nitrogen for RNA and protein processing. Sections were prepared for immunohistochemistry and immunofluorescence staining. Immunostaining was performed for α-SMA to identify muscularized pulmonary vessels, which were then counted per high-powered field as previously described.26 Western blots were performed on tissue and cell lysates as previously described.27 Semiquantitative lung fibrosis scoring28 and hydroxyproline microplate assay were performed as previously described.29
Quantitative reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated from frozen whole-lung tissue and human cell lysate using a RNEasy kit (Qiagen, Venlo, Netherlands) per the manufacturer’s recommendations, DNase treated, and prepared for quantitative RT-PCR. Specific transcript levels for HIF1-α were determined by normalization to 18S. Values are presented as mean normalized transcript level using the comparative Ct method (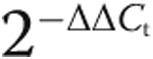 ).
).
Cell culture
Given the method of intraperitoneal delivery of bleomycin in this model and previous literature linking vascular endothelial BMPR2 function to the onset of PH,30 we chose to examine the effects of BMPR2 mutation in endothelial cells. Primary murine and human pulmonary microvascular endothelial cells (mPMVECs and hPMVECs, respectively) with and without BMPR2 mutation transfection were grown in culture as previously described31 (EBM-2 media; Lonza, Basel, Switzerland) and maintained in standard cell culture incubators (37°C, humidified, 5%, CO2). Native/wild-type and mutant PMVECs were grown to 80% confluence. In experiments with hypoxic exposure (fraction of inspired oxygen [FiO2]: 5%), cells were place in a hypoxia cabinet (Coy Lab Products, Grass Lake, MI) for either 6 or 24 hours, as stated below. The content of oxidized lipids was determined by the thiobarbituric acid reactive substances assay (R&D Systems, Minneapolis, MN), with and without the addition of 1 mM TEMPOL (Sigma-Aldrich).32
HRE activity was determined using a luciferase-based system. Pulmonary endothelial cells with BMPR2 mutation and native cells with no BMPR2 mutation were seeded at 70% confluency in 96-well plates. Cignal lenti HIF reporter was obtained from Qiagen. The lenti HIF reporter is made from vesicular stomatitis virus pseudotyped lentivirus particles expressing the firefly luciferase gene under the control of a minimal cytomegalovirus promoter and tandem repeats of the HRE. Cells were transfected with 10 multiplicity of infection lenti HIF reporter in antibiotic-free conditions using SureENTRY transduction reagent (Qiagen; 5 μg/mL) as per the manufacturer’s instructions. Twenty-four hours after transfection, the cells were switched into medium containing 10% fetal bovine serum. Doxycycline (300 ng/mL) was added to the medium to induce BMPR2 expression. Ninety-six hours after transfection, activity of firefly luciferase (indicative of HIF activity) was measured as relative light units using the Dual-Luciferase reporter assay system (Promega).
Statistics
Statistical analyses were performed using GraphPad Prism (GraphPad, La Jolla, CA). Differences among groups were assessed using one-way analysis of variance (ANOVA) or a Kruskal-Wallis rank ANOVA. Differences between pairs were assessed using a Mann-Whitney test. Survival differences were evaluated using a Fisher’s exact test. Results are presented as mean ± standard error of the mean; P < 0.05 was considered significant.
Results
Mice exposed to bleomycin have decreased levels of BMPR2 and inhibitor of differentiation 1 (ID-1) expression compared to vehicle exposure
In order to examine the response of BMPR2 and downstream signaling to bleomycin exposure, control wild-type mice were given intraperitoneal bleomycin and then euthanized to evaluate whole-lung homogenate levels of BMPR2 and ID-1, a canonical BMPR2-signaling cascade protein.33 Both BMPR2 and ID-1 RNA expression levels were decreased in response to bleomycin exposure (Fig. 1a), and BMPR2 protein was decreased by 48% in the bleomycin-exposed group (Fig. 1b).
Figure 1.

