

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a consequence of sedentary life style and high fat diets with an estimated prevalence of about 30% in western countries. It is associated with insulin resistance, obesity, glucose intolerance and drug toxicity. Additionally, polymorphisms within, e.g., APOC3, PNPLA3, NCAN, TM6SF2 and PPP1R3B, correlate with NAFLD. Several studies have already investigated later stages of the disease. This study explores the early steatosis stage of NAFLD with the aim of identifying molecular mechanisms underlying the etiology of NAFLD. We analyzed liver biopsies and serum samples from patients with high- and low-grade steatosis (also pre-disease states) employing transcriptomics, ELISA-based serum protein analyses and metabolomics. Here, we provide a detailed description of the various related datasets produced in the course of this study. These datasets may help other researchers find new clues for the etiology of NAFLD and the mechanisms underlying its progression to more severe disease states.
Subject terms: Non-alcoholic fatty liver disease, Metabolomics, Gene expression analysis, Systems biology
Background & Summary
With an estimated prevalence of about 30% in western countries, NAFLD is a major public health issue1. Sedentary life-style and excessive food consumption correlate with rate at which NAFLD cases appear. Epidemiologic studies showing a prevalence of the disease that differs between countries as well as between groups in the same country, appear to reflect an interplay of environmental and genetic factors in its etiology1. Additionally, polymorphisms in, e.g., APOC3, PNPLA3, NCAN, TM6SF2 and PPP1R3B, correlate with NAFLD2,3. Over-feeding directly induces insulin resistance4. Causality between steatosis and the metabolic syndrome of insulin resistance, obesity, and glucose intolerance, is still unresolved5. While the correlation between steatosis and insulin resistance is established there is debate about the relationship between steatosis and hepatic insulin resistance6. Samuel et al. showed that activated PKC-ϵ and JNK can induce insulin resistance via impaired IRS1 and IRS2 tyrosine phosphorylation in rats fed with high fat diet7. An investigation on the insulin-like growth factor (IGF) axis in the Nurses’ Health Study8 and another population study of 3863 people9 addressed connections between the IGF axis, insulin resistance, diabetes risk and NAFLD. IGFBP3 is associated with various cancers and up-regulation of IGF1 receptor (IGF1R) is considered an early event in hepatocarcinogenesis10. Thus, the IGF axis might play an important role in a direct development of carcinoma from steatosis without the formerly assumed intermediary phase of cirrhosis11.
The progression of NAFLD from mild steatosis up to severe steatohepatitis and even liver cirrhosis and hepatocellular carcinoma, varies widely between individual patients. Insulin resistance, dysregulation of cytokines as a basis for inflammation, and oxidative stress appear to foster progression to steatohepatitis12. A two-step progression from simple steatosis to steatohepatitis and fibrosis has been proposed13, and suggests that after fat accumulation in the liver due to insulin resistance, lipids are peroxidized with cytokines and Fas ligand induced by excessive ROS. However, this two-step progression has been questioned5. We found that in fibroblasts derived from steatosis patients AKT/mTOR signaling was reduced and that the insulin-resistant phenotype is exhibited not only by insulin-metabolizing central organs, e.g., the liver, but also by skin fibroblasts14. Transcriptome data identified a regulatory network orchestrated by the transcription factor SREBF1 and linked to a metabolic network of glycerolipid and fatty acid biosynthesis. The downstream transcriptional targets of SREBF1 which include the phosphatidic acid phosphatase LPIN1 and LDLR, were also involved.
Moreover, there is the possible involvement of ROS in disease progression. Houstis et al. 15 demonstrated that oxidative stress can induce insulin resistance and that anti-oxidants may ameliorate insulin resistance. Depletion of glutathione can improve insulin sensitivity in mice16. Glutathione is known as the body’s master antioxidant, protecting cells against damage caused by numerous reactive intermediates17. Detoxification of these reactive metabolites results in the consumption of glutathione either via oxidation or conjugation. Maintenance of the intracellular glutathione level is thereby a critical liver function, which could be impaired following insult/injury or in steatosis and steatohepatitis.
