

Abstract Abstract
Despite the important role played by the nonmuscle isoform of myosin light chain kinase (nmMLCK) in vascular barrier regulation and the implication of both nmMLCK and vascular endothelial growth factor (VEGF) in the pathogenesis of acute respiratory distress syndrome (ARDS), the role played by nmMLCK in VEGF-induced vascular permeability is poorly understood. In this study, the role played by nmMLCK in VEGF-induced vascular hyperpermeability was investigated. Human lung endothelial cell barrier integrity in response to VEGF is examined in both the absence and the presence of nmMLCK small interfering RNAs. Levels of nmMLCK messenger RNA (mRNA), protein, and promoter activity expression were monitored after VEGF stimulation in lung endothelial cells. nmMYLK promoter activity was assessed using nmMYLK promoter luciferase reporter constructs with a series of nested deletions. nmMYLK transcriptional regulation was further characterized by examination of a key transcriptional factor. nmMLCK plays an important role in VEGF-induced permeability. We found that activation of the VEGF signaling pathway in lung endothelial cells increases MYLK gene product at both mRNA and protein levels. Increased nmMLCK mRNA and protein expression is a result of increased nmMYLK promoter activity, regulated in part by binding of the Sp1 transcription factor on triggering by the VEGF signaling pathway. Taken together, these findings suggest that MYLK is an important ARDS candidate gene and a therapeutic target that is highly influenced by excessive VEGF concentrations in the inflamed lung.
Keywords: vascular endothelial growth factor (VEGF), MYLK, myosin light chain kinase (MLCK), Sp1, promoter, transcription factor, endothelial, permeability, ARDS
Acute respiratory distress syndrome (ARDS) is an inflammatory lung disease characterized by diffuse alveolar infiltration, hypoxemia, respiratory failure, and death due to multiorgan failure. Mortality rates in ARDS, the most severe clinical form of acute lung injury, are up to 60% of patients admitted,1 with ∼150,000–200,000 ARDS cases per year in the United States.2 Furthermore, ARDS constitutes a major healthcare burden in the United States due to intensive and often prolonged intensive care unit hospitalizations. In addition to these epidemiologic studies, race and sex differences in ARDS deaths in the United States over the past several decades have clearly demonstrated an increase in incidence and mortality due to sepsis and acute lung injury in African Americans and Latinos compared with non-Latino whites,3,4 highlighting the contribution of genetic components in disease susceptibility and severity.5-12
Increased lung vascular permeability is a major pathobiologic feature of ARDS and drives morbidity and mortality. At a mechanistic level, increased gaps between lung endothelium are directly related to the extent of lung fluid imbalance and lung edema. The nonmuscle isoform of myosin light chain kinase (nmMLCK) is an important component of endothelial cell (EC) gatekeeping machinery, as nmMLCK regulates actin cytoskeleton rearrangements and contraction.13 nmMLCK is a calcium/calmodulin (Ca2+/CAM)–dependent serine/threonine kinase, and its major function is to phosphorylate myosin light chain (MLC). The MYLK gene, located on 3q21, encodes three proteins (nonmuscle MLCK, smooth muscle MLCK, and telokin) via three independent promoters. The nmMYLK promoter, or the promoter specific to nmMLCK, regulates the full-length (210-kDa) nonmuscle MLCK protein isoform. The nmMLCK isoform is primarily expressed in nonmuscle tissues, including ECs. Phosphorylation of MLC at Thr14 and Ser15 by nmMLCK drives assembly of actin monomers into polymers or stress fibers and further signals the contraction of actin stress fibers. The important role played by nmMLCK during vascular barrier regulation has been highlighted in vivo; MYLK−/− mice have been shown to be protected from both lipopolysaccharide (LPS)–induced lung injury16 and ventilator-induced lung injury (VILI),17 whereas genetically engineered MYLKec/ec transgenic mice, in which overexpression of nmMLCK is restricted to the vascular endothelium, experience increased LPS-induced lung injury.18 In addition, up-regulation of the nmMLCK isoform is observed in lung tissues obtained from asthmatic patients where asthma is characterized by chronic inflammation of the airway,19 underscoring the important role played by nmMLCK in various inflammatory lung diseases.5,7,14-19
The exact signaling mechanisms that result in enhanced nmMLCK expression and activation are poorly understood. Vascular endothelial growth factor (VEGF) is a proangiogenic glycoprotein that is essential for a variety of cellular processes, including angiogenesis, cell proliferation, and cell migration.20-23 VEGF, originally named vascular permeability factor for its profound effects on vascular barrier function, was discovered as a tumor-secreted protein that led to venular hyperpermeability to circulating macromolecules24-26 and is known to increase lung EC permeability.27 Since its discovery, VEGF has been implicated in a wide range of inflammatory diseases, including pulmonary inflammation, such as ARDS, VILI, fibrosis, and asthma.28-32
While VEGF plays a critical role in organogenesis,33,34 its expression in lungs persists into adulthood and remains the predominant source of VEGF in adults.35,36 Given the physiological role played by VEGF in lung maintenance and homeostasis, the involvement of VEGF in the pathogenesis of ARDS has been well investigated.28,31,32 Several studies have shown that VEGF is present at higher levels in the plasma of ARDS patients than non-ARDS patients32,37 as well as phase-dependent increases in VEGF levels during the initial stage of ARDS.38 In addition, single-nucleotide polymorphisms (SNPs) in VEGFA are associated with high ARDS mortality rates.39
Despite the important role played by nmMLCK in vascular barrier regulation and the implication of both nmMLCK and VEGF in the pathogenesis of ARDS, the role played by nmMLCK in VEGF-induced vascular permeability is poorly understood. In this study, the role played by nmMLCK in VEGF-induced vascular hyperpermeability was investigated, and nmMLCK was found to play an important role in increasing VEGF-induced permeability via increased nmMYLK promoter activity, messenger RNA (mRNA) and protein expression, and enzymatic activity. In addition, the nmMYLK promoter is partially regulated by activation of the Sp1 transcription factor upon triggering of the VEGF signaling pathway. Taken together, these findings suggest that MYLK is an important ARDS candidate gene and a therapeutic target that is highly influenced by excessive VEGF expression and activity in the inflamed lung.
