

A coordinatively unsaturated single iron site confined in a graphene matrix shows an ultrahigh activity for catalytic oxidation.
Keywords: single atom site, coordinatively unsaturated iron, benzene oxidation, Graphene, non-precious catalyst
Abstract
Coordinatively unsaturated (CUS) iron sites are highly active in catalytic oxidation reactions; however, maintaining the CUS structure of iron during heterogeneous catalytic reactions is a great challenge. Here, we report a strategy to stabilize single-atom CUS iron sites by embedding highly dispersed FeN4 centers in the graphene matrix. The atomic structure of FeN4 centers in graphene was revealed for the first time by combining high-resolution transmission electron microscopy/high-angle annular dark-field scanning transmission electron microscopy with low-temperature scanning tunneling microscopy. These confined single-atom iron sites exhibit high performance in the direct catalytic oxidation of benzene to phenol at room temperature, with a conversion of 23.4% and a yield of 18.7%, and can even proceed efficiently at 0°C with a phenol yield of 8.3% after 24 hours. Both experimental measurements and density functional theory calculations indicate that the formation of the Fe═O intermediate structure is a key step to promoting the conversion of benzene to phenol. These findings could pave the way toward highly efficient nonprecious catalysts for low-temperature oxidation reactions in heterogeneous catalysis and electrocatalysis.
INTRODUCTION
Earth-abundant transition metal centers, such as coordinatively unsaturated (CUS) iron sites, can exhibit higher catalytic activity for reactions than precious metals. Yet, because of the instability of CUS sites, it is difficult to maintain the active structure of transition metal centers during a heterogeneous catalytic reaction. On the other hand, many successful examples can be found in enzymes such as cytochrome P-450 (1, 2), nitrogenase (3), and methane monooxygenase (4), as well as some homogeneous catalysts, where the organic ligands and proteins confine these CUS iron sites, making them highly active and stable (1, 5–7). In heterogeneous catalysis, however, preparation of the analogous CUS iron sites in supported catalysts with robust structures and high activity remains an attractive challenge (8–11). Our previous work demonstrated that the CUS ferrous sites, confined at the interface of precious metal Pt, are highly active and stable in activating oxygen at low temperatures (12, 13). However, the high cost of Pt prevents the commercialization of these catalysts. A major research thrust has been made to replace Pt with earth-abundant materials while maintaining the CUS ferrous structure. Graphene with a well-defined two-dimensional (2D) structure and high specific surface area shows high mechanical strength and thermal stability under realistic catalytic conditions (14, 15). Its unique structural and electronic properties render it a promising host to confine the CUS metal atoms in the matrix. Several recent works have demonstrated that the single metal atom can be successfully embedded in a graphene matrix through in situ electron beam irradiation in a transmission electron microscopy (TEM) system (16–18). However, the pure metal atoms in graphene are mobile under irradiation (16, 17), implying their instability under realistic catalytic conditions. Moreover, it is difficult to obtain a sufficient quantity for catalytic applications using the irradiation method. FeN4 centers with CUS Fe sites in organic macrocycles have been proven to be stable structures, whereas the supported FeN4 macrocycles on substrates tend to aggregate during catalytic reactions because of the weak interactions between these macrocycles and substrates (19). Therefore, one possible route to stabilizing the CUS Fe sites in the graphene matrix is via the introduction of N atoms as an “anchor,” because the C–N bond has been proven to be highly stable in N-doped graphene (20, 21).
Here, we report one strategy to achieve a highly dispersed single FeN4 center with CUS Fe sites confined in a graphene matrix at a large quantity via high-energy ball milling of iron phthalocyanine (FePc) and graphene nanosheets (GNs) under controllable conditions. High-energy ball milling has been demonstrated as a powerful method to cut and reconstruct the chemical bonds of materials or molecules with necessary energy input (20, 22–24). We prepared a series of graphene-embedded FeN4 (FeN4/GN) catalysts with different Fe content, that is, FeN4/GN-1.5 (1.5% Fe, see table S1), FeN4/GN-2.7, and FeN4/GN-4.0, by ball milling the composites of FePc and GN with appropriately chosen energies (see Materials and Methods for more details).
