

Summary
The integrity of chromatin, which provides a dynamic template for all DNA-related processes in eukaryotes, is maintained through replication-dependent and -independent assembly pathways. To address the role of histone deposition in the absence of DNA replication, we deleted the H3.3 chaperone Hira in developing mouse oocytes. We show that chromatin of non-replicative developing oocytes is dynamic and that lack of continuous H3.3/H4 deposition alters chromatin structure, resulting in increased DNase I sensitivity, the accumulation of DNA damage, and a severe fertility phenotype. On the molecular level, abnormal chromatin structure leads to a dramatic decrease in the dynamic range of gene expression, the appearance of spurious transcripts, and inefficient de novo DNA methylation. Our study thus unequivocally shows the importance of continuous histone replacement and chromatin homeostasis for transcriptional regulation and normal developmental progression in a non-replicative system in vivo.
Graphical Abstract
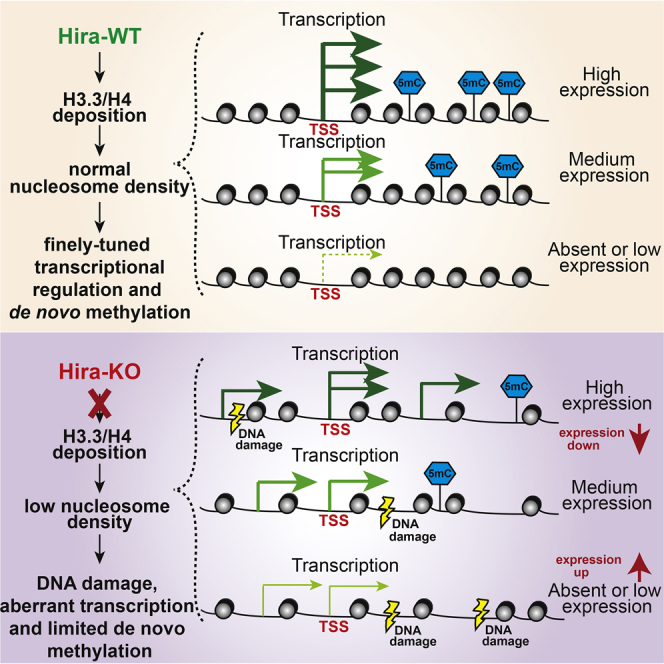
Highlights
-
•
Histone H3/H4 replacement is continuous and mediated by Hira during mouse oogenesis
-
•
Loss of Hira results in chromatin abnormalities and extensive oocyte loss
-
•
Hira depletion reduces histone load, which prevents normal transcriptional regulation
-
•
Hira-mediated histone replacement is required for normal 5mC deposition in oocytes
To address the extent to which basic cellular processes depend on replication-independent chromatin assembly in vivo, Nashun et al. deleted histone H3.3 chaperone Hira during mouse oogenesis. Their results demonstrate a critical relationship between continuing histone replacement, chromatin homeostasis, transcriptional regulation, and de novo DNA methylation.
Introduction
DNA in all eukaryotic organisms is bound by nucleosomes, forming a physiological chromatin context in which all molecular processes involving DNA operate. The integrity of the chromatin template is constantly compromised by fundamental biological processes, such as DNA replication, repair, and transcription, following which the normal chromatin structure is restored with the help of histone chaperone proteins (Gurard-Levin et al., 2014, Ransom et al., 2010). Advances in recent years have shed light on some of the molecular players involved in chromatin assembly and maintenance, separating (on the molecular level) DNA replication-dependent and -independent pathways (Burgess and Zhang, 2013). Histone proteins themselves are central to these processes: the expression and incorporation of canonical histones is tightly coupled to DNA replication; in contrast, histone variants can be incorporated into chromatin independent of the cell cycle (Maze et al., 2014).
While it has become increasingly clear that the activity of histone chaperone proteins is of critical importance during DNA replication and repair (Adam et al., 2013, Hoek and Stillman, 2003, Polo et al., 2006, Ransom et al., 2010), studies that have attempted to dissect the importance of histone replacement in the interphase nucleus have revealed only a limited contribution of histone chaperones and/or variants to transcriptional regulation (Banaszynski et al., 2013, Goldberg et al., 2010, Hödl and Basler, 2009, Sakai et al., 2009). These studies, however, have been complicated by the use of proliferative cell systems that could (at least partially) restore chromatin integrity through replication-coupled chromatin assembly (Banaszynski et al., 2013, Hödl and Basler, 2009, Sakai et al., 2009, Wyrick et al., 1999). Thus, the extent to which basic physiological processes, such as transcription, are dependent on or regulated by histone replacement remains unclear.
To overcome the limitations of these previous studies, we took advantage of the unique system presented by mammalian oogenesis. Over an extended time span and in the absence of DNA replication, postnatal mammalian oocytes execute the oogenesis-specific developmental program, involving widespread transcriptional changes and de novo DNA methylation, ultimately acquiring the competencies required for fertilization and embryogenesis (De La Fuente, 2006, Li and Albertini, 2013, Tomizawa et al., 2012).
To address the importance of histone turnover during this process, we have generated a mouse oocyte-specific knockout of the histone chaperone Hira. As opposed to Caf1, which is implicated in the replication-coupled deposition of canonical histones H3.1 and H3.2, Hira has been shown to deposit the H3.3 variant during replication-independent chromatin assembly (Ray-Gallet et al., 2002, Tagami et al., 2004). Our findings show that depletion of Hira in primordial oocytes causes a severe developmental defect associated with extensive oocyte death. On the molecular level, Hira depletion compromises the oocyte’s ability to replace histones H3 and H4. This causes significantly increased DNA accessibility, accumulation of DNA damage, and chromosome segregation defects. The lack of normal chromatin structure has a striking impact on transcriptional regulation: lack of normal H3.3/H4 replacement prevents the oocytes from maintaining full dynamic range of gene expression, and it leads to compromised transcriptional transitions normally associated with the oocyte development. Furthermore, we show that histone replacement is necessary for silencing of spurious transcription and, in the context of the oocyte, also for the efficient deposition of de novo DNA methylation.
Our results thus uniquely demonstrate the critical relationship between continuous histone replacement, the structural integrity of the chromatin template, the normal regulation of transcription, and the establishment of DNA methylation, in the context of an in vivo developmental system.
Results
Hira Is Responsible for H3.3/H4 Turnover in Postnatal Oocytes
In the mouse, female germ cells arrest in prophase of the first meiotic division at embryonic day (E)13.5. The female gonocytes subsequently resume growth in postnatal ovaries where they undergo oogenesis and progress through meiosis (De La Fuente, 2006). This process involves the following: (1) pronounced transcriptional changes as the developing oocytes progress from primordial to large antral follicle stages; (2) the deposition of de novo DNA methylation starting in the secondary follicle stage; (3) the silencing of transcription in the germinal vesicle (GV) stage; and, finally, (4) the condensation of chromosomes in the MII stage, resulting in an oocyte that is competent to undergo fertilization and support early embryonic development (Figure 1A).
Figure 1.

Incorporation of H3.3 in Developing Oocytes Is Driven by Hira
(A) Schematic illustration shows developmental stages and global transcriptional activity during oogenesis with indicated onset of Gdf9-Cre+ and Zp3-Cre+ expression (adapted from De La Fuente, 2006, Lan et al., 2004, Li and Albertini, 2013; and Tomizawa et al., 2012).
(B) Growing oocytes (postnatal day [P]14) were subjected to mRNA microinjection of Flag-tagged H3.1, H3.2, H3.3, or H2A.X. Incorporation of histone variants was visualized by anti-Flag antibody staining.
(C) Flag-H2A.X, Flag-H4, and Flag-H3.3 mRNA was microinjected into the cytoplasm of transcriptionally inert GV oocytes from Hiraf/f or Hiraf/fGdf9-Cre+ siblings. Following 5-hr incubation in the IBMX-containing medium, the oocytes were either fixed (GV) or transferred to IBMX-free medium to induce germinal vesicle breakdown (GVBD) and the incorporation of tagged histones was visualized as in (B). Dashed rectangles indicate the location of condensed chromosomes of the GVBD oocyte.