Methods
Reagents
Human recombinant VEGF165 was obtained from Sigma-Aldrich (St. Louis, MO). All other reagents were obtained from Sigma-Aldrich unless otherwise noted. Antibodies were obtained as follows: Texas Red phalloidin from Life Technologies (Grand Island, NY); nmMLCK (A20) and Sp1 from Santa Cruz Biotechnology (Santa Cruz, CA); and anti-diphosphorylated MLC and pan-MLC from Cell Signaling (Danvers, MA).
Cell culture and transfections
Human pulmonary artery ECs (Lonza, Walkersville, MD) were cultured as described elsewhere.40 Small interfering RNAs (siRNAs) were obtained from Dharmacon (Lafayette, CO; firefly luciferase siCONTROL#2, human nmMLCK custom siRNA sequence GGAAAGAGGUGACCAAUGUUU, and human Sp1 [6667] TARGETplus siRNA) and transfected into lung ECs using siPORTAmine (Dharmacon; silencing, 3 days; final concentration, 100 nM). The enhanced green fluorescent protein (EGFP)–tagged nmMLCK (wild-type isoform 1; hereafter, EGFP-nmMLCK) mammalian expression vector was constructed as described elsewhere.41,42 The EGFP-nmMLCK (5 μg/mL concentration for 24 hours) and all nmMYLK promoter constructs (5 μg/mL concentration for 24 hours; generation details are provided below) were transfected into lung ECs using Xfect (Clontech, Mountain View, CA) before stimulations were conducted as detailed in individual experiments.
Transendothelial electrical resistance (TER)
TER measurements were performed using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY), as described elsewhere.43 TER values from each microelectrode were pooled as discrete time points and plotted versus time as the mean ± SEM.
Immunofluorescence
Lung ECs were immunostained and imaged as described elsewhere.41 Fluorescence was as follows: blue, 461 nm (nuclei); red, 565 nm (F-actin); and green, 509 nm (EGFP-nmMLCK).
Western blotting
Lung EC protein was isolated and blotted as described elsewhere.44 Band intensity was quantified using ImageJ, normalized to β-actin or glyceraldehyde‐3‐phosphate dehydrogenase control, and expressed as arbitrary units.
Promoter plasmid generation and luciferase promoter assay
The nmMYLK promoter region was synthesized by GenScript (Piscataway, NJ) and cloned into pGL3-Basic vector (Promega, Madison, WI). This region of DNA included 2,535 bp of sequence upstream of the nmMYLK promoter (NM_053025.3) located on chromosome 3q21, containing 2,431 bp of 3′ untranslated region, 97 bp of exon 1, and 7 bp of intron 1. To create promoter deletion constructs, a series of primers was designed to amplify each length (−2,441, −2,111, −1,751, −1,471, −1,211, −831, −420, −271, −220, and −118 relative to the transcription start site). Luciferase promoter assays were performed as described elsewhere.11
mRNA extraction, complementary DNA (cDNA) synthesis, quantitative polymerase chain reaction (qPCR), and chromatin immunoprecipitation (ChIP) analysis
mRNA was isolated from lung ECs using RNA isolation kits (Promega). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). mRNA expression was measured using qPCR (1 μL of cDNA mixed with 2× SYBR Select Master Mix [Applied Biosystems] and 500 nM forward and reverse primers). qPCR was performed using iCycler (Bio-Rad, Hercules, CA; 50°C for 2 minutes, 95°C for 2 minutes, and 40 cycles of 95°C for 15 seconds and 60°C for 1 minute). ChIP analysis of DNA bound to Sp1 protein complexes was performed using the EZ-Magna ChIP assay (Millipore, Billerica, MA).
In silico promoter analysis
Genomatix software (Munich, Germany; http://www.genomatix.de) was used to predict transcription factor binding motifs on nmMYLK core promoter (−2,500 to 0). Sites were computationally predicted with predefined transcription factor binding modules45,46 in the Promoter Module Library (Genomatix).
Statistical analysis
Analysis of variance tests were used for all comparisons. Statistical significance was defined as P < 0.05.
Results
Dependence of VEGF-induced increased lung vascular permeability on nmMLCK
Disruption of lung EC barrier integrity was assessed using TER, a highly sensitive measurement of EC barrier integrity. VEGF stimulation (5–200 ng/mL) induced dose- and time-dependent reduction in TER (Fig. 1), indicating lung EC barrier permeability. VEGF stimulated time-dependent formation of actin stress fibers, a hallmark of loss of EC integrity, within lung ECs (Fig. 2A). Overexpression of nmMLCK, an essential component of endothelial barrier regulatory machinery, in lung ECs resulted in colocalization of nmMLCK along VEGF-induced stress fibers (Fig. 2A). In addition, nmMLCK enzymatic activity, as indicated by phosphorylation of its substrate MLC, is increased where MLC Thr14 and Ser15 are phosphorylated in a time-dependent manner after VEGF stimulation (Fig. 2B). TER measurement of lung ECs transfected with nmMLCK siRNA demonstrates an attenuation of VEGF-induced barrier disruption compared with control siRNA (Fig. 2C), highlighting the role played by nmMLCK in exacerbating the loss of barrier integrity caused by VEGF.