Bleomycin-induced pulmonary fibrosis results in decreased expression of bone morphogenetic protein receptor II (BMPR2) and downstream signaling molecule inhibitor of differentiation 1 (ID-1). a, Relative expression of BMPR2 and ID-1 was decreased in lungs of wild-type mice exposed to 33 days of bleomycin (Bleo; normalized to 18S). b, Immunoblot detailing decreased BMPR2 in the lungs of wild-type mice exposed to bleomycin. β-Act: β-actin. Asterisk indicates P < 0.05. N = 4 per group.
Bleomycin administration to mutant BMPR2 mice results in elevated RVSP with evidence of increased vascular remodeling
To evaluate the effect of bleomycin-induced fibrosis on the development of secondary PH, mutant BMPR2 and control mice were given intraperitoneal bleomycin and data collected on hemodynamic variables (Fig. 2a). After bleomycin treatment, there was a statistically significant increase in RVSP between control and mutant BMPR2 mice (30.0 ± 1.0 vs. 35.2 ± 1.9 mmHg, P = 0.03; Fig. 2b). The time point used was earlier than when mutant BMPR2 mice develop spontaneous PH. There was a significant increase in pulmonary vascular resistance (PVR) between the bleomycin-exposed groups, driven primarily by the difference in RVSP (111.1 ± 10.4 vs. 149.4 ± 11.3 dyn s/cm5, P = 0.04; Fig. 2c). However, these changes were not associated with cardiac remodeling, given the lack of significant increase in RV hypertrophy based on the Fulton index (RV∶(LV + S)) between control and BMPR2 bleomycin-exposed mice (13.5% ± 1.5% vs. 12.8% ± 1.9%, P = 0.70; Fig. 2d), as described in prior studies using BMPR2 transgenic mice.34 In contrast to the cardiac remodeling measurements, increased RSVP and PVR were associated with an increase in muscularized pulmonary vessel counts in bleomycin-treated mutant BMPR2 mice (Fig. 2e, 2f). This finding was reinforced qualitatively by lack of significant change in RV wall thickness and end-diastole cavity dimension on echocardiographic images, as visualized in Figure 2g.
Figure 2.

Mutant bone morphogenetic protein receptor II (BMPR2) mice exposed to bleomycin develop worse pulmonary hypertension compared to control, with no evidence of cardiovascular remodeling but with increased vascular remodeling. a, Time line of animal experiment with initiation of doxycycline (Dox) chow 7 days before starting twice weekly intraperitoneal (IP) bleomycin injections for 4 weeks. Echocardiogram was performed on day 32, with euthanasia for hemodynamic and histological data on day 33. b, Right ventricular systolic pressure (RVSP) was greater in mutant BMPR2 mice on exposure to bleomycin compared to bleomycin-treated controls. c, A similar change was noted in measurement of pulmonary vascular resistance (PVR). d, However, no change was noted in cardiac remodeling as determined by the right ventricle to left ventricle plus septum ratio (RV∶(LV + S); P = 0.70). e, f, There was an increase in muscularization of pulmonary vessels within the bleomycin-exposed mutant BMPR2 mice lungs compared to control through counting of muscularized pulmonary arteries per 10 high-powered fields (HPF). g, Qualitative two-dimensional echocardiogram images of parasternal short-axis view in vehicle- and bleomycin-exposed mice demonstrating RV enlargement in both groups of mice exposed to bleomycin compared to vehicle. No difference was noted between bleomycin-treated groups in RV wall thickness or cavity dimensions. PBS: phosphate-buffered saline. Asterisk indicates P < 0.05. N = 8 per group, except N = 4 for control vehicle.
