

Abstract
Surface initiated atom transfer radical polymerization (ATRP) of substituted styrenes leads to rapid synthesis of uniform and thick substituted polystyrene brushes (>100 nm in 1 hour) from gold surface. High growth rates were observed for styrenes substituted with electron withdrawing groups in meta/para positions. The effects seen in surface and solution polymerizations are similar for styrenes with electron withdrawing groups, and for electron donors in ortho and para positions. However, electron donors at meta sites have surprisingly fast growth rates, which may be due to steric inhibition of termination. The overall surface polymerization rates for substituted styrenes was analyzed and found to follow the Hammett relation with ρ = 0.51. The ratio of kp to kt, is as an indicator of the likelihood that a reaction will reach high degrees of polymerization before termination.
Keywords: ATRP, surface polymerization, substituent effect and Hammett relation
Graphical Abstract

Introduction
The development of controlled radical polymerizations such as Atom Transfer Radical Polymerization (ATRP),1,2 Reversible-Addition-Fragmentation Transfer Polymerization (RAFT)3 and Nitroxide-Mediated Polymerization (NMP),4,5 provide powerful methods for the growth of polymer brushes from surfaces. These controlled polymerization methods limit radical concentrations during polymerization, minimize bimolecular termination reactions and provide control over Mn and the polydispersity (PDI). However, compared to traditional radical polymerization, control comes at the expense of a substantial reduction in the polymerization rate. Rapid growth of brushes could expand the scope and applications for polymer brushes by greatly reducing the time required for their synthesis.
Previously, we reported the remarkably rapid growth of well-defined poly(tert-butyl acrylate) brushes under mild conditions (50 °C) using ATRP. The key aspect of the polymerization was the use of a highly active ATRP catalyst[Cu(I)1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Cu(I)-Me4Cyclam)] which provided 100 nm thick poly(tert-butyl acrylate) (PtBA) brushes on flat Au surfaces in just 5 minutes.6 Such polymerization rates are several orders of magnitude greater than typical controlled polymerizations from surfaces. The Cu(I)-Me4Cyclam) system also enabled rapid growth of thick brush layers from other types of vinyl monomers such as styrene, vinyl pyridine and methacrylates.6
The unusually rapid growth of PtBA films compared to other vinyl monomers can be rationalized as a combination of tBA’s fast propagation rate and reduced bimolecular coupling due to the steric bulk of the monomer. As shown in Scheme 1, sterically demanding monomers show a high propensity towards head to tail placement during polymer growth, and the same steric interactions would be expected to restrict termination by coupling (analogous to forming a head to head linkage). The generality of this phenomena can be seen in kp (propagation rate constant) and kt (termination rate constant) values for the methacrylate monomers.7,8 There are two important trends in the methacrylate data; kt decreases as the size of the ester moiety increases because of the steric crowding depicted in Scheme 1, but a more surprising result is that in some cases, kp also increases with the size of the ester group. Gilbert suggested that the increases in kp may be due to the increased momentum associated with collisions between higher molecular weight species.9 Monomers with both high kp and low kt values, i.e. a high kp/kt ratio, should be the prime candidates for rapid polymerizations from surfaces.
Scheme 1.

Steric constraints favor head to tail addition and low kt values.
The polymerization results for PtBA prompted us to examine other bulky monomers to see if they also polymerized rapidly and provide thick films. Thick polystyrene brushes are difficult to grow via surface initiated ATRP (eg. 40 nm thick films in 1 hour compared to 100 and 200 nm films in 1 hour for MMA or tBA, respectively6), consistent with its small kp/kt ratio (4.3 at 60 °C)7, though it is important to have thick polystyrene brushes for hydrophobic coating applications.10 We studied substituted styrene derivatives (Chart 1) to evaluate the effects of steric congestion and electronic effects on the polymerization rate under the same conditions used for tBA system. These monomers efficiently polymerize via free radical polymerization in solution,11–14 but as per our knowledge, there are no reports of their polymerization via surface-initiated ATRP or other controlled radical processes.
Chart 1.