RESULTS
The typical morphology of FeN4/GN is presented in Fig. 1 (A to D) and figs. S1 and S2, and was obtained using low-voltage (80 kV) spherical aberration–corrected HRTEM. One can see homogeneously dispersed small black dots in the graphene matrix. Some are tagged by the red arrows and circles in Fig. 1 (A to D) and fig. S2, which could be assigned as single Fe atoms. The structure of single Fe centers was further evidenced by sub-angstrom resolution HAADF-STEM images (Fig. 1, G and H, and fig. S3), which show the atomic size and homogeneous distribution of the bright dots within the graphene matrix. Through EELS atomic spectra of the bright dots (Fig. 1, H and I), one can clearly see the presence of both Fe and N elements in one bright dot, suggesting the formation of Fe–Nx bonding. This indicates that Fe atoms observed in Fig. 1 (A to D, G, and H) and figs. S1 to S3 should be bonded with N atoms in the surroundings and further contacted with the graphene matrix, as shown in the atomic models (Fig. 1E) for the experimental structures (Fig. 1D), which is also highly consistent with the density functional theory (DFT)–simulated HRTEM image (Fig. 1F). Note that some disordered structures can also be observed around Fe atoms in some areas (fig. S4), implying the introduction of defects in the graphene network around some iron atoms during the high-energy ball milling. X-ray diffraction (XRD) (fig. S5) and Raman spectra (fig. S6) further indicate that there is no characteristic structural information of FePc, Fe, or FeOx observed in FeN4/GN samples, implying a well-dispersed feature of these Fe sites in FeN4/GN samples, which is highly consistent with the TEM and HAADF-STEM analysis. To obtain more atomic and electronic structure information of FeN4 centers in the graphene matrix, we performed low-temperature scanning tunneling microscopy (LT-STM, 4 K). Figure 1J shows a typical atomic-resolution STM image of a single FeN4 center embedded in the graphene matrix. The iron center is resolved as a bright spot, whereas neighboring atoms exhibit a higher apparent height than other carbon atoms in the graphene matrix. STM simulation (Fig. 1K) of an FeN4 center embedded in the graphene lattice is in agreement with the measured STM image (Fig. 1J), suggesting that the iron center significantly modifies the density of states of adjacent atoms. The bright dot in Fig. 1J is attributed to the iron center, whose neighboring C and N atoms are also electronically rich and appear brighter than carbon atoms located further away. Accordingly, STM contours of the bright spot, the corresponding conductance spectra (Fig. 1L), and the stability of the bright spot during scanning tunneling spectroscopy (STS) measurements all suggest that the FeN4 center is in the plane of graphene and forms stable bonds with neighboring carbon atoms. FeN4 in macrocycles often exhibits sharp electronic states near the Fermi level, corresponding to their highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) levels (25–27). On graphene, because of the large gap between the HOMO and LUMO levels of the FeN4 center, such states are often 1.5 to 2 eV away from the Fermi level (28). In Fig. 1L, STS measurements across the FeN4 center also show a sharp resonance state at −0.63 eV below the Fermi level, suggesting that the iron center strongly interacts with the graphene lattice and thus introduces a new electronic state near the Fermi level, not seen in isolated FePc molecules or N-doped graphene (29). To the best of our knowledge, this is the first time the well-defined FeN4 atomic structure in graphene has been observed by combining HRTEM/STEM with LT-STM/STS.
Fig. 1. Structural analysis of graphene-embedded FeN4 (FeN4/GN) catalysts.