Several other studies exploring various aspects of NAFLD have been published. A recent publication by Moylan et al. showed that it is possible to discriminate mild versus severe fibrosis stages of NAFLD patients via their gene expression profiles18. Another study from Speliotes et al. investigated NAFLD via a genome-wide association study (GWAS) approach3. Besides the most prominent association of PNPLA3 this study reported several other associations including one at locus 19p13.11 which is in strong linkage disequilibrium with a recently found steatosis-linked polymorphism in TM6SF2, transmembrane6 superfamily member 2 (refs 19,20) . A knockdown of TM6SF2 in human hepatoma cell lines and in mice led to an increase in lipid droplet area while overexpression led to a decrease19.
Interestingly, the above mentioned genes associated with NAFLD in GWAS were not detected in a large-scale GWAS about obesity and insulin biology although the metabolic syndrome connects NAFLD and obesity21. Feldstein et al. found CK-18 as a non-invasive biomarker for NASH by comparison of plasma samples from patients with biopsy proven NAFLD22. Du Plessis et al. used analysis results from subcutaneous and visceral fat and liver biopsies to construct a model which predicts NAFLD liver histology23. This model involves the genes CCL2, DMRT2, GADD45B, IL1RN, and IL8. In contrast to the studies of Moylan et al. and Feldstein et al. our study highlights potential means of classifying distinct grades of Steatosis in NAFLD—the very early stage of the disease. Although it is evident that a complex interplay of genetic and environmental factors contribute to the development of steatosis, to date there has not been a systemic study of the disease employing a multi-omic approach- transcriptome, ELISA-based proteome and metabolome. Therefore, the intention of this study is to provide a more comprehensive view of steatosis based on transcriptomic, metabolomic and protein biomarker profiles. Additionally, this should lay down the foundation for follow-up systems biology-based studies.
In the current study we analyzed patient liver biopsies and associated serum samples, from patients with the insulin resistance phenotype confirmed by the HOMA-IR model24. Here, we describe these valuable data sets deposited in public repositories, which might support other researchers in identifying new clues for the etiology of NAFLD and the mechanisms underlying its progression to more severe disease states.
Methods
Patient recruitment, sample collection and clinical measurements
All patients participating in this study were recruited in the Multidisciplinary Obesity Research (MORE) project at the Medical University of Graz, Austria or at the Interdisciplinary Adipositas Center at the Kantonsspital St Gallen, Switzerland. Patients with morbid obesity who admitted into hospital for treatment by bariatric surgery (gastric banding, gastric bypass, sleeve gastrectomy) were invited to participate in the study and to sign the informed consent. The study was approved by the institutional review board of the Medical University of Graz (reg. IRB00002556 at the Office for Human Research Protections of the US Departments of Health and Human Services) under license 20–143 ex 08/09. All experiments were performed in accordance with approved guidelines. Written informed consent was obtained from all participants. In the course of the bariatric surgery, samples of blood, skin and a liver biopsy were taken. Out of 18 patients (Table 1), 9 liver biopsies were of high quality enabling their use in the transcriptome analyses. Serum plasma was available from all the patients. The overall experimental design of this study is illustrated in Fig. 1. A pathological diagnosis of the liver phenotype, including liver steatosis grading based on H&E morphology, was performed by an experienced, board certified pathologist (CL). We simplified Kleiner’s scoring scheme by condensing Steatosis grades 2 (34–66%) and 3 (> 66%) to our ‘high-grade’ while adopting grades 0 (‘none’) and 1 (‘low’)25. This simplification was made because the inter-patient-variability in this complex heterogeneous disease did not allow a more detailed grading on the omics levels. Two examples of liver biopsies are shown in Fig. 2a.
Table 1. Samples related to data sets in repositories (Data Citations 1–Data Citation 3).