Substituted styrene monomers polymerized in the study
Based on previously reported data from other types of radical polymerizations in solution,15–17 we expect that substituents on the aromatic ring of styrene may affect acceleration of polymerization rate, and enable fast growth of substituted polystyrene brushes from surfaces. Furthermore, we expected that such a study would provide a better understanding of the mechanism of surface initiated polymerizations (especially the correlation between surface polymerization rate and monomer structure) as there has been no systematic investigation of substituent effects in substituted styrenes in surface initiated polymerizations. Herein we report the rapid surface initiated polymerization of substituted styrene and the effect of various substituents on the polymerization kinetics. The data are discussed with regard to the Hammett equation:
Experimental Section
Materials
Unless otherwise noted, all chemicals were obtained from Aldrich and stored under nitrogen. 11-Mercapto-1-undecanol (MUD, 97%), 2-bromopropionyl bromide (2-BPB) (97%), anisole (99.7%), N,N-dimethylformamide (DMF, 99.8%), Cu(I)Br (99.999%), Cu(II)Br2 (99.999%), Me4Cyclam (99%), and 4,4′-dinonyl-2,2′-bipyridine (dnNbpy, 97%) were used as received. Triethylamine was vacuum-distilled from calcium hydride prior to use. tert-Butyl acrylate (tBA) (98%), MMA (99%), styrene (99%), 2-vinylanisole (95%), 4-bromostyrene, pentafluorostyrene, 4-methylstyrene and 4-tert-butyl styrene were passed through a 10 × 0.5 cm column of activated basic alumina followed by distillation to remove inhibitors. 3,5-Bis(trifluoromethyl) styrene (97%) and 3,5-dimethylstyrene were purified by the same method, but not distilled. After purification, the monomers and solvents were transferred to Schlenk flasks, degassed using three freeze-pump-thaw cycles, and then transferred into a drybox. 3,5-di-tert-butyl styrene, 3,5-dimethoxystyrene, 2,6-dimethoxystyrene, 4-vinylanisole and 4-(trifluoromethyl)styrene were prepared as previously reported.11–14,18
Characterization methods
Film thicknesses were measured using a rotating analyzer ellipsometer (model M-44, J. A. Woollam) at an incident angle of 75°. The data were analyzed using WVASE32 software, and thickness and refractive index determinations were performed on at least three spots on each substrate. The refractive index of the films was assumed to be 1.5 and then fitted with the film thickness. Reflectance Fourier Transform Infrared (reflectance FTIR) spectra were obtained from a Nicolet Magna-560 FTIR spectrometer with a MCT detector and a PIKE grazing angle (80°) attachment. Typically, 128 scans were collected for each spectrum.
Preparation of immobilized Initiator on gold substrates
Gold coated Si wafers, cleaned for 30 min in UV/O3, were washed with water and ethanol, and then transferred into a glove bag purged with N2. The wafers were immersed in a 1 mM ethanolic solution of MUD for 24 h to form a self-assembled initiator monolayer, and then the films were washed with ethanol and dried under a stream of N2. The ellipsometric thickness of the MUD layer was 10–15 Å. MUD-coated substrates were transferred to a drybox filled with N2 and were dipped in a 10 mL solution of 0.12 M triethylamine in anhydrous THF at 0°C. After 1 min, 10 mL of a solution of 2-BPB in anhydrous THF (0.1 M) was added dropwise to the solution to form the immobilized initiator layer. The reaction time was limited to 2–3 min, since thiol-terminated SAMs could be unstable in the presence of the acyl bromide. After rinsing with THF, the Au substrates were removed from the drybox, and then rinsed sequentially with ethyl acetate, ethanol and deionized water (Milli-Q) and dried under a stream of N2.
Surface-initiated polymerization of substituted styrenes
Note: For best results, solvents, initiators and monomers must be scrupulously purified and deoxygenated. In a N2-filled drybox, CuBr (5.7 mg, 0.04 mmol) CuBr2, (4.5 mg, 0.02 mmol), Me4Cyclam (10.3 mg, 0.04 mmol) and dnNbpy (16.4 mg, 0.04 mmol) were added to a round-bottom flask containing 20 mL of a degassed solution of monomer in DMF/anisole (DMF/anisole ~1:1 (v:v), [monomer] ~ 4 M). The mixture was well-stirred and heated with an oil bath to 50 °C until a transparent, light green solution formed. The prepared solution was then transferred into a small vial containing an initiator-modified Au substrate to start the surface-initiated polymerization. After a predetermined reaction time at 55 °C, the substrate was removed from the vial, washed with ethyl acetate and THF sequentially, and then dried under a flow of N2 in the drybox. The same conditions were used for the polymerization of other monomers.