In order to determine whether there were differences in susceptibility to lung fibrosis in mutant BMPR2 mice compared to controls, Masson trichrome stains of histologic sections were evaluated for fibrosis scoring, and hydroxyproline assays were done from lung tissue. After bleomycin, both groups had marked fibrotic remodeling in the lung but to a similar degree. Specifically, there was no significant difference in observed fibrosis between control and mutant BMPR2 mice after exposure to bleomycin as determined by blinded scoring of trichrome-stained lung sections (1.3 ± 0.3 vs. 1.4 ± 0.2, P = 0.90; Fig. 3a, 3b). However, there was a significant difference between lung collagen content as determined by the hydroxyproline assay between control (453 ± 25 μg) and BMPR2 (575 ± 26 μg) mice (right middle lobe of the lung was utilized for all measurements; P < 0.05; Fig. 3c). Given the lack of difference in histologic evaluation of fibrosis, this collagen content difference may have been due to the differences in perivascular fibrotic remodeling. The role of BMPR2 signaling in vessel remodeling has been described previously.35
Figure 3.

Mutant bone morphogenetic protein receptor II (BMPR2) mice had a similar level of lung fibrosis following bleomycin compared to control, with increased collagen content. a, Representative microscopy images of trichrome-stained lung sections of groups as listed, showing no difference in lung architecture at baseline and similar fibrosis after exposure to bleomycin. Magnification ×20. b, Graph representing no difference in control versus BMPR2 groups with respect to fibrosis score before and after intraperitoneal bleomycin treatment (P = 0.90). c, However, there was significantly increased collagen content in the right lower lobe of harvested lungs by microplate hydroxyproline assay in the bleomycin BMPR2 group compared to control. PBS: phosphate-buffered saline. Asterisk indicates P < 0.05. N = 8 per group, except N = 4 for control vehicle.
BMPR2 mutation results in increased HIF1-α activation in mice and cell culture
Whole-lung lysates from mutant BMPR2 mice exposed to bleomycin were found to have greatly increased HIF1-α protein (25-fold) compared to controls, without change in RNA expression, suggesting a posttranslational HIF stabilization in the bleomycin-exposed mutant BMPR2 mice (Fig. 4a, 4c, 4d). CTGF, a HIF-regulated mediator that is known to influence extracellular matrix remodeling, was found to have increased RNA and protein expression in response to bleomycin administration in lungs of mutant BMPR2 mice compared to controls (Fig. 4b–4d). We next sought to examine the upregulation of hypoxic-mediated CTGF RNA and protein expression in hPMVECs with BMPR2 mutation. The hPMVECs with BMPR2 mutation exposed to acute hypoxia (FiO2 5% for 6 hours) displayed a similar significant increase in CTGF expression compared to native controls (Fig. 4e, 4f).
Figure 4.

Mutant bone morphogenetic protein receptor II (BMPR2) mice exposed to bleomycin had increased hypoxia response element (HRE) gene expression, similar to human pulmonary microvascular endothelial cells carrying the BMPR2 mutation exposed to hypoxia. By quantitative polymerase chain reaction on whole-lung preparations, relative expression of hypoxia-inducible factor (HIF)1-α (a) was unchanged (normalized to 18S) on exposure to bleomycin, as contrasted with increased expression of connective tissue growth factor (CTGF; b). c, d, In whole-lung lysates, both proteins were significantly increased in mutant BMPR2 mice exposed to bleomycin (25-fold for HIF1-α). N = 8 per group, except N = 4 for control group. e, f, A similar association was seen in human pulmonary microvascular endothelial cells with BMPR2 mutation exposed to acute hypoxia (FiO2 5% for 6 hours), with increased expression of CTGF. Asterisk indicates P < 0.05. N = 4 per group.
To further dissect the BMPR2-HIF relationship, we transfected control and mutant BMPR2 mPMVECs with an HRE-dependent luciferase vector. Mutant-BMPR2-expressing cells had a doubling of luciferase activity under normoxic conditions, demonstrating that mutant BMPR2 expression alone can activate HRE pathways (Fig. 5a). In order to investigate the mechanism for BMPR2-mediated increase in HIF signaling, control and BMPR2 mPMVECs were exposed to hypoxia (FiO2 5% for 24 hours) with and without the addition of the antioxidant TEMPOL (1 mM). HIF1-α level was increased in those cells expressing the mutant BMPR2 protein on exposure to hypoxia, an effect that was abolished with addition of TEMPOL, suggesting a role for increased ROS contributing to BMPR2-mediated HIF expression (Fig. 5b). This observation paralleled the decrease in ROS production (as measured by malondialdehyde concentration) in mutant BMPR2 cells exposed to hypoxia (same conditions) with or without the addition of TEMPOL (Fig. 5c).