| ID | gender | Age | BMI | % steatosis | grouping by pathologist | steatosis grouping | medical centre | liver illumina array rep.1 (GSE46300) | liver illumina array rep.2 (GSE46300) | serum NMR data | serum ELISA data |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H0004 | f | 54 | 47 | 10% | obese, low steatosis | Graz (Austria) | GSM1128362 | GSM1128363 | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0007 | f | 33 | 51 | 40% | obese, high steatosis | Graz (Austria) | GSM1128364 | GSM1128365 | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0008 | m | 61 | 46 | 40% | obese, high steatosis | obese, high steatosis | Graz (Austria) | GSM1128366 | GSM1128367 | MTBLS174 | 10.6084/m9.figshare.1333564 |
| H0009 | f | 48 | 49 | 5–10% | obese, low steatosis | obese, low steatosis | Graz (Austria) | GSM1128368 | GSM1128369 | MTBLS174 | 10.6084/m9.figshare.1333564 |
| H0011 | f | 58 | 45 | 70% | obese, high steatosis | obese, high steatosis | Graz (Austria) | GSM1128370 | GSM1128371 | MTBLS174 | 10.6084/m9.figshare.1333564 |
| H0012 | f | 50 | 35 | 0 | obese, low steatosis | obese, low steatosis | Graz (Austria) | GSM1128372 | GSM1128373 | no | no |
| H0018 | f | 35 | 41 | 30–40% | obese, high steatosis | obese, high steatosis | Graz (Austria) | GSM1128374 | GSM1128375 | MTBLS174 | 10.6084/m9.figshare.1333564 |
| H0021 | m | 49 | 41 | 0% | no steatosis | Graz (Austria) | GSM1128376 | GSM1128377 | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0022 | m | 45 | 49 | 40% | obese, high steatosis | Graz (Austria) | GSM1128378 | GSM1128379 | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0024 | m | 29 | 44 | 50% | obese, high steatosis | Graz (Austria) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0025 | f | 53 | 46 | 15–20% | obese, low steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0026 | f | 46 | 39 | 0% | no steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0027 | m | 44 | 42 | 50% | obese, high steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0028 | f | 28 | 43 | 20% | obese, low steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0029 | f | 40 | 39 | <5% | no steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0030 | m | 22 | 45 | 30% | obese, low steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0031 | m | 22 | 41 | 0% | no steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0033 | f | 44 | 43 | 40% | obese, high steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 | |
| H0034 | m | 50 | 42 | 10% | obese, low steatosis | St Gallen (Switzerland) | no | no | MTBLS174 | 10.6084/m9.figshare.1333564 |
Figure 1. Scheme of experiments for multi-omics comparison of steatosis grades.

The scheme shows how the distinct severities of non-alcoholic fatty liver disease (NAFLD) are compared in terms of transcriptomics, metabolomics and potentially relevant parts of the proteome. Liver biopsies were taken from NAFLD patients and classified by pathologists as low-grade (5–33% steatosis area) and high-grade (>33% steatosis area). The transcriptome of liver biopsies were assessed on Illumina HumanHT-12 v4 BeadChips and on RT-PCR. Serum samples of these NAFLD patients and from healthy persons were taken and investigated at the protein level employing ELISA assays and at the metabolome level via Nuclear Magnetic Resonance (NMR).
Figure 2. Histopathological and transcriptome characterization of liver tissue.

(a) Liver tissue with only marginal pathological changes (H9, low-grade steatosis group). The hepatocytes are arranged in one cell thick plates, separated by sinusoids. They contain only few small isolated fat valuoles (H&E stained section). Hepatocytes of the intermediate and central lobular areas contain macrovesicular fat (image to the right, H8, steatosis group, hepatocytes with fatty change are indicated by arrow heads; H&E stained section). (b) Hierachical clustering of the transcriptomes of patient liver samples. We identified three clusters: high (>33%) steatosis (cyan), low (5–33%) steatosis (magenta) and heterogeneous clusters of high, low and no steatosis (grey). (c) Quantitative QRT-PCR confirmation of genes differentially expressed in high versus low steatotic livers. The columns represent the mean of four biological replicates (high steatosis) versus two biological replicates (low steatosis). Error bars indicate standard errors of the mean. Array-derived and RT-PCR-derived columns are depicted in dark grey and red respectively. (d) Heatmap of genes differentially expressed in high versus low steatotic livers and genes found in literature and in genome-wide association studies.
Illumina bead chip hybridization and data analysis
Microarray experiments were carried out on the Illumina BeadStation 500 platform (Illumina, San Diego, CA, USA). Briefly, 500 ng DNase-treated total RNA were used as input for amplification and biotin labeling reactions (Illumina TotalPrep RNA Amplification Kit, Ambion) prior to hybridization of the resulting cRNAs onto Illumina HumanHT-12_v4_BeadChips, washing, Cy3-streptavidin staining and scanning according to the manufacturer’s instructions.