Results and Discussion
Synthesis of Substituted Polystyrene Brushes
Scheme 2 outlines the synthesis of substituted styrene polymer brushes grown from gold substrates. The experimental procedure is similar to that described by Bao et al. for the rapid growth of tert-butyl acrylate from gold surfaces.6 A self-assembled monolayer was prepared on the gold surface using MUD and converted into an initiator monolayer by reacting its terminal hydroxy group with 2-bromopropionyl bromide. Initiator immobilization was apparent from the appearance of a carbonyl peak at 1743 cm−1 in the reflectance FTIR spectrum (spectrum a, Figure S1). Substrates were transferred into a dry box, and the polymerizations were performed inside the drybox to avoid contamination from oxygen. The catalyst system was a mixture of Cu(I)Br/Me4Cyclam and Cu(II)Br2/(dnNbpy)2 in 1:1 (v/v) solutions of DMF and anisole. The Cu(II) complex ensures deactivation of active radicals and provided some control over the rapid polymerization. Polymerizations of substituted styrenes were run at 55 °C for 8 hours under identical conditions, then the substrates were sonicated and washed with ethyl acetate and THF. The IR reflectance spectra of the dried films (Figures S1 and S2) confirmed formation of polymer brushes by the appearance of characteristic peaks that correspond to the substituents on the styrene ring.
Scheme 2.

Surface initiated polymerization of styrene derivatives
Figures 1 and 2 show the evolution of film thickness with time for the polymerization of substituted styrenes. Figure 1 shows surface polymerization data for ortho and meta substituted monomers while Figure 2 shows comparable data for para-substituted styrenes. The polymerization rates of 3,5-disubstituted styrenic monomers at 55 °C were noticeably high resulting 100–350 nm thick layers in 4 hours.19 In the early stages of polymerization, the nonlinear relationship between the film thickness and time suggests a relatively high concentration of radicals leading to some termination as well as a high polymerization rate. However, after >2 hours, growth in film thickness with time was roughly linear, indicating that some level of control was established during the polymerization. Polymerizations of ortho-substituted monomers were sluggish, likely due to steric effects, and none reached 100 nm thick films in 8 hours.
Figure 1.

Evolution of the ellipsometric brush thickness with time for the polymerization of substituted styrenes from gold substrates. 3,5-bis-(trifluoromethyl)styrene (■), 3,5-dimethoxystyrene (◆), 3,5-dimethylstyrene (△), 3,5-di-tert-butylstyrene (empty circle), styrene (◇), 2-(trifluoromethyl)styrene (□),2-methoxystyrene (filled circle),2,6-dimethoxystyrene (▲).
Figure 2.

Evolution of the ellipsometric brush thickness with time for the polymerization of 4-substituted styrenes from gold substrates. 4-(trifluoromethyl)styrene (■), 4-bromostyrene (□), styrene (◇), 4-tert-butylstyrene (◆),4-methylstyrene (△), 4-methoxystyrene (▲).
The IR spectra confirm the growth of polymers from the Au surfaces. Furthermore, the spectral intensities agree well with the kinetic data. In Figure S1, spectrum c shows the four peaks (overtones and combinations) from 2000-1700 cm−1 characteristic of mono-substituted benzene rings (styrene). Strong methyl C-H stretching bands at 3000-2850 cm−1 and the characteristic doublet for tert-butyl groups, a strong peak at 1370 cm−1 and a weaker signal at 1390 cm−1 confirms the growth of 3,5-di-tert-butylstyrene in spectrum b. The strong, broad, asymmetric (1280-1220 cm−1) and symmetric (1050-1000 cm−1) stretching modes for aryl ethers confirm the attachment of 2-methoxy and 2,6-dimethoxystyrene(spectrum d and e)and in spectrum f, a sharp strong peak at 1350 cm−1 (C-F stretching)identifies the CF3 group of poly(3,5-bis-(trifluoromethyl) styrene).