Figure 1.
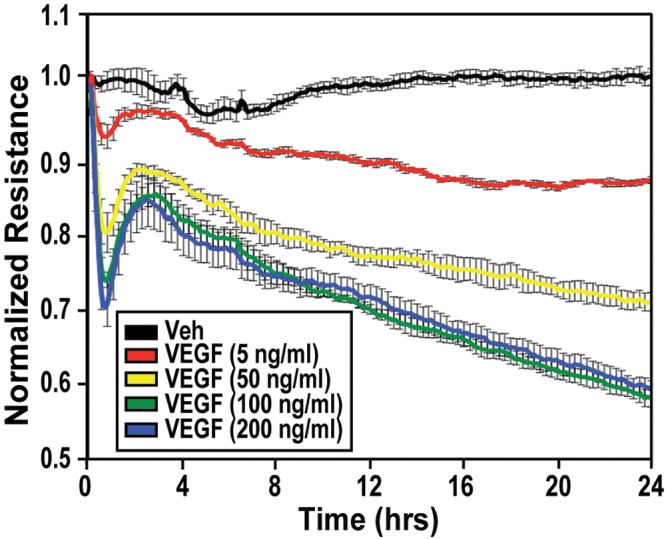
Dose- and time-dependent loss of endothelial cell (EC) barrier integrity due to vascular endothelial growth factor (VEGF). Lung ECs were plated on gold microelectrodes for transendothelial electrical resistance (TER) measurements and stimulated at time = 0 with phosphate‐buffered saline vehicle (black), 5 ng/mL VEGF (red), 50 ng/mL VEGF (yellow), 100 ng/mL VEGF (green), or 200 ng/mL VEGF (blue). The TER tracings represent resistance (Ω) measurements over time (normalized to 1), where decreased resistance indicates increased permeability. Three independent TER experiments were performed, with representative TER tracings shown. Rapid cytoskeletal changes were monitored in all subsequent experiments at earlier time points (5 minutes to 6 hours), while long-term promoter activity and messenger RNA or protein levels were monitored at 24 hours to allow for transcriptional and translational changes. Veh: vehicle.
Figure 2.

Dependence of VEGF-induced permeability on nmMLCK. A, Enhanced green fluorescent protein (EGFP)–tagged nmMLCK (EGFP-nmMLCK) was overexpressed42 in lung endothelial cells (ECs) and then challenged with vehicle or VEGF (100 ng/mL) for 5 or 30 minutes. EGFP-nmMLCK (green) initially migrates to the cell periphery (5 minutes), which is followed by translocation and colocalization with VEGF-induced actin stress fibers (red) at 30 minutes. Three independent experiments were performed, with representative images shown. B, Confluent lung ECs were stimulated with vehicle or VEGF (100 ng/mL) for 1, 3, 6, or 24 hours. Western blot analysis was conducted with phospho–myosin light chain (MLC-pp; specific for Thr14 and Ser15), pan-MLC, and pan–glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) antibodies as indicated. Increased MLC phosphorylation after VEGF stimulation peaks at 1 hour. Three independent experiments were performed, with representative blots shown. C, Lung ECs were plated on gold microelectrodes for transendothelial electrical resistance (TER) measurements prior to nmMLCK silencing by means of small interfering RNAs (siRNA). Lung ECs were next stimulated with vehicle or VEGF (100 ng/mL, 24 hours). VEGF-induced EC barrier disruption was significantly attenuated by nmMLCK siRNA (sinmMLCK) but not by control siRNA (siCONT). Inset shows Western blot demonstrating nmMLCK protein silencing. Four independent TER experiments were performed, with pooled data represented as bar graphs. *P < 0.05 versus control siRNA. nmMLCK: nonmuscle isoform of myosin light chain kinase; VEGF: vascular endothelial growth factor; Veh: vehicle.
Increased nmMLCK gene and protein expression due to VEGF stimulation
We previously demonstrated that nmMLCK expression is up-regulated in asthmatic lung tissues in both preclinical mouse models and human patients;19 therefore, we further examined whether VEGF stimulation regulates expression of nmMLCK in lung ECs. After VEGF stimulation, lung ECs demonstrated an approximate fivefold increase in nmMLCK mRNA expression (Fig. 3A). nmMLCK protein expression also increased significantly after VEGF stimulation (Fig. 3B). In addition, transfection of the full-length 2.5-kb nmMYLK promoter (−2.5 kb/−2,562 bp) construct in lung ECs followed by VEGF stimulation resulted in significantly increased nmMYLK promoter activity compared with that in unstimulated lung ECs (Fig. 3C). The combined data on increased expression of mRNA and protein levels as well as promoter activity confirm the overall increase in MYLK gene activation by VEGF in lung ECs.
Figure 3.