Transciptomics data analysis
Illumina data was processed via R/Bioconductor26 and packages lumi27, limma28 and biomaRt. Background-corrected log2-transformed data was normalized via quantile normalization from the lumi package.
qRT-PCR
Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out to confirm the microarray-derived data. Reactions were carried out on the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Data analysis was carried out using the ABI PRISM SDS 2.2.1 software (Applied Biosystems) and Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). GAPDH-normalized, relative mRNA levels of each gene (high steatosis versus low steatosis) were calculated based on the 2-ΔΔCT Method. Primer sequences for QRT-PCR validation are described in Table 2.
Table 2. Primer sequences for QRT-PCR validation of genes differentially expressed between high-grade and low-grade steatosis.
| Gene | Fwd | Rev | Product size |
| ACADSB | CACCATTGCAAAGCATATCG | GCAAGGCACTTACTCCCAAC | 117 |
| AGPAT2 | GGGGCGTCTTCTTCATCA | TTGAGGTTCTCCCTGACCAT | 91 |
| ECHS1 | AACCTTTGCCACTGATGACC | CAAGCAGAGGTGTGAAGCAG | 112 |
| IGF1 | TGCAGGAGGGACTCTGAAAC | AGCTGCGTGATATTTGAAAGG | 111 |
| IGFBP2 | CTCCCTGCCAACAGGAACTG | TCTTGCACTGTTTGAGGTTGTACAG | 147 |
| IGFBP3 | CAACTGTGGCCATGACTGAG | CCTGACTTTGCCAGACCTTC | 92 |
| INSIG1 | CAACACCTGGCATCATCG | CTCGGGGAAGAGAGTGACAT | 118 |
| KRT18 | GAGGTTGGAGCTGCTGAGAC | CAAGCTGGCCTTCAGATTTC | 99 |
| LIPA | CATCTGTGTGAAGCCAAAGC | AATCCCTGAGCTGAGTTTGC | 112 |
| PLIN2 | GCTGAGCACATTGAGTCACG | TGGTACACCTTGGATGTTGG | 102 |
ELISA-based assay for biomarkers
ELISA measurements from plasma samples were carried out using the Ciraplex platform (Aushon Biosystems, Billerica, MA, US). Commercial assays were purchased and measurements were carried out according to instructions provided by the manufacturer. The following 29 targets were analyzed either as single-plex assays or as multiplex assay: hFGFb; hGROa; hLIF;hIFNg; hIL1b; IL4; IL5; hIL6; hIL10; hIL12p70; hIL13; hTNFa; hI309; hIL8; hIP10; hMCP4; hMIP1a; hMIP1b; hCRP; hLeptin; hPAIactive; hResistin; hIGFBP1; hIGFBP3; hIGFBP2; hMIF; hApoA1; hCRP; hAcrp30.
NMR sample preparation
Frozen plasma samples were thawed at room temperature and shaken before use. According to standard methodologies a total of 300 μl of buffer (70 mM Na2HPO4; 20% (v/v) D2O; 6.15 mM NaN3; 6.64 mM TMSP; pH 7.4) was added to 300 μl of each serum sample. A total of 450 μl of this mixture was transferred into a 4.25 mm NMR tubes (Bruker BioSpin) for analysis.
NMR spectra acquisition and processing
NMR spectra from 18 plasma samples from morbidly obese patients that underwent different type of bariatric surgery and additionally have developed steatosis were collected (Table 1). 1H-NMR spectra were acquired using a Bruker spectrometer (Bruker Biospin). Unsupervised and supervised methods were used in order to identify a disease-related metabolomic profile that might contain a signature of steatosis.
Data Records
Data record 1
The microarray experiments discussed in this publication were carried out on the Illumina BeadStation 500 platform (Illumina, San Diego, CA, USA). The data have been deposited in NCBI's GEO and are accessible through GEO Series accession number GSE46300 (Data Citation 1).