(D) Lack of Hira in growing (P14, left) or GV-stage (GV, right) oocytes from Hiraf/fGdf9-Cre+ is shown (Hiraf/f siblings were used as controls).
(E) Deletion of Hira leads to dramatically reduced H3.3 in chromatin. Growing and GV oocytes from Hiraf/f, H3.3B-EGFP, or Hiraf/fGdf9-Cre+, H3.3B-EGFP mice. (Top) Oocytes were subject to paraformaldehyde (PFA) fixation directly after collection. (Bottom) Oocytes were pre-extracted by Triton X-100 prior to PFA fixation to reveal chromatin-associated H3.3. A minimum of 12 oocytes were examined. DNA was stained with DAPI (in blue). Scale bar, 10 μm. See also Figures S1 and S2.
We first addressed the capacity of developing oocytes to incorporate histones by microinjection of mRNA for tagged versions of histones. In agreement with the absence of replication during oocyte maturation, growing oocytes did not show any incorporation of canonical histones H3.1 and H3.2 (Figure 1B; Akiyama et al., 2011). In contrast, microinjected Flag-tagged H3.3 and H2A.X, previously shown to be enriched in the developing oocyte (Akiyama et al., 2011, Nashun et al., 2010), were readily incorporated into the chromatin of growing oocytes (Figure 1B). We thus conclude that H3.3 is the only H3 subtype incorporated during postnatal oocyte development. In further support of this observation, oocytes isolated from our H3.3B-EGFP knockin mice (Figure S1G) confirmed that both growing and GV oocytes contain high levels of nuclear H3.3 (Figure 1E). Growing oocytes are transcriptionally highly active, which could explain the observed histone turnover. However, we also detected a high level of chromatin incorporation for Flag-tagged H2A.X, H4, and H3.3 upon injection of tagged histone mRNA into transcriptionally inactive GV-stage oocytes (Figure 1C), demonstrating that the chromatin in the mature GV oocyte is still dynamic even after global transcription has ceased.
Previous studies have shown that the majority of H3.3 incorporation is critically dependent on the presence of histone chaperone Hira (Goldberg et al., 2010, Pchelintsev et al., 2013). In agreement with the observed H3.3 incorporation, Hira is expressed and shows nuclear localization during postnatal oocyte development (Figure 1D). To investigate whether Hira is specifically required for the observed H3.3 incorporation during oogenesis, we took advantage of Gdf9- and Zp3-driven expression of Cre recombinase and deleted Hira in the primordial and primary stages of follicle development, respectively (Figures 1A and S1A). We confirmed that both Gdf9-Cre- and Zp3-Cre-deleted oocytes undergo genetic recombination (Figure S1B; data not shown) and are depleted of Hira mRNA, protein and complex (Figures 1D and S1C–S1F).
The absence of Hira severely decreased H3.3 content and completely abolished the incorporation of microinjected tagged H3.3 in both growing and GV-stage oocytes (Figures 1B, 1C, and 1E). The observed impairment of H3.3 incorporation occurred despite the presence of an alternative ATRX/DAXX chaperone complex that has been well characterized to incorporate H3.3 (Burgess and Zhang, 2013, Maze et al., 2014). Of note, the presence and the localization of this complex in the GV oocyte were not affected by Hira deletion (Figure S2A), and we could observe H3.3B-EGFP loci overlapping with ATRX staining in Hiraf/f Gdf9-Cre+, H3.3B-EGFP GV oocytes at higher laser detection intensity (Figure S2C). The severe reduction of H3.3 incorporation in Hiraf/f Gdf9-Cre+ oocytes thus demonstrates that the ATRX/DAXX chaperone complex cannot compensate for the lack of Hira, as was also observed in mouse embryonic stem cells (ESCs) (Goldberg et al., 2010). The lack of Hira and H3.3 incorporation also was not compensated for by incorporation of the canonical histones H3.1 or H3.2 (Figure 1B), and it coincided with the lack of detectable incorporation of histone H4 (Figure 1C).
Taken together, these findings demonstrate that the histone variant H3.3, rather than H3.1 or H3.2, is actively incorporated into chromatin during oocyte development, and confirm that Hira is responsible for the incorporation of the majority of H3.3 in conjunction with histone H4 (Figures 1B–1E and S2B). Of note, although Hira-deficient oocytes failed to incorporate tagged H3 and H4 histones, the incorporation of tagged H2A.X was readily detectable (Figures 1B and 1C), confirming that distinct molecular pathways are implicated in the deposition of H3/H4 and H2A/H2B histones (Burgess and Zhang, 2013).
Hira Depletion Results in a Severe Ovarian Phenotype, Associated with Extensive Oocyte Death and the Failure to Support Zygotic Reprogramming or Embryonic Development
Assessment of females with oocyte-specific Hira deletion revealed that loss of Hira led to a severe ovarian phenotype. Significantly lower numbers of MII oocytes could be recovered following superovulation (Figure 2A); Zp3-Cre-driven deletion of Hira led to a more than 50% reduction in the number of ovulated MII oocytes, and this effect was even more dramatic upon earlier Gdf9-Cre-driven deletion (Figure 2A). Furthermore, GV oocytes (Figure 1A) isolated from both Hiraf/f Zp3-Cre+ and Hiraf/f Gdf9-Cre+ ovaries showed dramatically reduced competency to complete the GV-to-MII transition during in vitro maturation (Figure S2E).
Figure 2.

Hira Is Essential for Progression through Oogenesis and Acquisition of Developmental Competence
(A) Number of ovulated oocytes recovered per female after hormonal stimulation. Numbers of females scored are indicated in the columns. Error bars indicate SEM. ∗∗∗p < 0.001; statistical analysis was carried out using two-tailed unpaired Student’s t test.
(B) Ovaries of Hiraf/f, Hiraf/fGdf9-Cre+, and Hiraf/fZp3-Cre+ females were collected from mice born on the same date and ovarian images were taken in a single picture.
(C) Representative images show H&E staining of 3-week (top) and 6-week (bottom) ovarian sections (follicles indicated by arrows).
(D) Maternal Hira depletion results in defects in chromosome condensation and segregation. Representative bright-field images of MII oocytes recovered from two Hiraf/f and two Hiraf/fGdf9-Cre+ mice at 3 weeks of age are shown. Arrows (left) indicate oocytes with defective asymmetric division normally associated with first polar body extrusion (left). The oocytes were fixed and stained with DAPI (right). Arrows and arrowheads (right) indicate chromosome bridges and lagging chromosomes, respectively. Quantification of normal and abnormal MII oocytes is shown (bottom right).
(E) Early developmental arrest of parthenogenetic embryos with maternal Hira depletion. Representative images show embryos (left); quantification of developmental progression is shown (right). Oocytes lacking Hira do not progress beyond the two-cell stage. Embryo numbers are indicated above each column. Error bars indicate SEM. See also Figures S2, S3, and S4.
The severity of Hira deletion was particularly obvious upon Gdf9-Cre-driven knockout. Hiraf/f Gdf9-Cre+ ovaries were significantly smaller (Figures 2B and S2F); and, although we could recover some GV oocytes from 3-week-old females, the ovaries of older mice contained only very few oocytes, which were predominantly at early developmental stages (Figures 2C and S2G). Further analysis of the superovulated Hiraf/f Gdf9-Cre+ MII oocytes revealed frequent defects in chromosome alignment and segregation and instances of aberrant polar body extrusion (Figure 2D).