Increase in nmMLCK messenger RNA (mRNA) and protein levels and in nmMYLK promoter activity due to vascular endothelial growth factor (VEGF). Confluent lung endothelial cells (ECs) treated with VEGF (100 ng/mL, 24 hours) showed a marked increase in nmMLCK mRNA levels (A) compared with vehicle, and confluent lung ECs treated with VEGF (100 ng/mL, 24 hours) also showed a marked increase in nmMLCK protein levels (B) compared with vehicle. Three independent experiments were performed, with bar graphs representing fold change in pooled densitometry measurements. *P < 0.05 versus vehicle. C, Full-length nmMYLK promoter (nmMYLK-Luc, −2.5 kb/−2,562 bp) and pGL3 control luciferase constructs were overexpressed in lung ECs and then treated with VEGF (100 ng/mL) for 24 hours. VEGF dramatically increased nmMYLK promoter activity compared with the pGL3 control construct, as detected by luciferase reporter activity. Three independent experiments were performed, with bar graphs representing pooled promoter activity readings as relative light units (RLU). *P < 0.05 versus vehicle nmMYLK-Luc. GAPDH: glyceraldehyde‐3‐phosphate dehydrogenase; nmMLCK: nonmuscle isoform of myosin light chain kinase; Veh: vehicle.
Transcriptional regulation of the nmMYLK promoter by VEGF
On the basis of the findings that nmMYLK promoter activity is dramatically increased upon VEGF stimulation in lung ECs, we sought to determine the mechanisms of nmMYLK promoter up-regulation by VEGF stimulation. Utilizing in silico bioinformatics software (Genomatix) to assess predicted transcription factor binding motifs in the nmMYLK promoter, potential transcription binding factors for the nmMYLK promoter were identified. Among the total of >750 motifs predicted, 36 transcription factors were identified that are implicated with or known to be associated with the VEGF pathway (Fig. 4A). To determine the smaller region of the promoter that is critical for promoter activation in response to VEGF, various lengths of nmMYLK promoter fragments were cloned in front of the luciferase gene in a pGL3-Basic vector lacking eukaryotic promoter elements (Fig. 4B), and the promoter activity of the various fragments were analyzed using a luciferase reporter assay. The full-length 2.5-kb promoter fragment increased the relative luciferase activity (luciferase/renilla) by approximately threefold in the presence of VEGF compared with that in untreated lung ECs (Fig. 4B). However, while the 0.4-kb fragment significantly increased relative luciferase activity after VEGF stimulation (Fig. 4B), further deletion of the promoter to a 0.3-kb fragment showed a loss of relative luciferase activity induced by VEGF (Fig. 4B). These results indicate that VEGF stimulation in lung ECs causes an increase in nmMYLK promoter activity and that the critical VEGF-responsive sequence lies between −450 and −331 bp from MYLK exon 1.
Figure 4.

nmMYLK promoter VEGF-associated transcription factor binding motif predictions and identification of a VEGF-responsive nmMYLK promoter region. A, Schematic depiction of predicted transcription factor binding sites (using Genomatix software) on the nmMYLK promoter (2.5 kb upstream of the transcription start site and noncoding exon 1). Thirty-six transcription factor binding modules are shown on top. Red indicates known VEGF effectors. B, Full-length nmMYLK promoter (−2.5 kb/−2,562 bp), nmMYLK promoter fragments (−0.4 kb/−450 bp, −0.3 kb/−331 bp), and pGL3 control luciferase constructs were overexpressed in lung endothelial cells and then treated with VEGF (100 ng/mL) for 24 hours. VEGF dramatically increases nmMYLK promoter activity in the 2.5-kb (full-length) and 0.4-kb fragments compared with that in the pGL3 control construct, as detected by luciferase reporter activity. However, the 0.3-kb fragment results in decreased luciferase activity compared with both the 2.5-kb (full-length) and the 0.4-kb fragment, indicating that the critical region for nmMLCK transcription by VEGF is between −0.4 kb/−450 bp and −0.3 kb/−331 bp on the nmMYLK promoter. Three independent experiments were performed, with bar graphs representing pooled promoter activity readings as relative light units (RLU). *P < 0.05 versus vehicle. TSS: transcription start site; VEGF: vascular endothelial growth factor; Veh: vehicle.
On the basis of transcription factor binding motif predictions and the identification of a critical VEGF-responsive region of the nmMYLK promoter, the binding of a predicted transcription factor, Sp1, on the nmMYLK promoter between −450 and −331 bp was examined using the ChIP technique (Fig. 5A). Sp1 is a ubiquitously expressed C2H2-type zinc finger DNA-binding transcription factor that is involved in such biological processes as cell growth, differentiation, apoptosis, angiogenesis, and immune response.47 Sp1 recognizes the sequence aactgGGGCggtgcagg and has a high affinity for GC-rich sequences. ChIP analysis using an Sp1 antibody demonstrated enrichment of a nmMYLK promoter fragment containing Sp1 binding sites from VEGF-treated lung ECs compared with that in control cells (Fig. 5B). ChIP analysis using a negative control immunoglobulin G antibody resulted in negligible enrichment of nmMYLK promoter sequence with or without VEGF (Fig. 5B).
Figure 5.