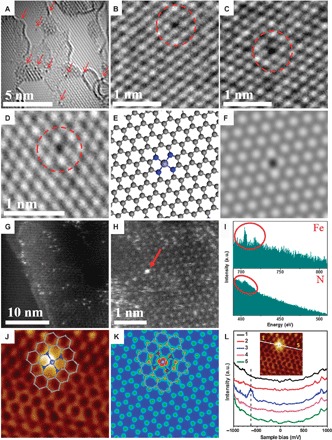
(A to D) High-resolution transmission electron microscopy (HRTEM) images of FeN4/GN-2.7. The area with arrows and the dashed circles shows some typical single Fe atoms in the nanosheets. (E and F) Atomic models (E) and the corresponding simulated HRTEM images (F) for the structures in (D), where the FeN4/GN structures have been optimized. (G and H) High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of FeN4/GN-2.7. (I) The electron energy loss spectroscopy (EELS) atomic spectra of Fe and N elements from the bright dots as shown by the red arrow in (H). The red circles show Fe and N signals, respectively. a.u., arbitrary units. (J) Low-temperature scanning tunneling microscopy (LS-STM) image of FeN4/GN-2.7, measured at a bias of 1.0 V and a current (I) of 0.3 nA (2 nm × 2 nm). (K) Simulated STM image for (J). The inserted schematic structures represent the structure of the graphene-embedded FeN4. The gray, blue, and light blue balls in (E), (J), and (K) represent C, N, and Fe atoms, respectively. (L) dI/dV spectra acquired along the white line in the inset image. U, 1.0 V; I, 0.3 nA; modulation frequency, 500 Hz; amplitude, 20 millivolts peak to peak; RC, 7 Hz.
X-ray absorption fine structure (XAFS) spectroscopy was used to further probe the chemical state and coordination structure of these confined Fe centers. As shown in Fig. 2A, the Fe K-edge of XANES in FeN4/GN samples exhibits a near-edge structure similar to that of the original FePc but is very different from those of Fe foil and Fe2O3, indicating that the valence state of Fe remains the same with FePc, which can be further confirmed by Fe 2p XPS analysis (fig. S7B). EXAFS of the Fe K-edge (Fig. 2B) shows that the magnitude of the FT spectra of the FeN4/GN samples also closely resembles the original FePc reference curve (30, 31). From the shape and amplitude of the first strong peak (with phase shift correction) in the FT plot, one can see that the bonding environment in the first shell of FeN4/GN samples is the same as that of FePc, suggesting that one Fe site connects four N atoms as the FeN4 structure in its precursor FePc. Furthermore, the N 1s XPS (Fig. 2E) reveals that the intensity of pyrrolic Nα (400.4 eV) (bonding with Fe) is almost unchanged whereas that of pyridinic Nβ (398.6 eV) (bonding with carbon on the outside macrocycle) is significantly reduced compared with FePc; this indicates that part of the pyridinic Nβ species has been destroyed during ball milling whereas pyrrolic Nα species are well retained in FeN4/GN samples. We further investigated the C K-edge XAS spectra of FeN4/GN samples to study the macrocyclic structure change during ball milling. As shown in Fig. 2C, the FeN4/GN samples show a strong π* and σ* band structure, indicating that the graphene matrix is still graphitized. It can be seen that the intensities of B and C features, considered as contributions predominantly from carbon atoms of the pyrrole rings (32–34), have been obviously reduced in FeN4/GN samples compared with the FePc sample, indicating that some parts of the carbon atoms in the outside macrocyclic structure have also been destroyed. Meanwhile, the N K-edge XAS spectra of FeN4/GN samples (Fig. 2D) show that the intensity of the π* band (at ca. 398 eV) significantly decreases relative to that of the σ* band (at ca. 406 eV) for N, suggesting that the number of C═N bonds was significantly reduced and the FeN4 structure remains almost unchanged.
Fig. 2. Chemical state and coordination information of FeN4/GN catalysts.
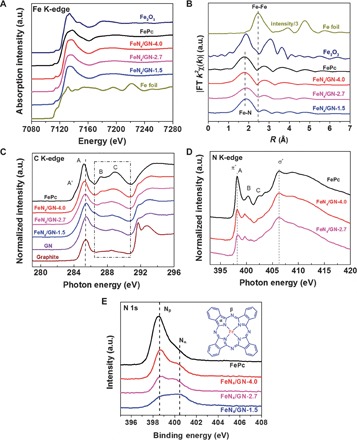
(A and B) Fe K-edge x-ray absorption near-edge structure (XANES) (A) and Fourier transform (FT) extended x-ray absorption fine structure (EXAFS) (B) signals of FeN4/GN samples with various Fe content in comparison to FePc, Fe foil, and Fe2O3. (C and D) C K-edge (C) and N K-edge (D) x-ray absorption spectroscopy (XAS) spectra of FeN4/GN samples with various Fe content in comparison to that of FePc. (E) N 1s x-ray photoelectron spectroscopy (XPS) spectra of FeN4/GN samples with various Fe content in comparison to FePc. The inserted schematic structures represent the FePc molecule, where the pyrrolic N with Fe bonding is denoted as Nα and the pyridinic N with carbon bonding on the outside macrocycle is denoted as Nβ.