In line with a severe ovarian phenotype and in agreement with recently published reports (Inoue and Zhang, 2014, Lin et al., 2014, Loppin et al., 2005), the surviving Hira-depleted oocytes were unable to either reprogram the paternal pronucleus or support early embryonic development after fertilization. In agreement with the observed enrichment of H3.3 and Hira in the paternal pronucleus of control zygotes (Figures S3A–S3C), fertilized Hiraf/f Gdf9-Cre+ oocytes could not fully de-condense the male pronucleus (Figure S3D), incorporate H3.3 (Figure S3E), or support normal zygotic development (Figure S3F). Failure to incorporate H3.3 occurred despite the efficient removal of protamines (Figure S4A), indicating that protamine removal and chromatinization of the paternal genome are mechanistically distinct (Inoue and Zhang, 2014, Lin et al., 2014). Although the histone load of the paternal pronucleus was severely reduced upon maternal Hira deletion, we could detect a residual amount of histones on the paternal DNA by immunofluourescent staining (Figure S4B). However, it remains to be addressed whether the residual histones were inherited from sperm or whether they were incorporated in a Hira-independent manner. In agreement with the severe chromatinization defect, the partially decondensed paternal pronucleus of zygotes in which Hira had been maternally deleted failed to undergo DNA demethylation (Figure S4C) and could not provide a normal template for zygotic replication or transcription (Figures S4D and S4E; Lin et al., 2014).
Apart from the failure to support normal reprogramming of the paternal pronucleus following fertilization, Hira-depleted oocytes showed severe maternal defects. The ATP content was significantly reduced in Hira-depleted MII oocytes (Figure S2H), and, upon parthenogenetic activation, only a fraction of these oocytes reached the two-cell stage and none progressed further, contrary to the control oocytes that efficiently supported development of parthenogenetic blastocysts (Figure 2E).
Hira-Depleted Oocytes Show Increased DNA Accessibility and Accumulation of DNA Damage
Considering the pronounced oocyte phenotype, we set out to understand the molecular mechanisms by which Hira regulates oocyte development. We hypothesized that, in the absence of H3/H4 incorporation, the chromatin structure in Hira-depleted oocytes would be severely affected. In agreement with this, the histone content of Hira-depleted GV-stage oocytes was severely reduced (Figures 3A and S2B), and the resulting chromatin displayed altered structure and increased DNase I sensitivity (Figures 3B and 3C). Moreover, in accordance with the predicted role of histones in protecting genetic material from environmental stress and mutagenesis (Ljungman and Hanawalt, 1992), Hira-depleted oocytes showed clear signs of DNA damage both by increased levels of γ-H2AX (Figure 3D) and upregulation of DNA damage-responsive genes (Figure 3E).
Figure 3.

Hira Deletion Leads to Increased DNA Accessibility and Accumulation of DNA Damage
(A) Hira deletion leads to reduced histone load in the Hiraf/fGdf9-Cre+ GV oocytes. The graph shows the quantification of the IF (pan histone antibody staining) signal normalized to DNA content (DAPI).
(B) Structural alteration of chromatin in Hiraf/fGdf9-Cre+ GV oocytes. Extensive chromatin de-condensation was observed in more than half of the examined Hiraf/fGdf9-Cre+ GV oocytes. DNA was stained with DAPI and pseudocolored in gray.
(C) Lack of Hira leads to increased DNase I accessibility. Schematic illustration shows in vivo DNase I TUNEL assay (left); see also the Supplemental Experimental Procedures. The fraction of oocytes with positive TUNEL signal is indicated in brackets.
(D) Lack of Hira leads to the accumulation of DNA damage. The γ-H2A.X staining of growing oocytes (P16, left) and GV oocytes (right) is shown.
(E) Gene set enrichment analysis (GSEA) compares genes upregulated after DNA damage (Kyng_DNA_Damage_Up gene set) and the ranked list of gene expression changes in Hiraf/fGdf9-Cre+ relative to Hiraf/f MII oocytes. NES, normalized enrichment score; FDR, false discovery rate (both were calculated in the GSEA program). DNA was stained with DAPI (in blue). Scale bar, 10 μm.
Continuing Histone Replacement Mediated by Hira Is Required to Maintain the Full Dynamic Range of Gene Expression
The importance of Hira and continuous H3.3 replacement has been previously assessed in the context of proliferating mouse pluripotent ESCs (Banaszynski et al., 2013, Goldberg et al., 2010). These studies surprisingly revealed that Hira deletion led to only minor transcriptional changes (Banaszynski et al., 2013, Goldberg et al., 2010). Considering the severity of the phenotype observed following the deletion of Hira in oocytes, we next asked whether the reduced H3/H4 incorporation and increased DNA accessibility (Figures 1B–1E, 3A, and 3C) affected the transcriptional program of the Hira-depleted oocytes. Surprisingly, although the lack of maternal Hira severely affected the amount of transcription emanating from the paternal pronucleus following fertilization (Figure S4E; Lin et al., 2014), Hira-depleted and control growing oocytes showed similar global levels of transcription, as judged by EU incorporation (Figure 4A).
Figure 4.

Continuous Histone Replacement Mediated by Hira Is Required to Maintain the Full Dynamic Range of Gene Expression
(A) Detection of newly synthesized RNA by EU incorporation in growing (P14) and GV (P20) Hiraf/f and Hiraf/fGdf9-Cre+ oocytes is shown. Scale bar, 10 μm.
(B) Comparison between obtained FPKM values and absolute copy number per cell for ERCC spike-in RNA in Hiraf/f and Hiraf/fGdf9-Cre+ MII oocytes revealed no significant difference.
(C) Principal component analysis of scRNA-seq data derived from Hiraf/f and Hiraf/fGdf9-Cre+ MII oocytes is shown.
(D and E) Selected gene ontology (GO) terms significantly enriched for among differentially upregulated (D, edgeR, FDR < 0.1) and differentially downregulated (E, edgeR, FDR < 0.1) genes in Hiraf/fGdf9-Cre+ MII oocytes are shown. x axis represents Benjamini-Hochberg adjusted p value.
(F) Number of annotated genes detected, as computed by HTSeq program, is shown.
(G) Relative proportion of genes distributed among ten equally sized expression level bins. All genes detected in our RNA-seq experiment were divided into ten equal bins based on their expression levels in Hiraf/f oocytes. The gene number in each bin was counted for Hiraf/f or Hiraf/fGdf9-Cre+, and the percentage was calculated relative to all genes detected in a given sample.
(H) Boxplot shows gene expression levels of differentially upregulated (edgeR, FDR < 0.1), downregulated (edgeR, FDR < 0.1), and all annotated genes in Hiraf/fGdf9-Cre+ MII oocytes.
(I) Boxplots show distribution of gene expression fold change for each gene expression level quintile (based on Hiraf/f gene expression levels).
(J) Variance of gene expression within a given Hiraf/f or Hiraf/fGdf9-Cre+ sample. In all cases, error bars indicate SEM. Statistical analysis was carried out using two-tailed unpaired Student’s t test (F and G), Kruskal-Wallis with Dunn’s post hoc test (H) or F test (J); ns, non-significant; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. See also Figure S6.
To address the role of Hira in transcription during oocyte development in depth, we carried out single-cell RNA sequencing (scRNA-seq) on four Hiraf/f Gdf9-Cre+ and four Hiraf/f control oocytes. We chose to analyze ovulated MII oocytes that had successfully completed their first meiotic division to avoid inconsistencies due to differences in staging or developmental progression. Of note, consistent with the identical patterns of EU incorporation in the Hiraf/f Gdf9-Cre+ and control growing oocytes (Figure 4A), normalization to the ERCC RNA spike-ins included in our scRNA-seq library preparations revealed no significant differences in the total quantity of poly-adenylated RNA between Hiraf/f Gdf9-Cre+ and control MII oocytes (Figure 4B).
In agreement with the observed severe developmental phenotype, the initial data analysis revealed pronounced transcriptional differences upon deletion of Hira (Figures 4C and S6A). Expression analysis identified 2,101 differentially expressed genes (false discovery rate [FDR] < 0.1) between Hiraf/f Gdf9-Cre+ and control oocytes (1,274 upregulated in Hiraf/f Gdf9-Cre+ oocytes relative to controls; 827 downregulated), representing over 15% of all annotated transcripts detected (Table S1). Gene ontology (GO) analysis of differentially upregulated genes showed enrichment for cell membrane, biological adhesion molecules, and cell junctions (Figure 4D). In contrast, GO analysis on differentially downregulated genes confirmed enrichment for processes important in late-stage oocytes, including chromosome, nucleolus, mitochondria, and pronucleus (Figure 4E).