Figure 5.

Mutant bone morphogenetic protein receptor II (BMPR2) expression promotes hypoxia response element (HRE) activity and hypoxia-inducible factor (HIF) expression, an effect that is attenuated with antioxidant administration. a, Using HRE luciferase systems, mutant BMPR2 cells had greater HRE activity than native cells. N = 5 in each group. b, Mutant BMPR2 cells have increased HIF expression upon exposure to hypoxia (FiO2 5% for 24 hours) compared to controls, which is markedly decreased with exposure to the antioxidant TEMPOL. c, A similar trend is shown with respect to increased reactive oxygen species (ROS) production, as determined by malondialdehyde (MDA) concentration, in mutant BMPR2 cells exposed to hypoxia, which is also markedly decreased with exposure to the antioxidant TEMPOL. One asterisk indicates P < 0.05; two asterisks indicate P < 0.001. N = 4 per group.
Discussion
In these studies, mice expressing mutant BMPR2 had similar levels of bleomycin-induced lung fibrosis compared to control littermates but developed more severe PH with an associated increase in HIF expression. This increase in HIF activity related to mutant BMPR2 expression appears to be facilitated, at least in part, by production of ROS (Fig. 6). The downstream hypoxic signaling related to mutant BMPR2 expression was corroborated in hPMVECs, showing an increase in HRE activity in cells exposed to hypoxia. Together, our findings link BMPR2 and HIF signaling to secondary PH, adding to prior studies connecting BMPR2 signaling and the vascular endothelium with various other chronic lung diseases, including asthma,36 sarcoidosis,37 and chronic obstructive pulmonary disease.38 It is important to note that at the harvest time point examined, the murine model of BMPR2 mutation would not be expected to develop spontaneous PAH—this usually occurs 6 weeks after doxycycline induction, at the earliest.23
Figure 6.

Summary of bone morphogenetic protein receptor II (BMPR2) mutation influence on development of pulmonary hypertension associated with pulmonary fibrosis, potentially mediated through reactive oxygen species (ROS) production and stabilization of hypoxia-inducible factor (HIF)1-α.
HIF is the most important mediator of the cellular response to hypoxia and has been implicated in the development of PH in several models of PH such as monocrotaline-treated rats,39 chronic hypoxia,19 and fawn-hooded rats.40 HIF has previously been shown to be expressed in immunohistochemistry stains of lung vasculature in patients with PH.41 Additionally, epigenetic downregulation of superoxide dismutase 2 causing an increase in mitochondrial ROS production42 and normoxic stabilization of HIF1-α has been speculated to play a role in the development of PAH.15 Our finding of a large increase in HIF protein within the lung tissue of mutant BMPR2 mice exposed to bleomycin is a novel discovery that demonstrates HIF association with worsening development of PH associated with lung fibrosis.