The above results demonstrated that the FeN4 centers have been successfully embedded into the matrix of GNs via high-energy ball milling of FePc and GN. In one proposed mechanism, described in fig. S8, the outside macrocyclic structure of FePc can be destroyed during the ball milling, the residual isolated FeN4 centers will interact with the graphene at the defected site, and the adjacent carbon atoms of FeN4 can further reconstruct with the high energy of ball milling, finally leading to the formation of the FeN4 centers embedded into the graphene matrix. Our previous work indicated that isolated Fe atoms embedded within a silicide matrix showed high activity and long-term stability toward direct conversion of methane to ethylene and hydrocarbons (35). Thus, this graphene-confined single CUS iron site is expected to have high performance for catalytic reactions.
The direct catalytic conversion of benzene to phenol is one of the most active topics in fundamental and applied research (36–39). Different catalysts including Ti-containing zeolites, palladium membranes, and transition metal (such as Fe, Cu, and V)–based oxides or chelates have been widely investigated for the direct conversion of benzene to phenol. This reaction is usually carried out at 50° to 140°C, because it is very difficult to directly proceed at room temperature owing to the highly stable C–H bond of benzene (36, 40–42). Here, we found that the FeN4/GN samples showed a high activity and selectivity for phenol at room temperature. The oxidation of benzene was conducted at 25°C with hydrogen peroxide as the oxidant. With the increase of the Fe content in graphene, the activity and the yield of phenol first increased quickly and then decreased (Fig. 3A and table S2). This trend in performance with the Fe content in graphene can be attributed to the observation that a moderate amount of FeN4 can promote both the dispersion of FeN4 centers in graphene and their bonding with graphene, whereas a higher content of FeN4 will lead to the agglomeration of FeN4. The optimized FeN4/GN-2.7 catalyst has a turnover frequency of 84.7 hour−1 for benzene conversion within the initial 5 min (Fig. 3B) and can achieve a benzene conversion of 23.4% and phenol yield of 18.7% in 24 hours (Fig. 3B and table S3). Some residual FePc dissolved in reaction solution may have contributed to converting benzene to phenol, but the contribution should be minor because the FeN4/GN-2.7 sample shows significantly better activity compared with the FePc monomer, despite the latter having more Fe sites. For comparison, in a blank experiment without catalyst, no obvious activity was observed over a 24-hour run (table S2). When the graphite flake (GF) and GN were used as the catalyst, only low conversions of benzene were observed (Fig. 3A and table S2), that is, 0.6% for GF and 5.4% for GN under the same conditions. Considering that the edges and defects of graphene may contribute to the activity as reported previously by Deng et al. (20), it is reasonable that the GN exhibited a higher conversion of benzene than GF because the GNs have more edges and defects. In addition, the low-temperature O2 temperature-programmed desorption (TPD) measurement shows that the FeN4/GN-2.7 has a significantly higher adsorption capacity of O2 compared with GN and GF (fig. S9). O2 can be easily adsorbed on the FeN4 structure in metal porphyrin or phthalocyanine according to previous studies (43, 44), which indicates that the FeN4/GN-2.7 had more active sites. Furthermore, we found that the FeN4/GN-2.7 catalyst can even proceed efficiently at 0°C with a phenol yield of 8.3% under 24 hours (table S4) and can remain stable after six cycles (fig. S10), further supporting its excellent catalytic performance.
Fig. 3. The performance and reaction process of the catalytic oxidation of benzene to phenol over FeN4/GN catalysts.