Surprisingly, we detected significantly more annotated transcripts (7.7% increase, p value < 0.01) in Hiraf/f Gdf9-Cre+ oocytes relative to controls (Figure 4F). The increased number of transcripts was not evenly distributed across all expression levels (Figure 4G); rather, deletion of Hira resulted in relatively fewer transcripts with very low (first decile) or very high (ninth and tenth deciles) expression (Figure 4G). Furthermore, differentially down- and upregulated genes showed higher and lower than expected expression, respectively (Figure 4H); and, while the genes normally not expressed or expressed at very low levels (bottom 20%) were upregulated upon Hira deletion (Figure 4I; gene set enrichment analysis [GSEA], FDR < 0.001; data not shown), genes with very high expression in normal oocytes (top 20%) were specifically downregulated in Hiraf/f Gdf9-Cre+ oocytes (Figure 4I; GSEA, FDR < 0.001; data not shown). This also was evident by the greatly reduced variance in expression levels of annotated transcripts within the Hira-depleted oocytes (Figure 4J; p value < 0.001), suggesting that Hira is necessary to maintain the full dynamic range of gene expression. Thus, contrary to previous findings in proliferating pluripotent ESCs, Hira appears to be critical for normal transcriptional regulation during oocyte development.
The major transcriptional transition during oogenesis occurs between primordial stage oocytes (when Gdf9-driven Cre is first expressed) and primary stage oocytes (when Zp3-driven Cre is first expressed), during which the major wave of gene activation is observed (Pan et al., 2005). To test whether the observed transcriptional defects are due to Hiraf/f Gdf9-Cre+ oocytes failing to execute this transcriptional activation or to other dominant secondary effects, we additionally carried out scRNA-seq on four Hiraf/f Zp3-Cre+ and three Hiraf/f control oocytes (Figure S6; Table S3). Remarkably, a nearly identical transcriptional phenotype was observed in Hiraf/f Zp3-Cre+ and Hiraf/f Gdf9-Cre+ oocytes: differentially down- and upregulated genes showed higher and lower than average expression in the control oocytes, respectively (Figure S6E), and genes normally not expressed or expressed at very low levels (bottom 20%) were upregulated upon Hira deletion (Figure S6F), while genes with very high expression in normal oocytes (top 20%) were specifically downregulated in Hiraf/f Gdf9-Cre+ oocytes (Figure S6F). Similar to the effect observed in Hiraf/f Gdf9-Cre+ oocytes, the variance in expression levels of annotated transcripts within the Hiraf/f Zp3-Cre+ oocytes was greatly reduced (Figure S6G; p value < 0.001).
Hira-Mediated H3.3/H4 Deposition Is Essential for Transcriptional Transitions Associated with the Oocyte Developmental Program
The developing oocyte is a unique experimental system, as it allows us to assess the importance of continuing histone H3.3/H4 replacement not only for steady-state transcription, but also for affecting transcriptional transitions associated with development, such as activation or repression of dynamically regulated genes. To address this, we used K-means clustering of published expression data (Pan et al., 2005; GEO: GSE3351) to identify eight clusters of genes that show distinct expression patterns over the course of oocyte development (Figures S5A and S5B), and we compared these to the expression profiles in Hiraf/f Gdf9-Cre+ and control oocytes. Remarkably, genes downregulated upon Hira depletion (Hiraf/f Gdf9-Cre+) were specifically enriched for genes whose transcription is activated either between the earliest stages of oocyte development (Figure 5A, cluster 4, primordial and primary follicle stages; GSEA, FDR < 0.001) or between early and late oogenesis (Figure 5A, cluster 5; GSEA, FDR < 0.001). In contrast, genes upregulated upon Hira deletion were most significantly enriched for clusters of genes that all uniquely show downregulation of expression between early and late oogenesis (Figure 5A, cluster 2; FDR = 0.017) or between primordial and primary follicle stages (Figure S5B, clusters 6 and 8). Of note, similar phenotypes were observed upon Zp3-Cre-driven deletion of Hira (Figure S6H).
Figure 5.

Hira Is Essential for Transcriptional Transitions Associated with the Oocyte Developmental Program and Is Required for Repression of Aberrant Transcription
(A) GSEA comparing genes of selected expression clusters during oogenesis (Figure S5B) and the ranked list of gene expression changes in Hiraf/fGdf9-Cre+ MII oocytes relative to Hiraf/f oocytes. NES and FDR both were calculated in the GSEA program. For each cluster, each dot represents mean-normalized gene expression for consecutive stages of oocyte development.
(B) Numbers of total transfrags and total annotated transfrags in Hiraf/f and Hiraf/fGdf9-Cre+ MII oocytes (as computed by CuffCompare program) are shown.
(C) Numbers of total annotated transfrags, unannotated entirely intronic transfrags, and unannotated entirely intergenic transfrags in Hiraf/f and Hiraf/fGdf9-Cre+ MII oocytes (as computed by CuffCompare program) are shown. In all cases, error bars indicate SEM. Statistical analysis was carried out using two-tailed Student’s t test (∗p < 0.05). See also Figures S5 and S6.
To confirm that these observations were not an artifact of the reduced dynamic range observed in the Hira-depleted MII oocytes, we carried out a similar GSEA analysis on only median expressed genes (i.e., genes in the second, third, or fourth expression quintile). Cluster 2 (genes progressively downregulated during oogenesis) remained enriched among upregulated genes in Hiraf/f Gdf9-Cre+ oocytes (GSEA, FDR = 0.08; data not shown); similarly, cluster 4 (genes upregulated in early oogenesis) still showed enrichment (although this was less pronounced) among genes downregulated in the absence of Hira (GSEA, FDR = 0.26; data not shown). To further test the direct link between normal transcriptional activation and Hira, we carried out differential expression analysis between Hiraf/f Gdf9-Cre+ and Hiraf/f Zp3-Cre+ MII oocytes. Given the same observed reduction of the dynamic range of transcription in both systems (Figures 4G, 4J, and S6G), we hypothesized that, if normal transcriptional activation was dependent on Hira, genes that are specifically upregulated between primordial (when Gdf9-driven Cre is first expressed) and primary (when Zp3-driven Cre is first expressed) stage oocytes should show higher expression in Hiraf/f Zp3-Cre+ MII oocytes. Indeed, this hypothesis was confirmed by the GSEA analysis (Figure S6I; GSEA, FDR < 0.001).
Taken together, our data bring to light the critical role of Hira (and H3.3/H4 turnover) for affecting changes to the transcriptional state. Histone H3.3/H4 replacement is required for normal transcriptional activation of genes associated with developmental progression of oocytes, and, surprisingly, it is also necessary for genes to become or to remain transcriptionally silent.
Hira Depletion Leads to Aberrant Transcription from Regions Not Normally Transcribed within the Genome
Nucleosomes have been hypothesized to constitute a barrier for transcription (Teves et al., 2014). Considering the failure of Hira-deleted oocytes to silence genes, we asked whether the increased DNA accessibility in the absence of continuous H3.3/H4 replacement may lead to aberrant transcription from regions not normally transcribed within the genome. To address this possibility, we used a previously described approach that allows for the identification of unannotated transcripts originating entirely from intronic or intergenic regions (Trapnell et al., 2010). Consistent with our previous mapping, we observed an increase (7.3%, p value < 0.05) in the number of annotated transcripts detected in Hiraf/f Gdf9-Cre+ oocytes (Figure 5B). Remarkably, a much larger increase was observed in the total number of transcribed fragments (37.7%, p value < 0.05; Figure 5B), with the largest increases observed in unannotated transcripts originating entirely from intronic (112.6% increase, p value < 0.05) and intergenic (92.0% increase, p value < 0.05) regions (Figure 5C) of the Hira-deleted oocytes. Of note, while expression of unannotated intergenic transcripts was clearly detectable (90% transcripts with >1 fragments per kilobase of transcript per million mapped reads [FPKM]) and often at high level (8% transcripts with >10 FPKM) in individual knockout samples, expression of a given unannotated intergenic transcript was only ever observed in one biological replicate, reflecting stochastic events consistent with spurious transcription (Table S2). Also of note, a nearly identical phenotype was observed upon Zp3-Cre-driven deletion of Hira (Figures S6D, S6J, and S6K; Table S4).