It is known that BMPR2 can lead to production of ROS contributing to cell injury, angiogenesis, and aberrant fibrogenic repair in the development of membranous nephropathy.33 However, it is only recently that we have grown to appreciate the role that BMPR2 plays in the development of pulmonary vascular oxidative injury, specifically through mitochondrial dysregulation,43 with at least one study suggesting that ROS formation may play a direct role in the pathogenesis of human PAH.44 mutant BMPR2 expression has also been shown to lead to stabilization of ROS in the setting of acute changes in oxygen tension,45 with resulting worsening of the PH phenotype. Interestingly, in endothelial cells derived from patients with PAH, baseline DNA damage was higher compared to control patient samples, with evidence of increased ROS production.46 Our data are thus consistent with prior work describing HIF stabilization in the presence of increased ROS production,47,48 providing a potential mechanism that explains in part how BMPR2 signaling contributes to the development of secondary PH.49-51
To explore a potential mechanism linking BMPR2-mutation-mediated induction of HIF to increased PH, we looked for downstream mediators of the HRE that may influence extracellular matrix remodeling. Since HIF is known to directly mediate CTGF,52-54 a member of the CCN family of proteins that coordinates extracellular matrix remodeling on exposure to several stimuli of cellular stress, we demonstrate an increase in CTGF expression in our in vivo model, as well as in vitro endothelial cell culture experiments. In addition, previous mouse studies have implicated upregulation of CTGF signaling within vascular smooth muscle cells as playing a key role in the development of PH in both monocrotaline-treated rats55 and pulmonary-artery-banded mice.56
Thus, our identification of HIF stabilization in both a mouse and a human model of BMPR2 mutation represents a new pathway for development of novel pharmacologic treatments for PH57,58 associated with chronic parenchymal lung disease, such as pulmonary fibrosis. This discovery is an important first step in an evolving area of research, though it prompts several questions for future research, including the role of endothelial cell BMPR2 signaling in the in vivo model of fibrosis-associated PH, through either (a) influencing paracrine signaling to surrounding smooth muscle cells59,60 or (b) progenitor cell recruitment and reprogramming.61 In addition, this study suggests that therapy directed at hypoxia-mediated targets shown to influence the development of PH, such as BMP antagonist gremlin 1,62 may prove beneficial in treating human subjects with disease. These future experiments will help define the agent and method of delivery for any potential HIF-targeting therapies for disease.
In conclusion, we have shown that bleomycin-exposed mice with mutant BMPR2 expression develop a more severe PH phenotype associated with increased HIF stabilization, which is likely facilitated by ROS production. Together, these findings represent a novel pathway for development of therapeutics for secondary PH.
Acknowledgment
The excellent professional assistance of John H. Newman, MD, is acknowledged and appreciated.
Source of Support: National Institutes of Health (NIH) grants NHLBI HL105479 (WEL), HL85317 (TSB), HL92870 (TSB), HL095797 (JDW), HL87738 (AJB), and HL121174 (JPF); NIH grant NCRR UL1 RR024975; and an American Thoracic Society/Pulmonary Hypertension Association Fellowship Research Grant (AJB).
Conflict of Interest: None declared.
References
- 1.Patel NM, Lederer DJ, Borczuk AC, Kawut SM. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest 2007;132:998–1006. [DOI] [PubMed]
- 2.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006;129:746–752. [DOI] [PubMed]