Alteration of VEGF-induced permeability by Sp1 regulation of the nmMYLK promoter. A, Schematic depiction of the nmMYLK promoter, approximately 400 bp upstream of the transcription start site (TSS), with locations of potential transcription factor binding sites, including putative Sp1 binding sites (red bars), indicated. The promoter region analyzed for Sp1 binding by chromatin immunoprecipitation (ChIP) analysis is indicated by the green line. B, Quantitative polymerase chain reaction was performed with primers specific to the nmMYLK promoter region of interest, as indicated in A (green arrows). ChIP analysis using an Sp1 antibody demonstrated significant enrichment of the nmMYLK promoter sequence containing Sp1 binding sites from VEGF-treated (100 ng/mL, 24 hours) lung endothelial cells (ECs) compared with that in vehicle-treated cells (after normalization to the total chromatin input). ChIP analysis using a negative control immunoglobulin G (IgG) antibody resulted in negligible enrichment of the nmMYLK promoter sequence, with or without VEGF. Three independent experiments were performed, with bar graphs representing percent total chromatin input. *P < 0.05 versus vehicle. C, Confluent lung ECs treated with VEGF (100 ng/mL, 24 hours) showed a marked increase in nonmuscle isoform of myosin light chain kinase (nmMLCK) messenger RNA (mRNA) levels (compared with vehicle) that was attenuated with Sp1 silencing by means of small interfering RNA (siRNA). Three independent experiments were performed, with bar graphs representing fold change in pooled densitometry measurements. *P < 0.05 versus vehicle. D, Lung ECs were plated on gold microelectrodes for transendothelial electrical resistance (TER) measurements before Sp1 was silenced by means of siRNA, and then lung ECs were stimulated with vehicle or VEGF (100 ng/mL, 24 hours). VEGF-induced barrier disruption was attenuated by Sp1 siRNA (siSp1) but not by control siRNA (siCONT). Four independent TER experiments were performed, with pooled data represented as bar graphs. *P < 0.05 versus control siRNA. Inset shows Western blot demonstrating Sp1 protein silencing for C and D. HPRT: hypoxanthineguanine phosphoribosyltransferase; VEGF: vascular endothelial growth factor; Veh: vehicle.
The importance of Sp1 in EC barrier integrity was validated by the fact that silencing Sp1 by means of siRNA significantly attenuated VEGF-induced increases in nmMLCK mRNA expression in lung ECs (Fig. 5C). In addition, silencing Sp1 by means of siRNA significantly attenuated VEGF-induced vascular permeability in lung ECs by TER measurements (Fig. 5D). Taken together, these results confirm that Sp1 binds to the critical region of nmMYLK promoter and that Sp1 increases VEGF-induced vascular permeability by increasing expression of nmMLCK.
Discussion
Despite efforts to improve our understanding of the development of various pulmonary inflammatory diseases such as ARDS, the molecular mechanisms underlying these pathophysiologies remain unclear. The regulation of vascular permeability is a well-orchestrated process in maintaining fluid homeostasis that requires finely tuned responses to various physiological stimuli. ECs form a tight cell monolayer lining the inner vessel, and this wall is the key gatekeeper separating the bloodstream and tissue interstitium.48,49 Failure to maintain such regulation leads to detrimental inflammation in lungs and development of conditions such as ARDS, VILI, and asthma. In an effort to better understand the etiology of pulmonary inflammation and to develop novel pharmacotherapies, our group previously used genomic-intensive approaches to identify potential ARDS- and VILI-susceptibility candidate genes50-52 and identified the MYLK gene as a candidate gene for sepsis-associated and trauma-associated ARDS.5,15 Our previous in vivo studies in an LPS-induced lung injury model demonstrated that nmMLCK-dependent MLC phosphorylation resulted in increased actomyosin contraction and reorganization within ECs, leading to disruption of vascular barrier integrity53 and subsequently causing alveolar flooding and profound vascular leakage.54-56 Consistent with the highly multifunctional nature of the MYLK gene product in inflammation, our group has also previously identified MYLK coding SNPs as risk variants in three asthmatic populations of African descent (i.e., Chicago Collaborative Study on the Genetics of Asthma [CSGA], Baltimore CSGA, and Barbados).14,15 Furthermore, a recent study by Wang et al.19 demonstrated that nonmuscle isoforms of MYLK gene products are up-regulated during asthmatic inflammation. Taken together, these studies underscore the importance of elucidating the exact functional role played by nmMLCK in barrier regulation and inflammatory lung injury. VEGF, originally named vascular permeability factor, also has profound effects on vascular permeability via junctional protein structure remodeling and cytoskeleton reorganization. Similar to nmMLCK, VEGF has been associated with the pathophysiology of ARDS and asthma.31,32
Despite the important role played by nmMLCK in vascular barrier regulation and the implication of both nmMLCK and VEGF in the pathogenesis of ARDS, the role played by nmMLCK in VEGF-induced vascular permeability is poorly understood. In this study, we initially sought to examine whether VEGF-induced vascular permeability is nmMLCK dependent by using an in vitro permeability assay. Measurement of electrical resistance across the cultured lung EC monolayer demonstrated VEGF-induced decreases in TER, indicating disruption of vascular barrier and paracellular gap formation between lung ECs. The formation of contractile actin stress fibers was also observed in lung ECs treated with VEGF, in addition to colocalization of overexpressed nmMLCK with stress fibers. Increased enzymatic activity of nmMLCK after VEGF stimulation in lung ECs was also indicated by phosphorylation of MLC on Thr14 and Ser.15 The important role played by nmMLCK in VEGF-induced vascular permeability was highlighted when VEGF-induced vascular permeability was attenuated in lung ECs in which nmMLCK was silenced. These observations demonstrated that nmMLCK is in part responsible for deregulation of the EC barrier, corresponding with previous findings of nmMLCK involvement in increasing vascular permeability induced by several other known agonists.57-59
Recent studies have demonstrated that nmMLCK expression levels are altered in affected tissues isolated from patients with pulmonary inflammatory diseases, such as asthma.19 In addition, abnormal levels of VEGF have been implicated in a variety of pulmonary inflammatory diseases, including ARDS and asthma.29,32 On the basis of these links between pulmonary inflammation, nmMLCK expression levels, and VEGF, we sought to examine whether activation of VEGF signaling has a direct effect on expression of nmMLCK in pulmonary tissue under inflammation. We demonstrated that activation of the VEGF signaling pathway in lung ECs increases nonmuscle MYLK gene product at both mRNA and protein levels. This increase in nmMLCK mRNA and protein expression is a result of increased nmMYLK promoter activity. Further examination of the nmMYLK promoter using truncated promoter constructs demonstrated that the −0.4- to −0.3-kb region of the nmMYLK promoter is important for VEGF-induced nmMYLK promoter activation.