Continuing Histone Replacement Is Essential for Efficient De Novo Methylation during Oogenesis
Progression through oocyte development is accompanied by the accumulation of DNA methylation. During this process, the largely hypomethylated genome of primordial oocytes acquires DNA methylation through the activity of the de novo DNA methyltransferase Dnmt3a and its co-factor Dnmt3l (Kaneda et al., 2004, Smallwood and Kelsey, 2012, Smallwood et al., 2011). Acquisition of DNA methylation in oocytes is known to be positively correlated with transcription, which could relate to the recruitment of Dnmt3a/3l to gene bodies through binding of its PWWP domain to H3K36me3, a histone modification associated with transcriptional elongation (Chotalia et al., 2009, Dhayalan et al., 2010, Kobayashi et al., 2012, Veselovska et al., 2015). Although H3K36me3-driven DNA methylation in gene bodies has recently been attributed to the enzymatic activity of Dnmt3b in mouse ESCs (Baubec et al., 2015, Yang et al., 2014), Dnmt3a is responsible for the accumulation of oocyte DNA methylation as Dnmt3b is absent in growing oocytes (Kaneda et al., 2004, Shirane et al., 2013). Given the widespread transcriptional changes observed in the Hira knockout oocytes, we set out to examine the potential impact of Hira deletion on de novo DNA methylation in the oocyte.
Although the total amount of transcription was not altered upon Hira deletion (Figures 4A and 4B), the observed reduced histone load was connected with lower levels of global H3K36me3 in Hira-depleted GV oocytes (Figures 3A and S7A). Interestingly, initial immunofluorescence (IF) detection of 5mC indicated that Hiraf/f Gdf9-Cre+ GV oocytes also contain lower levels of DNA methylation (Figure S7B). Further quantitative assessment by liquid chromatography-mass spectrometry (LC-MS) revealed a dramatic reduction of 5mC in Hira-deleted oocytes, with a more pronounced effect following earlier Gdf9-Cre+-driven deletion (Figure 6A). Importantly, the reduction of DNA methylation occurred despite the transcriptional upregulation of Dnmt3a and without statistically significant changes in the expression of other DNA methyltransferases (Figures S5C, S7C, and S7D).
Figure 6.

Continuous Histone Replacement Is Required for Efficient De Novo Methylation during Oogenesis
(A) Reduced total 5mC was measured by LC-MS in GV (Hiraf/f and Hiraf/fGdf9-Cre+) and MII (Hiraf/f and Hiraf/fZp3-Cre+) oocytes.
(B) Global levels of DNA methylation quantified by scBS-seq in the CpG (CG) and non-CpG (CHH/G) contexts are shown.
(C) Example shows CpG methylation quantified over 3-kb sliding windows (1.5-kb steps) for published GV datasets (Shirane et al., 2013; WGBS, top), Hiraf/f, and Hiraf/fGdf9-Cre+ (scBS-seq).
(D) Distribution shows the 3-kb genomic windows in the indicated bins of DNA methylation in Hiraf/f oocytes (top, horizontal, percentage indicating the proportion of methylation bins) and their corresponding DNA methylation values in Hiraf/fGdf9-Cre+ oocytes (bottom, vertical columns).
(E) Pie chart distribution shows the 3-kb genomic windows presenting statistically significant (chi-square test, p < 0.01) changes in Hiraf/fGdf9-Cre+ versus Hiraf/f (percentage indicates the proportion of each segment).
(F) Effect of Hira deletion on DNA methylation is global and independent of the genomic context. DNA methylation at CpGs (five reads coverage) was determined and averaged for each genomic context.
(G) CpG islands (CGIs) methylated in Hiraf/f oocytes are globally hypomethylated in Hiraf/fGdf9-Cre+ oocytes. CGI methylation was defined for each genotype, and only CGIs hypermethylated in Hiraf/f (>80%) are displayed, for both Hiraf/f (blue) and Hiraf/fGdf9-Cre+ (red), and ordered on the x axis based on their genomic location.
(H) Effects on DNA methylation in Hiraf/fGdf9-Cre+ oocytes are more pronounced at highly expressed genes. DNA methylation was quantified for genes binned into expression percentile based on the scRNA-seq data (boxplot with plus signs representing mean values and horizontal bars representing median values).
(I) Comparison between DNA methylation and gene expression differences in Hiraf/f and Hiraf/fGdf9-Cre+ oocytes. See also Figure S7.
To further understand the relationship among Hira deletion, transcriptional changes, and global changes in DNA methylation, we profiled DNA methylation genome-wide by bisulphite sequencing. Because of the very limited availability of Hira-deleted oocytes, we performed single-cell bisulphite sequencing (scBS-seq) on 14 individual Hiraf/f Gdf9-Cre+ MII oocytes and 14 individual Hiraf/f control MII oocytes, followed by in silico merging of the individual datasets, as previously described (Smallwood et al., 2014). We first confirmed that our scBS-seq dataset represented an accurate depiction of oocyte methylation, as we observed a high correlation (R = 0.96) between our merged scBS-seq control oocytes and an independent whole-genome bisulphite sequencing (WGBS) dataset of pooled oocytes (Shirane et al., 2013; DNA Data Bank of Japan: DRA000570; Figures 6C and S7E). In agreement with the LC-MS measurements and IF data, our WGBS analysis confirmed the dramatic reduction in DNA methylation in both CpG (42.7% decrease) and CHH (73.2% decrease) contexts (Figures 6B and S7F). Further analysis revealed that only 3.8% of the 3-kb genomic windows classed as methylated in wild-type oocytes (>80% methylation) remained methylated to the same extent in Hira-depleted oocytes, with the majority of methylated (>80%) regions showing less than 50% DNA methylation in Hira-depleted oocytes (Figure 6D). At the whole-genome level, 40.2% of 3-kb genomic windows displayed a statistically significant loss of DNA methylation in Hiraf/f Gdf9-Cre+ oocytes (Figure 6E; chi-square test, p < 0.001). This extensive reduction of DNA methylation was observed across all types of genomic regions, including genic regions, CpG islands, maternally methylated imprint control regions, and repetitive elements (Figures 6F, 6G, S7G, and S7H).
In agreement with previously published observations (Kobayashi et al., 2012, Veselovska et al., 2015), we observed a positive correlation between transcription and the level of DNA methylation in control oocytes (Figure 6H). Although the methylome of Hiraf/f Gdf9-Cre+ oocytes was severely reduced in comparison to control oocytes, they too retained a positive correlation between transcription and DNA methylation across most of the transcriptional range (Figure 6H); however, this correlation was abolished for highly expressed genes (Figure 6H). Consequently, the effect on DNA methylation in Hira-deleted oocytes was more pronounced at highly expressed genes compared to median expressed genes (Figure S7I). Surprisingly, beyond the observed relationship between transcription level and DNA methylation described above, on average, DNA methylation was reduced both at genes whose expression was unaffected by Hira deletion and also at genes differentially expressed between control and Hiraf/f Gdf9-Cre+ oocytes, regardless of whether they were up- or downregulated (Figure 6I). Cumulatively, these observations suggest that, although de novo methylation is still targeted to transcribed regions in Hira-depleted oocytes, this process appears to be inefficient, clearly indicating that transcription is not sufficient to ensure normal levels of de novo DNA methylation in this system.