Data record 2
Metabolomic raw data from Nuclear magnetic resonance (NMR) measurements have been deposited at the MetaboLights database (http://www.ebi.ac.uk/metabolights) of the European Bioinformatics Institute (EBI) under MTBLS174 (Data Citation 2).
Data record 3
ELISA measurements have been deposited at figshare (http://www.figshare.com) (Data Citation 3).
Technical Validation
Transcriptomic data
Microarray data passed the proprietary Illumina quality controls. All samples were investigated in duplicates. Fig. 2b shows that—as would be expected—the duplicates cluster together demonstrating the validity of experiments in terms of whole-genome gene expression. The Pearson correlation coefficients of all samples versus each other were calculated with the intention to detect outliers. However, all correlation coefficients were greater than 0.98 and all correlation coefficients of duplicates even greater than 0.99 so that all samples passed this quality check (Table 3). Genes with significant differential gene expression were selected for validation via RT-PCR experiments (Fig. 2c). Genes were termed differentially expressed if the multiple-testing-corrected limma28 P-value was less than 0.05, the ratio was less than 0.75 or greater than 1.33 and the gene was expressed (detection P-value less than 0.05) in at least one of both cases. Furthermore, we analysed clusters of genes differentially expressed in high versus low steatotic livers together with genes found in literature19,23 and genome-wide association studies3 (Fig. 2d). This analysis confirms high similarity between duplicates and clustering—to some extent but not fully—according to steatosis grade. Fig. 3a shows a plot of the first two components of the Principal Component Analysis (PCA) of the microarray data.
Table 3. Pearson correlation coefficients of transcriptome data of all samples versus each other.
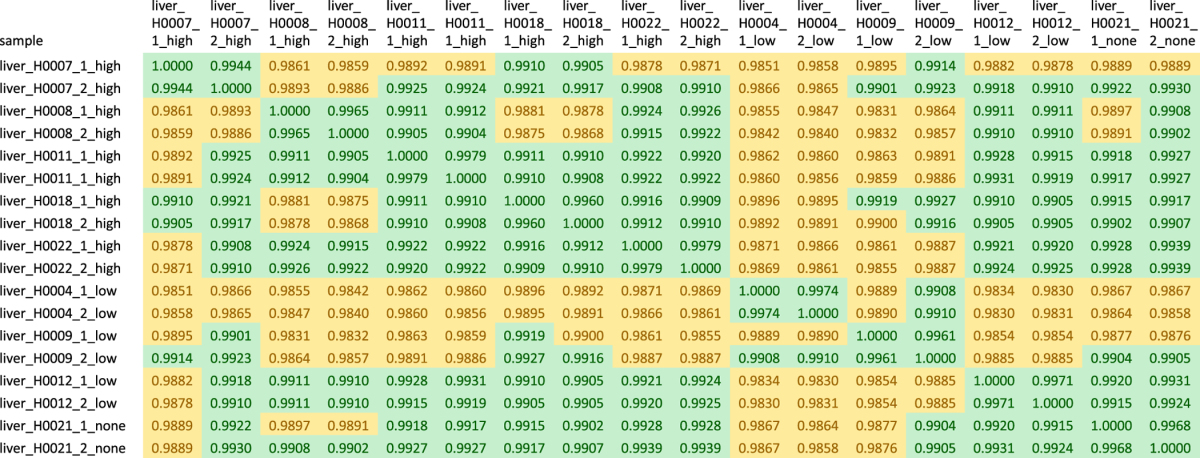
Figure 3. Transcriptomic and metabolomic profiles.