In Figure S2, strong peaks between 1280-1220 cm−1 in spectrum b (C-O-C stretching of an alkyl aryl ether) confirm the growth of poly(4-methoxystyrene). A sharp strong peak at 1350 cm−1 (C-F stretching) identifies the CF3 group in poly(4-trifluoromethyl styrene) (spectrum c). The spectrum of poly(4-methyl styrene) (d) is similar to that of poly(styrene) (a), except for the 2 peaks between 2000-1700 cm−1 (overtones and combinations) consistent with a 1,4-disubstituted benzene ring. The strong, narrow peak seen at 1000 cm−1 in spectrum e is characteristic of aromatic C-Br stretching as expected for poly(4-bromostyrene). In spectrum f, the characteristic doublet for tert-butyl groups, a strong peak at 1370 cm−1 and a weaker signal at 1390 cm−1 confirms the growth of poly(p-tert-butylstyrene)
Substituent effects on polymerization rate
The data in Figures 1 and 2 show that monomers with electron withdrawing groups polymerize faster than monomers with electron donating groups, irrespective of the substitution pattern on the aromatic ring, excluding ortho-substituted monomers where steric effects restrict propagation and termination. We also failed to grow α-methyl styrene from gold surfaces (data not shown) due to similar steric reasons. Monomers with bulky electron-donating groups (tert-butyl) at meta or para positions polymerized faster than styrene and ortho-substituted monomers. Surprisingly, monomers with electron donating groups at meta sites polymerized faster than styrene or their ortho/para substituted analogues, irrespective of their steric demand. The observed film growth ratesvaried in the order of m-di-CF3, p-CF3>m-dimethoxy>m-dimethyl, m-di-tert-butyl >p-tert-butyl >p-Br > H >p-methyl >o-methoxy>o-dimethoxy, and p-methoxy. The results are consistent with the solution polymerization data obtained from conventional radical polymerization as well as controlled radical polymerizations such as ATRP17 and radical polymerization initiated by TEMPO/BPO.15,16
To analyze the rate data, we constructed a Hammett plot for the para-substituted monomers, similar to the method used to analyze solution polymerizations.17,20 The Hammett analysis is limited to para-substituted styrenes because Hammett constants (σ) are known only for para-substituted styrenes. The propagation and termination rate constants for surface polymerization can be estimated from a model proposed by the Wirth group.21
The initiation and propagation steps of living radical polymerization have been well documented and adopted for ATRP systems as shown in Scheme 3.
Scheme 3.

The initiation, propagation and termination steps of ATRP
According to this mechanism, the disappearance of monomer only occurs during propagation (step 2 of Scheme 3).
| (1) |
If [R·] is constant, the monomer concentration can be reduced to first-order kinetics.
| (2) |
However, [R·] usually is not a constant due to termination, especially in case of surface polymerization which is clearly visible in the data of Figures 1 and 2. According to elegant Wirth’s model, termination happened only by coupling for surface-initiated ATRP as concentration of Cu(II) does not significantly change for a surface with tiny amount of attached initiators and also ignoring possible contributions from surface fouling or side reactions such as radical transfer to solvent, the rate of termination can be expressed as:
| (3) |
| (4) |
Recognizing that monomer-to-initiator ratio is so high that monomer concentration changes are negligible simplifies the solution of Equation 2.
| (5) |
Substituting Equation 4 into Equation 5 reduces conversion to a simple nonlinear time dependence:
| (6) |
Realizing that the ellipsometric thickness is proportional to [M]0 − [M], i.e. no polymerization in solution, the data of Figure 2 were fit to Equation 6. The fits to the data, as shown for 4-bromostyrene (Figure 3), are reasonable and allow extraction of the apparent rate constants for propagation (kpapp = [M]0kp [R·]0) and termination (ktapp = [R·]0kt). The data obtained for different para-substituted styrenes are tabulated in Table 1.
Figure 3.