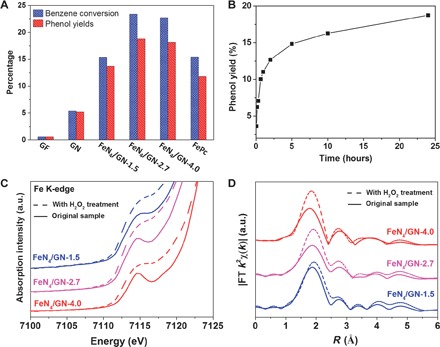
(A) The performance of the direct oxidation of benzene to phenol by FeN4/GN samples compared with GF, GN, and FePc. Reaction conditions: 50 mg of catalyst, 0.4 ml of benzene, 6 ml of H2O2 (30%), and 3 ml of CH3CN in a pressure vessel at 25°C for 24 hours. (B) The phenol yield of FeN4/GN-2.7 for the direct oxidation of benzene to phenol with different reaction times. (C and D) Fe K-edge XANES (C) and FT EXAFS (D) signals of FeN4/GN samples with H2O2 treatment in comparison to their corresponding original samples.
To gain further insights into the activity of FeN4/GN toward benzene oxidation, we carried out DFT calculations to investigate the reaction mechanism of the process (see Materials and Methods and figs. S11 and S12 for more details on calculation). A model of the FeN4 structure embedded in graphene was adopted according to experimental characterization. Figure 4A shows that the formation energy of the FeN4 center in graphene matrix (FeN4/GN) is significantly lower than that of the single-atom Fe in a pure graphene matrix (Fe/GN), suggesting that the N atoms can be used as an anchor to enhance the stability of Fe atoms in graphene, which supports the experimental results. Furthermore, the free energy profile and reaction pathway of benzene oxidation on the confined iron site are depicted in Fig. 4 (B and C). A H2O2 molecule can be easily dissociated on the confined iron site by forming an Fe═O intermediate and releasing one H2O molecule, followed by the dissociation of another H2O2 on the other side of the iron atom with an energy barrier of 0.55 eV by forming an O═Fe═O center. The O species of the O═Fe═O is active for the adsorption of the benzene molecule via the formation of a C–O bond with an energy barrier of 0.59 eV. In comparison, the direct adsorption of benzene on the O site of the Fe═O site is energetically unfavorable and needs an additional free energy of 0.86 eV (fig. S12). The benzene adsorbed on the O═Fe═O site can transform to phenol via the transfer of one adjacent H atom from C to O with a barrier of 0.35 eV. The Fe═O site can be regenerated in a reaction cycle after desorption of the phenol from the iron. The highest energy barrier in the reaction pathway occurs at the adsorption of benzene on the O═Fe═O site, which is only 0.59 eV and moderate for low-temperature reactions. The formation of Fe═O/O═Fe═O intermediates on the FeN4 center is also evidenced by XAFS analysis of the FeN4/GN samples after the H2O2 treatment. As shown in Fig. 3C and fig. S13, after the H2O2 treatment, the XANES of the Fe K-edge shows almost no energy shift, whereas the pre-edge peak, that is, the Fe 1s-to-3d transition in all FeN4/GN samples, would increase and broaden, probably because the formation of Fe═O leads to Fe 3d mixing with O 2p and thus destroys the D4h symmetry of FeN4 according to the literature (30, 45). The Fe═O interaction (relative to the Fe–N bonding) will increase the unoccupied state of Fe because of the electronegativity of O; thus, the pre-edge is more intense. EXAFS of Fe K-edge further confirmed the hypothesis. One can see that the amplitude of the first strong peak in the FT plot of these samples was significantly enhanced after the H2O2 treatment (Fig. 3D), suggesting that the coordination number of the Fe center sharply increases, which likely originates from the formation of Fe═O/O═Fe═O bonds during the reaction. EXAFS fitting of these results shows that these original FeN4/GN samples have an average coordination number of about 4 (fig. S14 and table S5), which is almost the same as that of FePc but less than the maximal coordination number of 6. Thus, the iron sites in these samples are also CUS. With further H2O2 treatment of the FeN4/GN sample, the coordination number of the iron site increases (table S5). These results supported the DFT calculation that the CUS iron site can effectively activate H2O2 and form Fe═O bonds. 57Fe Mössbauer spectra (fig. S15 and table S6) also indicate that the symmetrical O═Fe═O structure will significantly increase in FeN4/GN when treated with H2O2, whereas the O═Fe═O structure will decrease again when further treated with benzene during the catalytic reaction. The above experimental results indicate that the FeN4 centers play an important role in the adsorption and activation of oxygen, which are in agreement with reaction cycles resulting from DFT calculations.