- 3.Raghu G, Behr J, Brown KK, Egan JJ, Kawut SM, Flaherty KR, Martinez FJ, et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: a parallel, randomized trial. Ann Intern Med 2013;158:641–649. [DOI] [PubMed]
- 4.Idiopathic Pulmonary Fibrosis Clinical Research Network. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. New Engl J Med 2010;363:620–628. [DOI] [PMC free article] [PubMed]
- 5.Fang A, Studer S, Kawut SM, Ahya VN, Lee J, Wille K, Lama V, et al. Elevated pulmonary artery pressure is a risk factor for primary graft dysfunction following lung transplantation for idiopathic pulmonary fibrosis. Chest 2011;139:782–787. [DOI] [PMC free article] [PubMed]
- 6.Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, Ahmad F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol 2010;298:H1235–H1248. [DOI] [PMC free article] [PubMed]
- 7.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 2012;122:4306–4313. [DOI] [PMC free article] [PubMed]
- 8.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res 2006;98:209–217. [DOI] [PubMed]
- 9.Majka S, Hagen M, Blackwell T, Harral J, Johnson JA, Gendron R, Paradis H, et al. Physiologic and molecular consequences of endothelial BMPR2 mutation. Respir Res 2011;12:84. [DOI] [PMC free article] [PubMed]
- 10.Raamsteeboers AJ, Bogaard HJ, Vonk Noordegraaf A. Pulmonary arterial hypertension preceding idiopathic pulmonary fibrosis in a BMPR2 mutation positive patient. Eur Respir Rev 2014;23:147–149. [DOI] [PMC free article] [PubMed]
- 11.Grünig E, Weissmann S, Ehlken N, Fijalkowska A, Fischer C, Fourme T, Galiè N, et al. Stress Doppler echocardiography in relatives of patients with idiopathic and familial pulmonary arterial hypertension: results of a multicenter European analysis of pulmonary artery pressure response to exercise and hypoxia. Circulation 2009;119:1747–1757. [DOI] [PubMed]
- 12.Fantozzi I, Platoshyn O, Wong AH, Zhang S, Remillard CV, Furtado MR, Petrauskene OV, Yuan JX. Bone morphogenetic protein-2 upregulates expression and function of voltage-gated K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2006;291:L993–L1004. [DOI] [PubMed]
- 13.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 2011;183:152–156. [DOI] [PMC free article] [PubMed]
- 14.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 2012;110:1484–1497. [DOI] [PMC free article] [PubMed]
- 15.Kim GH, Ryan JJ, Marsboom G, Archer SL. Epigenetic mechanisms of pulmonary hypertension. Pulm Circ 2011;1:347–356. [DOI] [PMC free article] [PubMed]
- 16.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med 2014;189:314–324. [DOI] [PMC free article] [PubMed]
- 17.Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 2003;111:1519–1527. [DOI] [PMC free article] [PubMed]
- 18.Veith C, Marsh LM, Wygrecka M, Rutschmann K, Seeger W, Weissmann N, Kwapiszewska G. Paxillin regulates pulmonary arterial smooth muscle cell function in pulmonary hypertension. Am J Pathol 2012;181:1621–1633. [DOI] [PubMed]
- 19.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 1999;103:691–696. [DOI] [PMC free article] [PubMed]
- 20.West J, Harral J, Lane K, Deng Y, Ickes B, Crona D, Albu S, Stewart D, Fagan K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am J Physiol Lung Cell Mol Physiol 2008;295:L744–L755. [DOI] [PMC free article] [PubMed]
- 21.Johnson JA, Hemnes AR, Perrien DS, Schuster M, Robinson LJ, Gladson S, Loibner H, et al. Cytoskeletal defects in BMPR2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2012;302:L474–L484. [DOI] [PMC free article] [PubMed]
- 22.Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM Jr., Hunter MG, Bringardner BD, et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med 2007;176:78–89. [DOI] [PMC free article] [PubMed]
- 23.West J, Tada Y, Fagan KA, Steudel W, Fouty BW, Harral JW, Miller M, Ozimek J, Tuder RM, Rodman DM. Suppression of type II bone morphogenic protein receptor in vascular smooth muscle induces pulmonary arterial hypertension in transgenic mice. Chest 2005;128(6 suppl.):553S. [DOI] [PubMed]