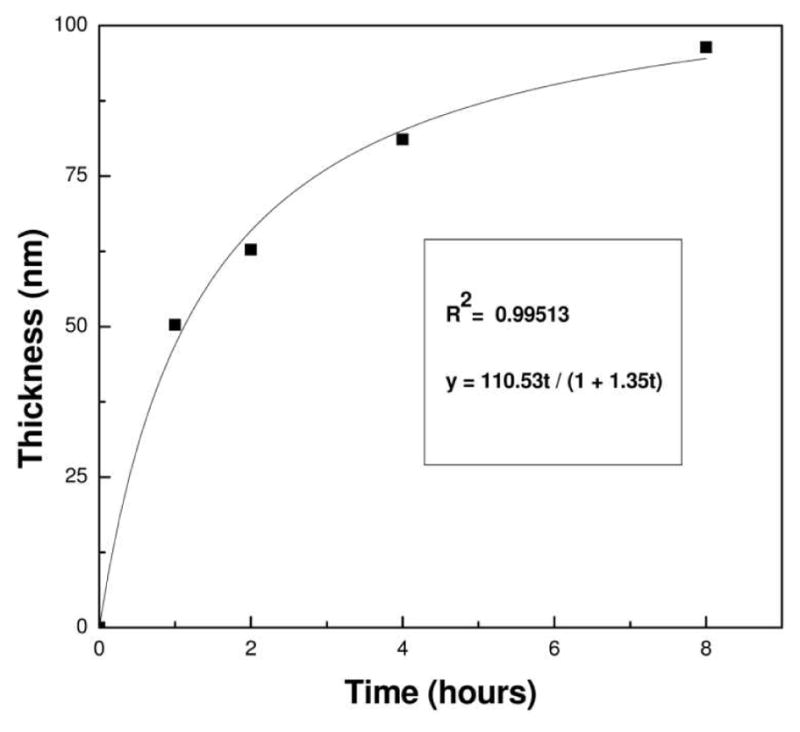
A representative example of the polymerization data for 4-bromostyrene fit to Equation 6. The polymer was grown from Au substrates at 55 °C.
Table 1.
Apparent Rate Coefficients in surface ATRP obtained from model and Absolute Propagation Rate Constants of Substituted Styrenes7
| Substituent | σ25 | kpapp (mol−1 Ls−1) | ktapp (s−1) | kpabs(mol−1 Ls−1) |
|---|---|---|---|---|
|
| ||||
| 4-CN | 0.66 | - | - | 219 |
| 4-CF3 | 0.54 | 649 | 7.23 | - |
| 4-Br | 0.23 | 111 | 1.35 | 186 |
| 4-Cl | 0.23 | - | - | 150 |
| 4-H | 0.00 | 106 | 1.82 | 110 |
| 4-Me | −0.17 | 76 | 2.03 | 84 |
| 4-CMe3 | −0.20 | 288 | 6.81 | - |
| 4-OMe | −0.27 | 9.0 | 0.59 | 71 |
Using the rate constants from fitting the data to Equation 6, we constructed a Hammett plot (Figure 4) relating the ratio of the apparent rate coefficients (kp app/kt app) (similar to analyzing product ratio, as applied by several authors to obtain Hammett plots)22–24 and the Hammett constant, σ, for different para substituents. By using the ratio of the rate constants, we eliminate the radical concentration [R·]0, and in principle, kp app/kt app should reflect the ratio of absolute rate coefficients. Thus,
Figure 4.

Hammett plot of (kp/kt) for surface-initiated ATRP of substituted styrenes (◆) and absolute kp values from conventional polymerizations (□) of substituted styrenes.7
where X and H refer to the substituted styrene and styrene respectively. For conventional radical polymerization of para-substituted styrenes, there is a linear correlation between log(kp app/kt app) and the Hammett constants σ for different substituents. The value of ρ for our surface ATRP data (ρ = 0.51) is similar to that of the conventional radical systems run in solution (ρ = 0.55, Figure 4),26 but much smaller than the ρ value for the solution ATRP (ρ = 1.5)17 reported by Matyjaszewski. The difference between the solution and surface ATRP results is the use of different ordinates. In solution ATRP, the correlation was established with regard to the apparent propagation rate constant which comprises [R·], rather than the ratio of the rate coefficients used for surface ATRP. Thus, the ρ values for surface ATRP and conventional (solution) radical polymerizations, which also use absolute rate coefficients as ordinate, are similar.