Fig. 4. Theoretical analysis of the FeN4/GN structure and the catalytic reaction process by DFT calculations.
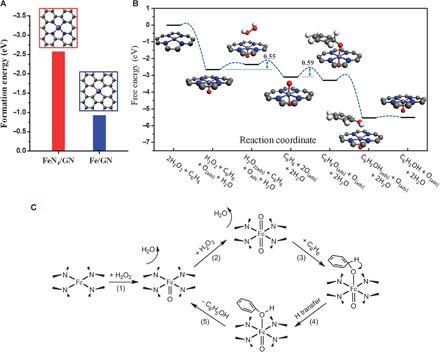
(A) The formation energies of FeN4/GN and Fe/GN structures. The formation energy is calculated as follows: EFe-embedded − EFe-bulk − E(N)GN, where EFe-embedded and EFe-bulk are the total energies of FeN4/GN and the Fe/GN structure and an Fe atom in Fe bulk, respectively, and E(N)GN is the total energy of the optimized structure of FeN4/GN or Fe/GN with the Fe atom removed from the system. (B) Free energy diagram of the oxidation of benzene to phenol on FeN4/NG. The gray, blue, light blue, red, and white balls represent C, N, Fe, O, and H atoms, respectively. (C) Scheme for the reaction mechanism of the oxidation of benzene to phenol on FeN4/NG.
The reaction pathway of benzene oxidation on the iron site of FePc was also calculated for comparison. Figure S12 shows that the reaction mechanism and free energy diagram of benzene oxidation on FePc is similar to that on FeN4/GN, but the dissociation energy of the first H2O2 forming Fe═O on FeN4/GN is lower than that on the FePc monomer. Bader charge analysis shows that the iron of FeN4/GN has an additional 0.14 electrons compared with that of FePc, leading to the O absorbed on FeN4/GN obtaining an additional 0.10 electrons compared with that on FePc, which then energetically favors the formation of the Fe═O bond on FeN4/GN. Therefore, the enhanced benzene oxidation activity of FeN4/GN can be attributed to both the intrinsic activity improvement of the active sites and the high dispersion of these CUS sites in FeN4/GN compared with bulk FePc.
DISCUSSION
In summary, the CUS single-iron site has been confined in the GN matrix through a one-step ball milling synthesis. The unique 2D structure of this catalyst provides a well-defined model for understanding the nature of the catalytic oxidation reaction on FeN4/GN catalysts by means of experiments and DFT calculations. In this system, the FeN4 center is highly dispersed and well stabilized by the graphene matrix, which subsequently enhances the activity and stability for the oxidation of benzene to phenol. This reaction can proceed efficiently at room temperature and even at temperatures as low as 0°C. DFT calculations indicate that the catalytic activity arises from the confined iron sites and the activation barriers are quite moderate for reactions to proceed at room temperature, in agreement with experimental results. These findings pave the way toward the design of highly efficient nonprecious catalysts for catalytic oxidation reactions at low temperatures.
MATERIALS AND METHODS
Raw materials
GFs (99.8%, metals basis) were purchased from Alfa Aesar. FePc (96%) was purchased from Acros Organics.
Synthesis of GNs
GNs were prepared following the same procedure used in our previous report (20). In a typical experiment, 2.0 g of GF and 60 g of steel balls (1 to 1.3 cm in diameter) were put into a hardened steel vial inside a glove box and purged with high-purity Ar (99.999%) for 20 min before the vials were sealed. Ball milling was carried out at 450 rpm for 20 hours.