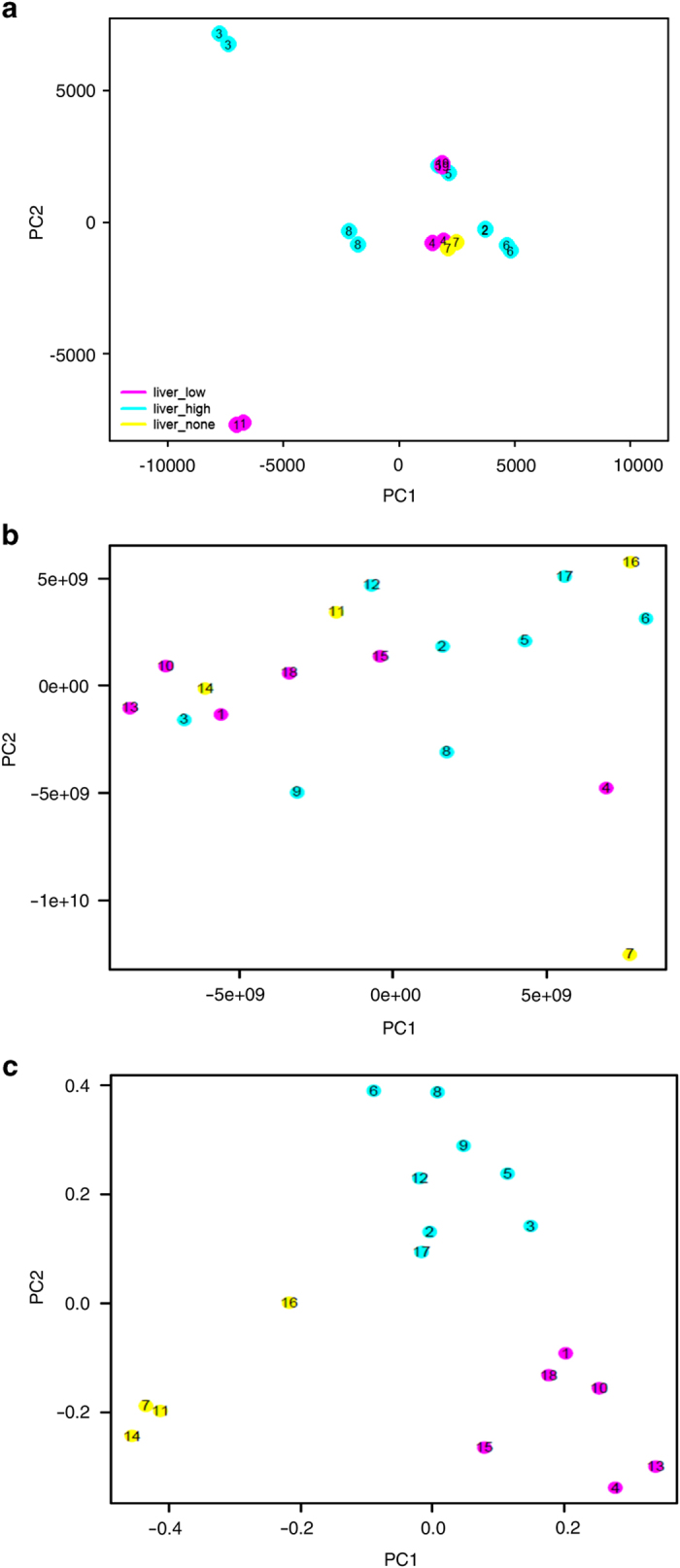
(a–c) Transcriptomics and metabolomics PCA plots. Distinct colours are used to aid visualizing patients with distinct levels of steatosis: yellow, patients with <5% no steatosis; magenta, patients with 5–33%, low level steatosis; cyan, patients with high steatosis >33%, high steatosis. (a) Unsupervised PCA plot for 18 liver biopsies, Illumina microarray data. (b) Unsupervised PCA plot for 18 plasma samples, metabolomics data. (c) Supervised discrimination analysis (pls/ca: partial least squares/canonical analysis) of metabolites in patient plasma samples. The correspondence between numbers in the plot and sample names in Table 1 is: 1=H0004, 2=H0007, 3=H0008, 4=H0009, 5=H0011, 6=H0018, 7=H0021, 8=H0022, 9=H0024, 10=H0025, 11=H0026, 12=H0027, 13=H0028, 14=H0029, 15=H0030, 16=H0031, 17=H0033, 18=H0034, 19=H0012.
ELISA-based assay for biomarkers
Samples below and above quantification limit as well as samples with coefficient of variation (cv) greater than 20% were marked in the measurements table (Data Citation 3). Independent validation of the ELISA-based measurements was checked by visual inspection of plots comparing disease states.