Based on the qualitative success of the simple Hammett equation similar to the solution polymerization via free radical pathway,20 we conclude that these reaction constants are roughly correlated with the Hammett parameters of the para substituent in surface polymerization by following the Hammett relation with ρ = 0.51. Such substituent effects are mainly due to an increase in kp/kt for monomers substituted with electron-withdrawing substituents in para position (Table 1). However, it does not exclude the possibility of a larger Keq (in addition to a larger kp) for electron withdrawing monomers due to the decrease in the bond dissociation energy (BDE) of the C-X bond by electron-withdrawing substituents and hence causes the additional enhancement of apparent polymerization rate coefficient with σ (similar to the solution polymerization).17 Of the monomers studied, only 4-methoxystyrene failed to grow a significant surface film. Similar observations was made by Matyzasjewski et al.17 in their solution ATRP of 4-methoxystyrene, where they failed to obtain high molecular weight polymer. They suggested that the active growing species has a cationic nature due to the presence of strong electron donating substituent. For this reason, we excluded 4-methoxystyrene from the Hammett plot.
Conclusion
Surface initiated atom transfer radical polymerization of substituted styrenes provides uniform and thick polymer brushes (>100 nm in 1 hour) from gold surfaces. Styrene substituted with electron withdrawing groups in meta and para positions exhibited high growth rates. The substituent effects seen in surface and solution polymerizations are similar for styrene with electron withdrawing groups, and for electron donors in ortho and para positions, but donors at meta sites have surprisingly fast growth rates, possible due to steric inhibition of termination. We also showed that the surface polymerization rate varies with different substituents follow the Hammett relation with ρ = 0.51. The ratio of kp to kt, acts as an indicator of the likelihood that a reaction will reach high degrees of polymerization before termination.
Supplementary Material
Highlights.
Suitably substituted styrenes can lead to rapid synthesis of uniform and thick polystyrene brushes (>100 nm in 1 hour) from gold surface via surface initiated ATRP.
High growth rates were observed for styrenes substituted with electron withdrawing groups in meta/para positions.
Electron donors at meta sites have surprisingly fast growth rates, which may be due to steric inhibition of termination.
Surface polymerization rates for substituted styrenes followed the Hammett relation with ρ = 0.51, similar to conventional radical polymerization in solution.
Acknowledgments
We are grateful to the U.S. National Institutes of Health(GM080511) for funding this work.
Footnotes
Associated content
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.matyjaszewski k, xia jh. atom transfer radical polymerization. chem rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 2.kamigaito m, ando t, sawamoto m. metal-catalyzed living radical polymerization. chem rev. 2001;101:3689–3745. doi: 10.1021/cr9901182. [DOI] [PubMed] [Google Scholar]
- 3.chiefari j, chong yk, ercole f, krstina j, jeffery j, le tpt, mayadunne rta, meijs gf, moad cl, moad g, rizzardo e, thang sh. living free-radical polymerization by reversible addition-fragmentation chain transfer: the raft process. macromolecules. 1998;31:5559–5562. [Google Scholar]
- 4.couvreur l, lefay c, belleney j, charleux b, guerret o, magnet s. first nitroxide-mediated controlled free-radical polymerization of acrylic acid. macromolecules. 2003;36:8260–8267. [Google Scholar]
- 5.grande d, guerrero r, gnanou y. cyanoxyl-mediated free-radical polymerization of acrylic acid: its scope and limitations. j polym sci pol chem. 2005;43:519–533. [Google Scholar]
- 6.bao zy, bruening ml, baker gl. rapid growth of polymer brushes from immobilized initiators. j am chem soc. 2006;128:9056–9060. doi: 10.1021/ja058743d. [DOI] [PubMed] [Google Scholar]