Considering the high levels of Dnmt3a and Dnmt3l in Hiraf/f Gdf9-Cre+ oocytes (Figures S7C and S7D), we reasoned that the observed effect was likely caused either by inefficient recruitment or reduced enzymatic activity on chromatin of the Dnmt3a/3l complex. We first asked whether chromatin association of Dnmt3a is grossly altered following Hira depletion in Hiraf/f Gdf9-Cre+ oocytes. In agreement with our RNA-seq and qPCR data, Hira knockout oocytes contained higher levels of Dnmt3a (Figures S7C, S7D, and S5C). Importantly, Dnmt3a remained associated with chromatin following Triton pre-extraction of soluble proteins, confirming that altered chromatin structure in Hira-deleted oocytes does not completely abolish recruitment of this enzyme (Figure S7J).
In this context, the PWWP domain of Dnmt3a has previously been shown to interact in vitro with H3K36me3 (Dhayalan et al., 2010). Although the recognition of this histone modification by Dnmt3b (and not by Dnmt3a) has recently been linked to gene body DNA methylation in mouse ESCs (Baubec et al., 2015), it is possible that reduced H3K36me3 levels observed in Hira-depleted GV oocytes contribute to the observed loss of DNA methylation in this system. In addition, using in vitro biochemical assays, it has recently been shown that the enzymatic activity of Dnmt3a is specifically enhanced by binding to the N-terminal tail of histone H3 lacking methylation at lysine 4 (H3K4me0) (Guo et al., 2015). Despite the lack of H3.3/H4 replacement, growing Hiraf/f Gdf9-Cre+ oocytes remained highly transcriptionally active (Figure 4A). It is, thus, conceivable that passage of the transcriptional machinery would result in histone H3 depletion and increased DNA accessibility, both of which we observed (Figures 3A and 3C). Although our developmental system precludes direct analysis of locus-specific histone turnover due to severely limited material, a very strong correlation had been observed previously between transcription levels and the degree of histone (and specifically H3.3) replacement, a relationship that is conserved among various model organisms including mouse (Deal et al., 2010, Kraushaar et al., 2013, Mito et al., 2005, Wirbelauer et al., 2005). In agreement with this, DNA methylation was most affected at genes with the highest level of transcription (Figures 6H and S7I). We also note that, while the combination of both compromised Dnmt3a/3l recruitment and reduced enzymatic activity would explain the reduction of DNA methylation in transcribed regions, reduced histone load is likely to underpin the methylation defect observed in other non-transcribed parts of the genome (Figure 6F; Guo et al., 2015).
Discussion
To address the biological significance of continuous histone replacement in a physiological context, we have generated a genetic deletion of the histone chaperone Hira in the early stages of mouse oogenesis. Developing mouse oocytes represent a unique experimental system as postnatal oocytes undergo developmental transitions, including major transcriptional changes and widespread de novo DNA methylation, in the absence of DNA replication. Our results show that the chromatin of developing oocytes is highly dynamic, with histone turnover being observed also in the transcriptionally inert GV-stage oocytes (Figures 1C and 4A). We demonstrate that constant histone replacement is necessary for the maintenance of normal chromatin homeostasis in vivo. Depletion of Hira during early oocyte development leads to a severe reduction of histone load, compromised developmental progression, and progressive oocyte loss (Figures 2, 3A, and S2G). We note that, albeit pronounced, our observed phenotype is milder in comparison to the recently reported oocyte death in H3.3 knockout mice (Tang et al., 2015). We attribute this difference to the presence of an alternative ATRX/DAXX H3.3 chaperone complex, which, although present in our system, cannot functionally replace Hira-driven H3.3 deposition (Figures S2A–S2C).
Our experiments document that, while lacking the normal ability to incorporate H3.3 and H4, Hira-deleted growing oocytes remain transcriptionally active, which results in chromatin with severely reduced histone content, increased DNase I sensitivity, and signs of DNA damage (Figures 3 and S2B). Reminiscent of the phenotype associated with Hira depletion in yeast (Blackwell et al., 2004, Greenall et al., 2006) and H3.3 depletion in mice (Bush et al., 2013, Lin et al., 2013) and Drosophila (Sakai et al., 2009), the compromised chromatin structure leads to chromosome segregation defects and aberrant first polar body extrusion (Figure 2D). However, the observed chromosome segregation defects in the Hira-depleted oocytes are not linked with aberrant CenpA incorporation, as previously suggested in somatic cells (Figure S2C; Bush et al., 2013).
Non-replicative Hira-depleted oocytes uniquely reveal the importance of histone replacement on transcriptional regulation in the absence of replication-coupled chromatin assembly. Although transcription can continue from histone-depleted chromatin, our study shows that the lack of histone replacement has a major impact on the dynamic range of gene expression. In the context of histone-depleted chromatin, genes can neither be efficiently silenced nor effectively activated. Additionally, the lack of normal histone occupancy leads to increased spurious transcription from otherwise not-transcribed regions of the genome, suggesting an evolutionarily conserved role for the Hira histone chaperone complex (Anderson et al., 2009).
Our results additionally reveal an unexpected connection among continuous H3.3 replacement, transcription, and de novo DNA methylation in developing oocytes. Following Hira depletion, more accessible chromatin with reduced histone load leads to significantly reduced DNA methylation (Figures 6A–6G). We note that, although in other systems DNA methylation changes result in pronounced transcriptional outcomes (Yang et al., 2014), reduced DNA methylation is not likely to contribute to the observed transcriptional changes in Hira-depleted oocytes, as Dnmt3l knockout oocytes lacking DNA methylation do not display any transcriptional phenotype (Kobayashi et al., 2012).
Our data suggest that the observed methylation phenotype cannot be attributed to downregulation of the Dnmt3a/3l complex, and, although we cannot exclude locus-specific effects on Dnmt3a/3l recruitment, the observed reduced DNA methylation is likely due to altered enzymatic activity of Dnmt3a in the absence of normal levels of chromatin-bound H3 (Figures 1C, 1E, and S2B). In this context, the N-terminal part of H3 has been shown in vitro to be required for allosteric activation of Dnmt3a catalytic activity (Guo et al., 2015). Our study thus provides the first support for this effect in an in vivo setting. The observed effect is more pronounced in highly expressed genes (Figure S7I), in agreement with the expected role of transcription in inducing nucleosome depletion in Hira-depleted oocytes. Furthermore, CpG island methylation is greatly reduced in Hira-depleted oocytes (Figure 6G) and methylation loss is more pronounced at regions with high CpG density (Figure S7K). As the Dnmt3a enzyme has been shown to operate in a non-processive manner (Dhayalan et al., 2010), stimulatory effect of the unmodified (H3K4me0) H3 tail might be necessary to ensure a high level of methylation at these regions.
We also considered that loss of Hira early during oocyte development (Gdf9-Cre-driven deletion) might lead to a variety of secondary effects that could contribute to the observed phenotypes. In this context it is important to note that deletion of Hira at a later time point during oogenesis (Zp3-Cre system) fully recapitulated the transcriptional and DNA methylation defects observed in Hiraf/f Gdf9-Cre+ oocytes (Figures 6A and S6), providing further support for our findings and indicating that the observed effects are not solely attributable to compromised development of the oocytes. Although we cannot exclude that some of the effects could be secondary to alterations in gene expression, the most parsimonious view is that the abnormal chromatin resulting from impaired H3.3/H4 deposition is responsible for the majority of the phenotypes we observed.