- 24.Hemnes AR, Maynard KB, Champion HC, Gleaves L, Penner N, West J, Newman JH. Testosterone negatively regulates right ventricular load stress responses in mice. Pulm Circ 2012;2:352–358. [DOI] [PMC free article] [PubMed]
- 25.Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, Maynard KB, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2014;189:325–334. [DOI] [PMC free article] [PubMed]
- 26.Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, Hemnes A, et al. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J 2012;26:2546–2557. [DOI] [PMC free article] [PubMed]
- 27.Fessel JP, Chen X, Frump A, Gladson S, Blackwell T, Kang C, Johnson J, et al. Interaction between bone morphogenetic protein receptor type 2 and estrogenic compounds in pulmonary arterial hypertension. Pulm Circ 2013;3:564–577. [DOI] [PMC free article] [PubMed]
- 28.Lawson WE, Polosukhin VV, Stathopoulos GT, Zoia O, Han W, Lane KB, Li B, et al. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol 2005;167:1267–1277. [DOI] [PMC free article] [PubMed]
- 29.Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA 2011;108:10562–10567. [DOI] [PMC free article] [PubMed]
- 30.Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood 2011;117:333–341. [DOI] [PubMed]
- 31.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2012;2:201–213. [DOI] [PMC free article] [PubMed]
- 32.Aviram M, Vaya J. Markers for low-density lipoprotein oxidation. Methods Enzymol 2001;335:244–256. [DOI] [PubMed]
- 33.Pache G, Schafer C, Wiesemann S, Springer E, Liebau M, Reinhardt HC, August C, Pavenstadt H, Bek MJ. Upregulation of ID-1 via BMP-2 receptors induces reactive oxygen species in podocytes. Am J Physiol Ren Physiol 2006;291:F654–F662. [DOI] [PubMed]
- 34.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res 2004;94:1109–1114. [DOI] [PubMed]
- 35.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, Dorfmüller P, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015;131:1006–1018. [DOI] [PubMed]
- 36.Kariyawasam HH, Xanthou G, Barkans J, Aizen M, Kay AB, Robinson DS. Basal expression of bone morphogenetic protein receptor is reduced in mild asthma. Am J Respir Crit Care Med 2008;177:1074–1081. [DOI] [PubMed]
- 37.Leng D, Huan C, Xie T, Liang J, Wang J, Dai H, Wang C, Jiang D. Meta-analysis of genetic programs between idiopathic pulmonary fibrosis and sarcoidosis. PLoS ONE 2013;8:e71059. [DOI] [PMC free article] [PubMed]
- 38.Thomashow MA, Shimbo D, Parikh MA, Hoffman EA, Vogel-Claussen J, Hueper K, Fu J, et al. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema: the Multi-Ethnic Study of Atherosclerosis Chronic Obstructive Pulmonary Disease study. Am J Respir Crit Care Med 2013;188:60–68. [DOI] [PMC free article] [PubMed]
- 39.Yan J, Shen Y, Wang Y, Li BB. Increased expression of hypoxia-inducible factor-1α in proliferating neointimal lesions in a rat model of pulmonary arterial hypertension. Am J Med Sci 2013;345:121–128. [DOI] [PubMed]
- 40.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Bonnet S, Haromy A, et al. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 2006;113:2630–2641. [DOI] [PubMed]
- 41.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol 2001;195:367–374. [DOI] [PubMed]
- 42.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 2007;104:1342–1347. [DOI] [PMC free article] [PubMed]
- 43.Lane KL, Talati M, Austin E, Hemnes AR, Johnson JA, Fessel JP, Blackwell T, et al. Oxidative injury is a common consequence of BMPR2 mutations. Pulm Circ 2011;1:72–83. [DOI] [PMC free article] [PubMed]
- 44.Flynn C, Zheng S, Yan L, Hedges L, Womack B, Fessel J, Cogan J, et al. Connectivity map analysis of nonsense-mediated decay-positive BMPR2-related hereditary pulmonary arterial hypertension provides insights into disease penetrance. Am J Respir Cell Mol Biol 2012;47:20–27. [DOI] [PMC free article] [PubMed]