Metabolomics
Assignment of all metabolites were done manually, signals were assigned on template one-dimensional NMR profiles by using matching routines of AMIX 7.3.2 (Bruker BioSpin) in combination with the BBIOREFCODE Version 2-0-0 reference database and published literature when available. Additional confirmation was done using data provided in our lab -database containing spectra of standard pure compounds. To assess which metabolites (i.e., NMR peaks) were significantly different between different sets a univariate paired Wilcoxon test was used. A P-value≤0.05 was considered statistically significant (P-value not corrected for multiple testing).
Robust validation of statistical analysis results was done using a cross-validation technique.
The accuracy of the classification was assessed by means of a single cross-validation scheme. The original data set was split into a training set (80% of the samples) and a test set (20% of the samples) prior to any step of statistical analysis. The number of PLS components was chosen on the basis of a 5-fold cross validation performed on the training set only, and the best model was used to predict the samples in the test set. The whole procedure was repeated 200 times with a Monte Carlo cross validation scheme, and the results averaged.
Figure 3b shows a plot of unsupervised discrimination analysis and Fig. 3c shows separation of steatosis grades in a plot of supervised discrimination analysis (pls/ca: partial least squares/canonical analysis) of metabolites in patient plasma samples. The clustering of Fig. 3c results from a supervised PLS/CA based only on the metabolomic NMR profiles. The algorithm takes into account the supervised information relative to the 3 steatosis groups.
Distribution plots
Figure 4a shows the distribution of percentage parenchymal involvement in steatotic patients derived from Table 1. The percentage is converted to a scale from zero to one and plotted with the kernel density function from the R statistical package. Fig. 4b–d display distributions separated into groups of age above/below median (median=45), body mass index (BMI) above/below median (median=43) and gender. Fig. 4d would suggest a slight tendency for more severe steatosis in males. A similar trend has been reported in a NAFLD study on Australian adolescents where 3.1% of males and only 2.2% of females had moderate to severe steatosis while 7.0% of males and 14.1% of females had mild steatosis29.
Figure 4. Distribution plots of percentage parenchymal involvement in steatosis.

(a) all patients. (b) Kernel density plot of patients above/below median age (median=45). (c) Kernel density plot of patients above/below median BMI (median=43). (d) Kernel density plot of male/female patients.
Usage Notes
All patients in this study underwent bariatric surgery. This should be taken into account when generalizing results although these are typical cases of morbid obesity which is connected to the metabolic syndrome including NAFLD. The sample size of these datasets—in particular the transcriptomics dataset—poses certain limits onto its usage. Due to its small size it will not enable rigorous analysis of gender effects. Therefore it would likely need to be combined with other data sources, such as data from Moylan et al.18 and Du Plessis et al.23.
Additional Information
How to cite this article: Wruck, W. et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci. Data 2:150068 doi: 10.1038/sdata.2015.68 (2015).
Supplementary Material
Acknowledgments
The authors acknowledge support from the German Federal Ministry of Education and Research (BMBF GRANT 0315717A), which is a partner of the ERASysBio+ initiative supported under the EU ERA-NET Plus scheme in FP7. Hans V. Westerhoff, & Samrina Rehman thank the EPSRC and BBSRC (and BBSRC-BRIC) for support of the Manchester Centre for Integrative Systems Biology, as well as various other funding agencies (EU-FP7 [SYNPOL #311815], H2020 [Epipredict, #642691 and Corbel #654248], NWO, the SysMO and ERASysBio funder communities), in particular the ERASysBio+ support by BBSRC. Additionally, James Adjaye and Wasco Wruck acknowledge support from the Medical faculty of the Heinrich Heine University Düsseldorf.
Footnotes
The authors declare no competing financial interests.