In addition, while it is important to consider that the observed transcriptional and methylation effects could be connected with the specific absence of the H3.3 variant upon Hira depletion in oocytes (Santenard et al., 2010), it is likely that the lack of normal chromatin structure due to the inability to continuously replace H3/H4 is the main cause underlying the described phenotype. Consistent with this view, the pronounced transcriptional phenotype observed in Hira-depleted oocytes is in stark contrast to the only limited transcriptional changes previously described in Hira knockout ESCs (Banaszynski et al., 2013, Goldberg et al., 2010). In that case, replication-dependent deposition of canonical H3 by the Caf1 complex was likely to compensate in the absence of Hira-mediated H3.3 deposition in rapidly dividing ESCs (Banaszynski et al., 2013, Goldberg et al., 2010). Indeed, H3.3 depletion does not lead to pronounced changes in nucleosome occupancy in proliferating ESCs (Banaszynski et al., 2013). In contrast, our findings document that post-replicative cells in vivo require continuous histone replacement in order to maintain an intact chromatin structure, which in turn is required for normal regulation of transcription and for de novo DNA methylation in the context of developing oocytes (Figure 7). Interestingly, the critical role of continuous histone replacement also has been demonstrated recently in post-mitotic neurons, where the lack of H3.3 incorporation had an impact on both transcription and physiological function of neurons (Maze et al., 2015, published while our manuscript was under review). Collectively, our studies thus provide new insights into the histone dynamics in vivo and highlight the importance of studying chromatin in a physiological context and in non-proliferating post-mitotic cells.
Figure 7.

Role of Hira in Transcriptional Regulation and De Novo DNA Methylation during Oogenesis
During oogenesis, normal chromatin structure and nucleosome density is maintained by Hira-mediated continuous H3.3/H4 replacement. This is required for fine-tuned transcriptional regulation and efficient de novo DNA methylation.
Experimental Procedures
Mice
Hira (Hiraf/f) mice were generated by the Wellcome Trust Sanger Institute. Oocyte-specific Hira depletion was achieved by crossing Hiraf/f mice with Gdf9-iCre or with Zp3-Cre mice, respectively. The H3.3B-EGFP knockin strain was generated in the MRC CSC transgenic facility and protamine 1-EGFP strain was generated by Dr. P. Pelczar. All animal experiments were carried out under a UK Home Office Project License in a Home Office-designated facility. Detailed information is available in the Supplemental Experimental Procedures.
Oocyte/Embryo Culture, mRNA Microinjection, IF, and H&E Staining
In vitro fertilization (IVF), in vitro maturation (IVM), parthenogenesis, mRNA microinjection, TUNEL assay, IF, and H&E staining were carried out as described previously (Hajkova et al., 2010, Nashun et al., 2010). Detailed information is available in the Supplemental Experimental Procedures.
scRNA-Seq
The scRNA-seq was performed on Hiraf/f, Hiraf/f Gdf9-Cre+, and Hiraf/f Zp3-Cre+ MII oocytes using the SMARTer Ultra Low Input RNA kit (Clontech Laboratories) with ERCC RNA spike-in mix 1 (Life Technologies). Detailed information is available in the Supplemental Experimental Procedures.
DNA Methylation Analysis
An scBS-seq described previously (Smallwood et al., 2014) was used to profile the DNA methylation landscape of Hiraf/f and Hiraf/f Gdf9-Cre+ oocytes. Ultra-sensitive LC-MS was used to determine overall 5-methylcytosine level. For detailed information, see the Supplemental Experimental Procedures.
Author Contributions
P.H. and B.N. conceived the study. B.N. performed the experiments and analyzed the data together with P.H. P.W.S.H. carried out scRNA-seq and analyzed the data with G.D. S.A.S. and S.J.C. carried out scBS-seq and S.A.S. analyzed the data with G.K. and P.W.S.H. R.A. carried out LC-MS experiments and analyzed the data. R.J.F. and V.S. provided original Hiraf/f mice and EGFP-H3.3 knockin vector. R.J.F. contributed to the data analysis and the interpretation of the experiments. E.N. conducted ESC targeting and generated EGFP-H3.3 chimeras. P.P. re-derived and provided the Protamine1-EGFP mice for this study. P.H., B.N., and P.W.S.H. wrote the manuscript with assistance from S.A.S., G.K., and R.J.F. P.W.S.H. and S.A.S. contributed equally to this work.
Acknowledgments
We are grateful to the members of the P.H. lab and to Naveenan Navaratnam for discussions and revision of the manuscript. We thank the MRC CSC microscopy facility (Dirk Dormann and Chad Whilding) for help with microscopy and imaging; Megan Woodberry, Emma Francis, Darran Hardy, and Justyna Glegola for mouse husbandry; and Heba Saadeh for bioinformatics support. We also thank the MRC transgenic facility (Zoe Webster) for help regarding microinjection. Work in the P.H. lab is supported by the MRC funding to P.H. (MC_US_A652_5PY70) and by FP7 EpiGeneSys network. G.K.’s lab is supported by grants from the MRC and Biotechnology and Biological Sciences Research Council (BBSRC). P.H. is a member of EMBO Young Investigator Programme. B.N. is a recipient of a Marie Curie International Incoming Fellowship (FP7).
Published: November 5, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2015.10.010.
Accession Numbers
The accession numbers for the sequencing reported in this paper are GEO GSE66931, GSE73382, and GSE66629.
Supplemental Information
The log fold change (LogFC), p value (PValue), and false discovery rate (FDR) were computed by EdgeR software; the FPKM values for each gene for each sample were computed using HTSeq and custom R script.
The transcript ID, transcript class, and FPKM for each transcript for each sample were computed using CuffCompare software.
The log fold change (LogFC), p value (PValue), and false discovery rate (FDR) were computed by EdgeR software; the FPKM values for each gene for each sample were computed using HTSeq and custom R script.
The transcript ID, transcript class, and FPKM for each transcript for each sample were computed using CuffCompare software.
References
- Adam S., Polo S.E., Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Akiyama T., Suzuki O., Matsuda J., Aoki F. Dynamic replacement of histone H3 variants reprograms epigenetic marks in early mouse embryos. PLoS Genet. 2011;7:e1002279. doi: 10.1371/journal.pgen.1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson H.E., Wardle J., Korkut S.V., Murton H.E., López-Maury L., Bähler J., Whitehall S.K. The fission yeast HIRA histone chaperone is required for promoter silencing and the suppression of cryptic antisense transcripts. Mol. Cell. Biol. 2009;29:5158–5167. doi: 10.1128/MCB.00698-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski L.A., Wen D., Dewell S., Whitcomb S.J., Lin M., Diaz N., Elsässer S.J., Chapgier A., Goldberg A.D., Canaani E. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–120. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T., Colombo D.F., Wirbelauer C., Schmidt J., Burger L., Krebs A.R., Akalin A., Schübeler D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- Blackwell C., Martin K.A., Greenall A., Pidoux A., Allshire R.C., Whitehall S.K. The Schizosaccharomyces pombe HIRA-like protein Hip1 is required for the periodic expression of histone genes and contributes to the function of complex centromeres. Mol. Cell. Biol. 2004;24:4309–4320. doi: 10.1128/MCB.24.10.4309-4320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess R.J., Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush K.M., Yuen B.T., Barrilleaux B.L., Riggs J.W., O’Geen H., Cotterman R.F., Knoepfler P.S. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin. 2013;6:7. doi: 10.1186/1756-8935-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotalia M., Smallwood S.A., Ruf N., Dawson C., Lucifero D., Frontera M., James K., Dean W., Kelsey G. Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev. 2009;23:105–117. doi: 10.1101/gad.495809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Fuente R. Chromatin modifications in the germinal vesicle (GV) of mammalian oocytes. Dev. Biol. 2006;292:1–12. doi: 10.1016/j.ydbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Deal R.B., Henikoff J.G., Henikoff S. Genome-wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science. 2010;328:1161–1164. doi: 10.1126/science.1186777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R.Z., Ragozin S., Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010;285:26114–26120. doi: 10.1074/jbc.M109.089433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenall A., Williams E.S., Martin K.A., Palmer J.M., Gray J., Liu C., Whitehall S.K. Hip3 interacts with the HIRA proteins Hip1 and Slm9 and is required for transcriptional silencing and accurate chromosome segregation. J. Biol. Chem. 2006;281:8732–8739. doi: 10.1074/jbc.M512170200. [DOI] [PubMed] [Google Scholar]