Further in silico analysis of the nmMYLK promoter revealed the presence of validated transcription factor binding sites for Sp1 within the −0.4- to −0.3-kb region upstream of the nmMYLK promoter transcription start site. The ubiquitous Sp1 transcription factor has high affinity for GC-rich sequences on the genome, which coincides with the presence of CpG islands on nmMYLK promoter. Binding of the Sp1 transcription factor on nmMYLK promoter was validated using ChIP technology, where VEGF stimulation of lung ECs increased Sp1 transcription factor binding to the −0.4-kb (−450 bp) to −0.3-kb (−331 bp) nmMYLK promoter region (predicted specifically at −366 bp). Silencing of the Sp1 protein attenuated VEGF-induced increased vascular permeability, suggesting that Sp1 plays a role as a transcriptional activator of the nmMYLK promoter.
As Sp3 competitively binds to Sp1 transcriptional factor binding sites, where they share the same consensus sequence, the role played by Sp3 in nmMYLK promoter regulation should be further examined. Another transcription factor binding site for hepatocyte nuclear factor 1 (HNF1), although not known to be associated with the VEGF pathway, was predicted to be located in the VEGF-responsive region of the nmMYLK promoter and should be examined to determine whether HNF1 also contributes to nmMYLK promoter regulation and subsequent VEGF-induced vascular permeability. Finally, the nmMYLK promoter should be further examined for promoter SNPs located in the VEGF-responsive region that may contribute to increased VEGF levels in ARDS patients or increased susceptibility of different populations to high levels of VEGF in the pathogenesis of ARDS.
In conclusion, these studies further detail the molecular mechanisms behind VEGF-induced vascular permeability and the important role that nmMLCK plays in regulating the vascular endothelial barrier through nmMYLK promoter regulation and subsequent increased nmMLCK expression. New insights into the regulation of the nmMYLK promoter via Sp1 transcription factor binding add a novel target, in addition to nmMLCK, for future acute lung injury therapeutic drug development.
Source of Support: This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants P01-HL058064 (JGNG) and R01-HL091889 (JGNG and TW).
Conflict of Interest: JGNG is the founder, president, and majority shareholder of Aqualung Therapeutics, which does not have any relevant conflicts of interest. All other authors: none declared.
References
- 1.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care 2005;11(1):43–49. [DOI] [PubMed]
- 2.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342(18):1334–1349. [DOI] [PubMed]
- 3.Brown LM, Kallet RH, Matthay MA, Dicker RA. The influence of race on the development of acute lung injury in trauma patients. Am J Surg 2011;201(4):486–491. [DOI] [PMC free article] [PubMed]
- 4.Moss M, Mannino DM. Race and gender differences in acute respiratory distress syndrome deaths in the United States: an analysis of multiple-cause mortality data (1979–1996). Crit Care Med 2002;30(8):1679–1685. [DOI] [PubMed]
- 5.Christie JD, Ma SF, Aplenc R, Li M, Lanken PN, Shah CV, Fuchs B, Albelda SM, Flores C, Garcia JG. Variation in the myosin light chain kinase gene is associated with development of acute lung injury after major trauma. Crit Care Med 2008;36(10):2794–2800. [DOI] [PubMed]
- 6.Flores C, Ma SF, Maresso K, Wade MS, Villar J, Garcia JG. IL6 gene-wide haplotype is associated with susceptibility to acute lung injury. Transl Res 2008;152(1):11–17. [DOI] [PubMed]
- 7.Gao L, Grant A, Halder I, Brower R, Sevransky J, Maloney JP, Moss M, et al. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol 2006;34(4):487–495. [DOI] [PMC free article] [PubMed]
- 8.Ma SF, Flores C, Wade MS, Dudek SM, Nicolae DL, Ober C, Garcia JG. A common cortactin gene variation confers differential susceptibility to severe asthma. Genet Epidemiol 2008;32(8):757–766. [DOI] [PMC free article] [PubMed]
- 9.Ma SF, Xie L, Pino-Yanes M, Sammani S, Wade MS, Letsiou E, Siegler J, et al. Type 2 deiodinase and host responses of sepsis and acute lung injury. Am J Respir Cell Mol Biol 2011;45(6):1203–1211. [DOI] [PMC free article] [PubMed]
- 10.Mitra S, Wade MS, Sun X, Moldobaeva N, Flores C, Ma SF, Zhang W, Garcia JG, Jacobson JR. GADD45a promoter regulation by a functional genetic variant associated with acute lung injury. PloS ONE 2014;9(6):e100169. [DOI] [PMC free article] [PubMed]
- 11.Sun X, Elangovan VR, Mapes B, Camp SM, Sammani S, Saadat L, Ceco E, et al. The NAMPT promoter is regulated by mechanical stress, signal transducer and activator of transcription 5, and acute respiratory distress syndrome–associated genetic variants. Am J Respir Cell Mol Biol 2014;51(5):660–667. [DOI] [PMC free article] [PubMed]