- 45.Fessel JP, Flynn CR, Robinson LJ, Penner NL, Gladson S, Kang CJ, Wasserman DH, Hemnes AR, West JD. Hyperoxia synergizes with mutant bone morphogenic protein receptor 2 to cause metabolic stress, oxidant injury, and pulmonary hypertension. Am J Respir Cell Mol Biol 2013;49:778–787. [DOI] [PMC free article] [PubMed]
- 46.Federici C, Drake KM, Rigelsky CM, McNelly LN, Meade SL, Comhair SA, Erzurum SC, Aldred MA. Increased mutagen sensitivity and DNA damage in pulmonary arterial hypertension. Am J Respir Crit Care Med 2015;192:219–228. [DOI] [PMC free article] [PubMed]
- 47.Shatrov VA, Sumbayev VV, Zhou J, Brune B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1α)accumulation via redox-dependent mechanisms. Blood 2003;101:4847–4849. [DOI] [PubMed]
- 48.Vaux EC, Metzen E, Yeates KM, Ratcliffe PJ. Regulation of hypoxia-inducible factor is preserved in the absence of a functioning mitochondrial respiratory chain. Blood 2001;98:296–302. [DOI] [PubMed]
- 49.Beppu H, Ichinose F, Kawai N, Jones RC, Yu PB, Zapol WM, Miyazono K, Li E, Bloch KD. BMPR-II heterozygous mice have mild pulmonary hypertension and an impaired pulmonary vascular remodeling response to prolonged hypoxia. Am J Physiol Lung Cell Mol Physiol 2004;287:L1241–L1247. [DOI] [PubMed]
- 50.Reynolds AM, Xia W, Holmes MD, Hodge SJ, Danilov S, Curiel DT, Morrell NW, Reynolds PN. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2007;292:L1182–L1192. [DOI] [PubMed]
- 51.Takahashi H, Goto N, Kojima Y, Tsuda Y, Morio Y, Muramatsu M, Fukuchi Y. Downregulation of type II bone morphogenetic protein receptor in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2006;290:L450–L458. [DOI] [PubMed]
- 52.Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of CTGF is directly mediated by HIF-1. Am J Physiol Ren Physiol 2004;287:F1223–F1232. [DOI] [PubMed]
- 53.Samarin J, Wessel J, Cicha I, Kroening S, Warnecke C, Goppelt-Struebe M. FoxO proteins mediate hypoxic induction of connective tissue growth factor in endothelial cells. J Biol Chem 2010;285:4328–4336. [DOI] [PMC free article] [PubMed]
- 54.Rimon E, Chen B, Shanks AL, Nelson DM, Sadovsky Y. Hypoxia in human trophoblasts stimulates the expression and secretion of connective tissue growth factor. Endocrinology 2008;149:2952–2958. [DOI] [PMC free article] [PubMed]
- 55.Lee YS, Byun J, Kim JA, Lee JS, Kim KL, Suh YL, Kim JM, et al. Monocrotaline-induced pulmonary hypertension correlates with upregulation of connective tissue growth factor expression in the lung. Exp Mol Med 2005;37:27–35. [DOI] [PubMed]
- 56.Friedberg MK, Cho MY, Li J, Assad RS, Sun M, Rohailla S, Honjo O, Apitz C, Redington AN. Adverse biventricular remodeling in isolated right ventricular hypertension is mediated by increased TGF-β1 signaling and is abrogated by angiotensin receptor blockade. Am J Respir Cell Mol Biol 2013;1019–1028. [DOI] [PubMed]
- 57.Fraisl P, Aragones J, Carmeliet P. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat Rev Drug Discov 2009;8:139–152. [DOI] [PubMed]
- 58.Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 2011;10:945–963. [DOI] [PMC free article] [PubMed]
- 59.Frank DB, Abtahi A, Yamaguchi DJ, Manning S, Shyr Y, Pozzi A, Baldwin HS, Johnson JE, de Caestecker MP. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res 2005;97:496–504. [DOI] [PubMed]
- 60.Talati M, West J, Zaynagetdinov R, Hong CC, Han W, Blackwell T, Robinson L, Blackwell TS, Lane K. BMP pathway regulation of and by macrophages. PLoS ONE 2014;9:e94119. [DOI] [PMC free article] [PubMed]
- 61.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, Yang J, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med 2009;180:780–787. [DOI] [PMC free article] [PubMed]
- 62.Ciuclan L, Sheppard K, Dong L, Sutton D, Duggan N, Hussey M, Simmons J, et al. Treatment with anti-gremlin 1 antibody ameliorates chronic hypoxia/SU5416-induced pulmonary arterial hypertension in mice. Am J Pathol 2013;183:1461–1473. [DOI] [PMC free article] [PubMed]