- Guo X., Wang L., Li J., Ding Z., Xiao J., Yin X., He S., Shi P., Dong L., Li G. Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature. 2015;517:640–644. doi: 10.1038/nature13899. [DOI] [PubMed] [Google Scholar]
- Gurard-Levin Z.A., Quivy J.P., Almouzni G. Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu. Rev. Biochem. 2014;83:487–517. doi: 10.1146/annurev-biochem-060713-035536. [DOI] [PubMed] [Google Scholar]
- Hajkova P., Jeffries S.J., Lee C., Miller N., Jackson S.P., Surani M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hödl M., Basler K. Transcription in the absence of histone H3.3. Curr. Biol. 2009;19:1221–1226. doi: 10.1016/j.cub.2009.05.048. [DOI] [PubMed] [Google Scholar]
- Hoek M., Stillman B. Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc. Natl. Acad. Sci. USA. 2003;100:12183–12188. doi: 10.1073/pnas.1635158100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A., Zhang Y. Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nat. Struct. Mol. Biol. 2014;21:609–616. doi: 10.1038/nsmb.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., Sasaki H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- Kobayashi H., Sakurai T., Imai M., Takahashi N., Fukuda A., Yayoi O., Sato S., Nakabayashi K., Hata K., Sotomaru Y. Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 2012;8:e1002440. doi: 10.1371/journal.pgen.1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar D.C., Jin W., Maunakea A., Abraham B., Ha M., Zhao K. Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol. 2013;14:R121. doi: 10.1186/gb-2013-14-10-r121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Z.J., Xu X., Cooney A.J. Differential oocyte-specific expression of Cre recombinase activity in GDF-9-iCre, Zp3cre, and Msx2Cre transgenic mice. Biol. Reprod. 2004;71:1469–1474. doi: 10.1095/biolreprod.104.031757. [DOI] [PubMed] [Google Scholar]
- Li R., Albertini D.F. The road to maturation: somatic cell interaction and self-organization of the mammalian oocyte. Nat. Rev. Mol. Cell Biol. 2013;14:141–152. doi: 10.1038/nrm3531. [DOI] [PubMed] [Google Scholar]
- Lin C.J., Conti M., Ramalho-Santos M. Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development. 2013;140:3624–3634. doi: 10.1242/dev.095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.J., Koh F.M., Wong P., Conti M., Ramalho-Santos M. Hira-mediated H3.3 incorporation is required for DNA replication and ribosomal RNA transcription in the mouse zygote. Dev. Cell. 2014;30:268–279. doi: 10.1016/j.devcel.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman M., Hanawalt P.C. Efficient protection against oxidative DNA damage in chromatin. Mol. Carcinog. 1992;5:264–269. doi: 10.1002/mc.2940050406. [DOI] [PubMed] [Google Scholar]
- Loppin B., Bonnefoy E., Anselme C., Laurençon A., Karr T.L., Couble P. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005;437:1386–1390. doi: 10.1038/nature04059. [DOI] [PubMed] [Google Scholar]
- Maze I., Noh K.M., Soshnev A.A., Allis C.D. Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014;15:259–271. doi: 10.1038/nrg3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I., Wenderski W., Noh K.M., Bagot R.C., Tzavaras N., Purushothaman I., Elsässer S.J., Guo Y., Ionete C., Hurd Y.L. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron. 2015;87:77–94. doi: 10.1016/j.neuron.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito Y., Henikoff J.G., Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat. Genet. 2005;37:1090–1097. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- Nashun B., Yukawa M., Liu H., Akiyama T., Aoki F. Changes in the nuclear deposition of histone H2A variants during pre-implantation development in mice. Development. 2010;137:3785–3794. doi: 10.1242/dev.051805. [DOI] [PubMed] [Google Scholar]
- Pan H., O’brien M.J., Wigglesworth K., Eppig J.J., Schultz R.M. Transcript profiling during mouse oocyte development and the effect of gonadotropin priming and development in vitro. Dev. Biol. 2005;286:493–506. doi: 10.1016/j.ydbio.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Pchelintsev N.A., McBryan T., Rai T.S., van Tuyn J., Ray-Gallet D., Almouzni G., Adams P.D. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013;3:1012–1019. doi: 10.1016/j.celrep.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo S.E., Roche D., Almouzni G. New histone incorporation marks sites of UV repair in human cells. Cell. 2006;127:481–493. doi: 10.1016/j.cell.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Ransom M., Dennehey B.K., Tyler J.K. Chaperoning histones during DNA replication and repair. Cell. 2010;140:183–195. doi: 10.1016/j.cell.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Gallet D., Quivy J.P., Scamps C., Martini E.M.D., Lipinski M., Almouzni G. HIRA is critical for a nucleosome assembly pathway independent of DNA synthesis. Mol. Cell. 2002;9:1091–1100. doi: 10.1016/s1097-2765(02)00526-9. [DOI] [PubMed] [Google Scholar]
- Sakai A., Schwartz B.E., Goldstein S., Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr. Biol. 2009;19:1816–1820. doi: 10.1016/j.cub.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santenard A., Ziegler-Birling C., Koch M., Tora L., Bannister A.J., Torres-Padilla M.E. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat. Cell Biol. 2010;12:853–862. doi: 10.1038/ncb2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirane K., Toh H., Kobayashi H., Miura F., Chiba H., Ito T., Kono T., Sasaki H. Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 2013;9:e1003439. doi: 10.1371/journal.pgen.1003439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood S.A., Kelsey G. De novo DNA methylation: a germ cell perspective. Trends Genet. 2012;28:33–42. doi: 10.1016/j.tig.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Smallwood S.A., Tomizawa S., Krueger F., Ruf N., Carli N., Segonds-Pichon A., Sato S., Hata K., Andrews S.R., Kelsey G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat. Genet. 2011;43:811–814. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood S.A., Lee H.J., Angermueller C., Krueger F., Saadeh H., Peat J., Andrews S.R., Stegle O., Reik W., Kelsey G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods. 2014;11:817–820. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagami H., Ray-Gallet D., Almouzni G., Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- Tang M.C., Jacobs S.A., Mattiske D.M., Soh Y.M., Graham A.N., Tran A., Lim S.L., Hudson D.F., Kalitsis P., O’Bryan M.K. Contribution of the two genes encoding histone variant h3.3 to viability and fertility in mice. PLoS Genet. 2015;11:e1004964. doi: 10.1371/journal.pgen.1004964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves S.S., Weber C.M., Henikoff S. Transcribing through the nucleosome. Trends Biochem. Sci. 2014;39:577–586. doi: 10.1016/j.tibs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Tomizawa S., Nowacka-Woszuk J., Kelsey G. DNA methylation establishment during oocyte growth: mechanisms and significance. Int. J. Dev. Biol. 2012;56:867–875. doi: 10.1387/ijdb.120152gk. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselovska L., Smallwood S.A., Saadeh H., Stewart K.R., Krueger F., Maupetit-Méhouas S., Arnaud P., Tomizawa S., Andrews S., Kelsey G. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol. 2015;16:209. doi: 10.1186/s13059-015-0769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirbelauer C., Bell O., Schübeler D. Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 2005;19:1761–1766. doi: 10.1101/gad.347705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick J.J., Holstege F.C., Jennings E.G., Causton H.C., Shore D., Grunstein M., Lander E.S., Young R.A. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature. 1999;402:418–421. doi: 10.1038/46567. [DOI] [PubMed] [Google Scholar]
- Yang X., Han H., De Carvalho D.D., Lay F.D., Jones P.A., Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The log fold change (LogFC), p value (PValue), and false discovery rate (FDR) were computed by EdgeR software; the FPKM values for each gene for each sample were computed using HTSeq and custom R script.
The transcript ID, transcript class, and FPKM for each transcript for each sample were computed using CuffCompare software.
The log fold change (LogFC), p value (PValue), and false discovery rate (FDR) were computed by EdgeR software; the FPKM values for each gene for each sample were computed using HTSeq and custom R script.
The transcript ID, transcript class, and FPKM for each transcript for each sample were computed using CuffCompare software.