- 12.Sun X, Ma SF, Wade MS, Acosta-Herrera M, Villar J, Pino-Yanes M, Zhou T, et al. Functional promoter variants in sphingosine 1-phosphate receptor 3 associate with susceptibility to sepsis-associated acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol 2013;305(7):L467–L477. [DOI] [PMC free article] [PubMed]
- 13.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol 1995;163(3):510–522. [DOI] [PubMed]
- 14.Flores C, Ma SF, Maresso K, Ober C, Garcia JG. A variant of the myosin light chain kinase gene is associated with severe asthma in African Americans. Genet Epidemiol 2007;31(4):296–305. [DOI] [PubMed]
- 15.Gao L, Grant AV, Rafaels N, Stockton-Porter M, Watkins T, Gao P, Chi P, et al. Polymorphisms in the myosin light chain kinase gene that confer risk of severe sepsis are associated with a lower risk of asthma. J Allergy Clin Immunol 2007;119(5):1111–1118. [DOI] [PubMed]
- 16.Rossi JL, Velentza AV, Steinhorn DM, Watterson DM, Wainwright MS. MLCK210 gene knockout or kinase inhibition preserves lung function following endotoxin-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 2007;292(6):L1327–L1334. [DOI] [PubMed]
- 17.Mirzapoiazova T, Moitra J, Moreno-Vinasco L, Sammani S, Turner JR, Chiang ET, Evenoski C, et al. Non-muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury. Am J Respir Cell Mol Biol 2011;44(1):40–52. [DOI] [PMC free article] [PubMed]
- 18.Moitra J, Evenoski C, Sammani S, Wadgaonkar R, Turner JR, Ma SF, Garcia JG. A transgenic mouse with vascular endothelial over-expression of the non-muscle myosin light chain kinase–2 isoform is susceptible to inflammatory lung injury: role of sexual dimorphism and age. Transl Res 2008;151(3):141–153. [DOI] [PMC free article] [PubMed]
- 19.Wang T, Moreno-Vinasco L, Ma SF, Zhou T, Shimizu Y, Sammani S, Epshtein Y, Watterson DM, Dudek SM, Garcia JG. Nonmuscle myosin light chain kinase regulates murine asthmatic inflammation. Am J Respir Cell Mol Biol 2014;50(6):1129–1135. [DOI] [PMC free article] [PubMed]
- 20.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 2004;25(4):581–611. [DOI] [PubMed]
- 21.Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer 2008;8(11):880–887. [DOI] [PMC free article] [PubMed]
- 22.McDonald DM. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med 2001;164(10 pt 2):S39–S45. [DOI] [PubMed]
- 23.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005;437(7058):497–504. [DOI] [PubMed]
- 24.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995;146(5):1029–1039. [PMC free article] [PubMed]
- 25.Dvorak HF, Dvorak AM, Manseau EJ, Wiberg L, Churchill WH. Fibrin gel investment associated with line 1 and line 10 solid tumor growth, angiogenesis, and fibroplasia in guinea pigs: role of cellular immunity, myofibroblasts, microvascular damage, and infarction in line 1 tumor regression. J Natl Cancer Inst 1979;62(6):1459–1472. [PubMed]
- 26.Dvorak HF, Orenstein NS, Carvalho AC, Churchill WH, Dvorak AM, Galli SJ, Feder J, Bitzer AM, Rypysc J, Giovinco P. Induction of a fibrin-gel investment: an early event in line 10 hepatocarcinoma growth mediated by tumor-secreted products. J Immunol 1979;122(1):166–174. [PubMed]
- 27.Becker PM, Verin AD, Booth MA, Liu F, Birukova A, Garcia JG. Differential regulation of diverse physiological responses to VEGF in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 2001;281(6):L1500–L1511. [DOI] [PubMed]
- 28.Barratt S, Medford AR, Millar AB. Vascular endothelial growth factor in acute lung injury and acute respiratory distress syndrome. Respiration 2014;87(4):329–342. [DOI] [PubMed]
- 29.Feltis BN, Wignarajah D, Zheng L, Ward C, Reid D, Harding R, Walters EH. Increased vascular endothelial growth factor and receptors: relationship to angiogenesis in asthma. Am J Respir Crit Care Med 2006;173(11):1201–1207. [DOI] [PubMed]
- 30.Kanazawa H, Hirata K, Yoshikawa J. Involvement of vascular endothelial growth factor in exercise induced bronchoconstriction in asthmatic patients. Thorax 2002;57(10):885–888. [DOI] [PMC free article] [PubMed]
- 31.Medford AR, Millar AB. Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): paradox or paradigm? Thorax 2006;61(7):621–626. [DOI] [PMC free article] [PubMed]
- 32.Thickett DR, Armstrong L, Christie SJ, Millar AB. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;164(9):1601–1605. [DOI] [PubMed]
- 33.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996;380(6573):435–439. [DOI] [PubMed]
- 34.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996;380(6573):439–442. [DOI] [PubMed]
- 35.Berse B, Brown LF, Van de Water L, Dvorak HF, Senger DR. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell 1992;3(2):211–220. [DOI] [PMC free article] [PubMed]