Data Citations
- Wruck W. 2014. NCBI GEO. GSE46300
- Wruck W. 2015. MetaboLights. MTBLS174
- Wruck W. 2015. Figshare. http://dx.doi.org/10.6084/m9.figshare.1333564
References
- Lazo M. & Clark J. M. The epidemiology of nonalcoholic fatty liver disease: a global perspective. Semin. Liver Dis. 28, 339–350 (2008). [DOI] [PubMed] [Google Scholar]
- Petersen K. F. et al. Apolipoprotein C3 Gene Variants in Nonalcoholic Fatty Liver Disease. N. Engl. J. Med. 362, 1082–1089 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speliotes E. K. et al. Genome-Wide Association Analysis Identifies Variants Associated with Nonalcoholic Fatty Liver Disease That Have Distinct Effects on Metabolic Traits. PLoS Genet 7, e1001324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha-Bonet D. et al. Overfeeding Reduces Insulin Sensitivity and Increases Oxidative Stress, without Altering Markers of Mitochondrial Content and Function in Humans. PLoS ONE 7, e36320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V., Bellentani S., Cortez-Pinto H., Day C. & Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 53, 372–384 (2010). [DOI] [PubMed] [Google Scholar]
- Farese R. V. Jr., Zechner R., Newgard C. B. & Walther T. C. The Problem of Establishing Relationships between Hepatic Steatosis and Hepatic Insulin Resistance. Cell Metab. 15, 570–573 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel V. T. et al. Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 279, 32345–32353 (2004). [DOI] [PubMed] [Google Scholar]
- Rajpathak S. N. et al. Insulin-like growth factor axis and risk of type 2 diabetes in women. Diabetes 61, 2248–2254 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völzke H. et al. Association between hepatic steatosis and serum IGF1 and IGFBP-3 levels in a population-based sample. Eur. J. Endocrinol. 161, 705–713 (2009). [DOI] [PubMed] [Google Scholar]
- Aleem E., Nehrbass D., Klimek F., Mayer D. & Bannasch P. Upregulation of the insulin receptor and type I insulin-like growth factor receptor are early events in hepatocarcinogenesis. Toxicol. Pathol. 39, 524–543 (2011). [DOI] [PubMed] [Google Scholar]
- Baffy G., Brunt E. M. & Caldwell S. H. Hepatocellular carcinoma in non-alcoholic fatty liver disease: An emerging menace. J. Hepatol. 56, 1384–1391 (2012). [DOI] [PubMed] [Google Scholar]
- Marra F., Gastaldelli A., Svegliati Baroni G., Tell G. & Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol. Med. 14, 72–81 (2008). [DOI] [PubMed] [Google Scholar]
- Angulo P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 346, 1221–1231 (2002). [DOI] [PubMed] [Google Scholar]
- Jozefczuk J. et al. A Systems Biology Approach to Deciphering the Etiology of Steatosis Employing Patient-Derived Dermal Fibroblasts and iPS Cells. Front. Physiol 3, 339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houstis N., Rosen E. D. & Lander E. S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948 (2006). [DOI] [PubMed] [Google Scholar]
- Findeisen H. M. et al. Glutathione Depletion Prevents Diet-Induced Obesity and Enhances Insulin Sensitivity. Obesity 19, 2429–2432 (2011). [DOI] [PubMed] [Google Scholar]
- Hayes J. D., Flanagan J. U. & Jowsey I. R. Glutathione Transferases. Annu. Rev. Pharmacol. Toxicol. 45, 51–88 (2005). [DOI] [PubMed] [Google Scholar]
- Moylan C. A. et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatol. Baltim. Md 59, 471–482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahali B. et al. TM6SF2: catch-22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology 148, 679–684 (2015). [DOI] [PubMed] [Google Scholar]
- Kozlitina J. et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 46, 352–356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shungin D. et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldstein A. E. et al. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: A multicenter validation study. Hepatology 50, 1072–1078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Plessis J. et al. Association of Adipose Tissue Inflammation With Histologic Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 149, 635–648, e14 (2015). [DOI] [PubMed] [Google Scholar]
- Matthews D. R. et al. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985). [DOI] [PubMed] [Google Scholar]
- Kleiner D. E. et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- Gentleman R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du P., Kibbe W. A. & Lin S. M. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008). [DOI] [PubMed] [Google Scholar]
- Smyth G. K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 3 doi:10.2202/1544-6115.1027 (2004). [DOI] [PubMed] [Google Scholar]
- Ayonrinde O. T. et al. Gender-specific differences in adipose distribution and adipocytokines influence adolescent nonalcoholic fatty liver disease. Hepatol. Baltim. Md 53, 800–809 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Wruck W. 2014. NCBI GEO. GSE46300
- Wruck W. 2015. MetaboLights. MTBLS174
- Wruck W. 2015. Figshare. http://dx.doi.org/10.6084/m9.figshare.1333564