- 7.brandup j, immergut eh, grulke ea., editors. polymer handbook. 4. john wiley and sons; new york: 1999. [Google Scholar]
- 8.odian g. principles of polymerization. 4. john wiley & sons, inc; new york: 2004. [Google Scholar]
- 9.heuts jpa, gilbert rg, radom l. a priori prediction of propagation rate coefficients in free-radical polymerizations: propagation of ethylene. macromolecules. 1995;28:8771–8781. [Google Scholar]
- 10.smiatek j, heuer a, wagner h, studer a, hentschel c, chi l. coat thickness dependent adsorption of hydrophobic molecules at polymer brushes. the journal of chemical physics. 2013;138:044904. doi: 10.1063/1.4789305. [DOI] [PubMed] [Google Scholar]
- 11.pillow jng, halim m, lupton jm, burn pl, samuel idw. a facile iterative procedure for the preparation of dendrimers containing luminescent cores and stilbene dendrons. macromolecules. 1999;32:5985–5993. [Google Scholar]
- 12.risch n, meyerroscher b, langhals m. 3,5-di-tert-butylstyrenes - synthesis of bulky polymer building-blocks. z naturforsch(b) 1994;49:141–144. [Google Scholar]
- 13.overberger cg, burns cm. cationic graft copolymerization of polystyrene onto poly-2,6-dimethoxystyrene by termination reaction. journal of polymer science part a-1-polymer chemistry. 1969;7:333. [Google Scholar]
- 14.kawamura t, uryu t, matsuzaki k. stereoregularity of polystyrene derivatives .1. poly(methylstyrene)s and poly(methoxystyrene)s obtained with ziegler catalyst, cationic catalyst, or radical initiators. makromolekulare chemie-macromolecular chemistry and physics. 1982;183:125–141. [Google Scholar]
- 15.imoto m, kinoshit m, nishigak m. vinyl polymerization .93. polar effects in radical polymerization of p-substituted styrenes. makromolekulare chemie. 1965;86:217. [Google Scholar]
- 16.kazmaier pm, daimon k, georges mk, hamer gk. nitroxide-mediated “living” free radical polymerization, substituent effects on the free radical polymerization of styrene. abstr pap am chem soc. 1996;211:422. [Google Scholar]
- 17.qiu j, matyjaszewski k. polymerization of substituted styrenes by atom transfer radical polymerization. macromolecules. 1997;30:5643–5648. [Google Scholar]
- 18.okuma k, sakai o, shioji k. wittig reaction by using dbu as a base. bull chem soc jpn. 2003;76:1675–1676. [Google Scholar]
- 19.samadi a, husson sm, liu y, luzinov i, kilbey sm. low-temperature growth of thick polystyrene brushes via atrp. macromol rapid commun. 2005;26:1829–1834. [Google Scholar]
- 20.coote ml, davis tp. propagation kinetics of para-substituted styrenes: a test of the applicability of the hammett relationship to free-radical polymerization. macromolecules. 1999;32:4290–4298. [Google Scholar]
- 21.xiao dq, wirth mj. kinetics of surface-initiated atom transfer radical polymerization of acrylamide on silica. macromolecules. 2002;35:2919–2925. [Google Scholar]
- 22.galardon e, le maux p, simonneaux g. cyclopropanation of alkenes, n-h and s-h insertion of ethyl diazoacetate catalysed by ruthenium porphyrin complexes. tetrahedron. 2000;56:615–621. [Google Scholar]
- 23.watkins al, hashiguchi bg, landis cr. highly enantioselective hydroformylation of aryl alkenes with diazaphospholane ligands. org lett. 2008;10:4553–4556. doi: 10.1021/ol801723a. [DOI] [PubMed] [Google Scholar]
- 24.yajima t, okada k, nagano h. substituent effect on the diastereoselectivity in the chelation-controlled radical reactions of gamma-(p-substituted-benzyloxy)-alpha-methylene esters with alkyl iodides. tetrahedron. 2004;60:5683–5693. [Google Scholar]
- 25.hansch c, leo a, taft rw. a survey of hammett substituent constants and resonance and field parameters. chem rev. 1991;91:165–195. [Google Scholar]
- 26.walling c, briggs er, wolfstirn kb, mayo fr. copolymerization .10. the effect of meta-substitution and para-substitution on the reactivity of the styrene double bond. J Am Chem Soc. 1948;70:1537–1542. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.