- 36.Monacci WT, Merrill MJ, Oldfield EH. Expression of vascular permeability factor/vascular endothelial growth factor in normal rat tissues. Am J Physiol 1993;264(4 pt 1):C995–C1002. [DOI] [PubMed]
- 37.Mirzapoiazova T, Kolosova I, Usatyuk PV, Natarajan V, Verin AD. Diverse effects of vascular endothelial growth factor on human pulmonary endothelial barrier and migration. Am J Physiol Lung Cell Mol Physiol 2006;291(4):L718–L724. [DOI] [PubMed]
- 38.Azamfirei L, Gurzu S, Solomon R, Copotoiu R, Copotoiu S, Jung I, Tilinca M, et al. Vascular endothelial growth factor: a possible mediator of endothelial activation in acute respiratory distress syndrome. Minerva Anestesiol 2010;76(8):609–616. [PubMed]
- 39.Zhai R, Gong MN, Zhou W, Thompson TB, Kraft P, Su L, Christiani DC. Genotypes and haplotypes of the VEGF gene are associated with higher mortality and lower VEGF plasma levels in patients with ARDS. Thorax 2007;62(8):718–722. [DOI] [PMC free article] [PubMed]
- 40.Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem 2004;279(23):24692–24700. [DOI] [PubMed]
- 41.Brown M, Adyshev D, Bindokas V, Moitra J, Garcia JG, Dudek SM. Quantitative distribution and colocalization of non-muscle myosin light chain kinase isoforms and cortactin in human lung endothelium. Microvasc Res 2010;80(1):75–88. [DOI] [PMC free article] [PubMed]
- 42.Wang T, Zhou T, Saadat L, Garcia JG. A MYLK variant regulates asthmatic inflammation via alterations in mRNA secondary structure. Eur J Hum Genet 2015;23(6):874–876. [DOI] [PMC free article] [PubMed]
- 43.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 2001;108(5):689–701. [DOI] [PMC free article] [PubMed]
- 44.Camp SM, Bittman R, Chiang ET, Moreno-Vinasco L, Mirzapoiazova T, Sammani S, Lu X, et al. Synthetic analogs of FTY720 [2-amino-2-(2-[4-octylphenyl]ethyl)-1,3-propanediol] differentially regulate pulmonary vascular permeability in vivo and in vitro. J Pharmacol Exp Ther 2009;331(1):54–64. [DOI] [PMC free article] [PubMed]
- 45.Frech K, Danescu-Mayer J, Werner T. A novel method to develop highly specific models for regulatory units detects a new LTR in GenBank which contains a functional promoter. J Mol Biol 1997;270(5):674–687. [DOI] [PubMed]
- 46.Klingenhoff A, Frech K, Quandt K, Werner T. Functional promoter modules can be detected by formal models independent of overall nucleotide sequence similarity. Bioinformatics 1999;15(3):180–186. [DOI] [PubMed]
- 47.Wierstra I. Sp1: emerging roles—beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 2008;372(1):1–13. [DOI] [PubMed]
- 48.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol 2001;91(4):1487–1500. [DOI] [PubMed]
- 49.Komarova Y, Malik AB. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 2010;72:463–493. [DOI] [PubMed]
- 50.Grigoryev DN, Finigan JH, Hassoun P, Garcia JG. Science review: searching for gene candidates in acute lung injury. Crit Care 2004;8(6):440–447. [DOI] [PMC free article] [PubMed]
- 51.Grigoryev DN, Ma SF, Irizarry RA, Ye SQ, Quackenbush J, Garcia JG. Orthologous gene-expression profiling in multi-species models: search for candidate genes. Genome Biol 2004;5(5):R34. [DOI] [PMC free article] [PubMed]
- 52.Simon BA, Easley RB, Grigoryev DN, Ma SF, Ye SQ, Lavoie T, Tuder RM, Garcia JG. Microarray analysis of regional cellular responses to local mechanical stress in acute lung injury. Am J Physiol Lung Cell Mol Physiol 2006;291(5):L851–L861. [DOI] [PubMed]
- 53.Jacobson JR, Dudek SM, Singleton PA, Kolosova IA, Verin AD, Garcia JG. Endothelial cell barrier enhancement by ATP is mediated by the small GTPase Rac and cortactin. Am J Physiol Lung Cell Mol Physiol 2006;291(2):L289–L295. [DOI] [PubMed]
- 54.Goldblum SE, Hennig B, Jay M, Yoneda K, McClain CJ. Tumor necrosis factor α–induced pulmonary vascular endothelial injury. Infect Immun 1989;57(4):1218–1226. [DOI] [PMC free article] [PubMed]
- 55.Hirano S, Rees RS, Yancy SL, Welsh MJ, Remick DG, Yamada T, Hata J, Gilmont RR. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of HSP27 in vivo. Cell Biol Toxicol 2004;20(1):1–14. [DOI] [PubMed]
- 56.Penn MS, Chisolm GM. Relation between lipopolysaccharide-induced endothelial cell injury and entry of macromolecules into the rat aorta in vivo. Circ Res 1991;68(5):1259–1269. [DOI] [PubMed]
- 57.Garcia JG, Schaphorst KL. Regulation of endothelial cell gap formation and paracellular permeability. J Investig Med 1995;43(2):117–126. [PubMed]
- 58.Petrache I, Birukov K, Zaiman AL, Crow MT, Deng H, Wadgaonkar R, Romer LH, Garcia JG. Caspase-dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF-α-mediated bovine pulmonary endothelial cell apoptosis. FASEB J 2003;17(3):407–416. [DOI] [PubMed]
- 59.Wadgaonkar R, Linz-McGillem L, Zaiman AL, Garcia JG. Endothelial cell myosin light chain kinase (MLCK) regulates TNFα-induced NFκB activity. J Cell Biochem 2005;94(2):351–364. [DOI] [PubMed]