Synthesis of FeN4/GN
A combined mass of 2.0 g of FePc and GN composites with a desired ratio and 60 g of steel balls (1 to 1.3 cm in diameter) was ball-milled following the same procedure as the GN synthesis. A series of FeN4/GN samples with different Fe content were prepared, that is, FeN4/GN-1.5 [1.5% Fe, see inductively coupled plasma (ICP) data in table S1, the same below], FeN4/GN-2.7, and FeN4/GN-4.0 from the precursor FePc and GN with a ratio of 15, 30, and 45%, respectively. The utility ratio of FePc in the final FeN4/GN catalysts is around 90% according to the Fe content analysis using inductively coupled plasma atomic emission spectroscopy (ICP-AES).
Characterization
HRTEM was carried out using an image spherical aberration–corrected TEM system (FEI Titan 80-300). An acceleration voltage of 80 kV was chosen to achieve enough resolution while maintaining the structure of the graphene. STEM and EELS were performed on a JEOL ARM200F equipped with double aberration correctors and a cold field emission gun operated at 80 kV. STEM images were recorded using a HAADF detector with a convergence angle of 30 mrad and a collection angle between 90 and 370 mrad. Under these conditions, the spatial resolution is ca. 0.08 nm. STM and STS were acquired using a commercial Createc LT-STM system with base pressures below 7.0 × 10−11 mbar. The sample was dispersed in petroleum ether and further dripped on the surface of HOPG (highly oriented pyrolitic graphite). The sample was then transferred to the Createc LT-STM system. Before imaging, the sample was degassed at ~450 K to remove impurities absorbed on the surface. STM experiments were performed at liquid He temperatures at a constant current mode using an electrochemically etched W tip. ICP-AES was conducted in Shimadzu ICPS-8100. The samples for ICP-AES analysis were first heated at 600°C for 12 hours in air, then treated with hydrochloric acid in Teflon-lined autoclaves at 120°C for 12 hours, and finally transferred to volumetric flasks. During XPS measurements, Mg Kα radiation (1253.6 eV) with a power of 200 W and a pass energy of 50.0 eV was used. XAS measurements were conducted at the SGM (11ID-1) beamline of the Canadian Light Source (CLS). Fe K-edge XAFS spectra of the catalysts were recorded at the SXRMB (06B1-1) beamline of the CLS and the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF). 57Fe Mössbauer spectroscopy analysis was conducted on a Topologic 500A spectrometer with a proportional counter. 57Co(Rh) was used as the radioactive source, and the Doppler velocity of the spectrometer was calibrated with α-Fe foil. The spectra were fitted with appropriate superpositions of Lorentzian lines using the MossWinn 3.0i program. XRD was performed on a Rigaku D/MAX 2500 diffractometer with Cu Kα radiation (λ = 1.5418 Å) at 40 kV and 200 mA. Raman spectroscopy was performed on a Jobin Yvon LabRAM HR 800 instrument with a 532-nm excitation laser at a power of 0.7 mW. O2 TPD measurements were carried out using the AutoChem II 2920 with a flowing 5% O2/He stream (50 ml min−1) at −50°C. The samples were pretreated with He at 250°C for 1 hour to remove the adsorbed gaseous impurities before the TPD test.
Catalytic benzene oxidation evaluation
Benzene oxidation reaction was carried out in a 50-ml Teflon-lined stainless steel reactor with 0.4 ml of benzene, 6 ml of H2O2 (30%), and 3 ml of CH3CN at 25° or 0°C. After the reaction, an additional 20 ml of CH3CN was added to transfer the products and 0.2 ml of toluene was also added as an internal standard. The products were analyzed with Agilent 1260 Infinity HPLC using a Unitary C-18 column. Before the analysis, the products were filtered by a syringe with a filter head.
DFT calculations
DFT calculations were performed using the Vienna Ab-initio Simulation Package (46–48). The projector augmented-wave pseudopotentials and a cutoff energy of 400 eV for the plane-wave basis set were adopted (49, 50). The generalized gradient approximation method with Perdew-Burke-Ernzerhof functionals for the exchange-correlation term was used (51, 52). The Monkhorst-Pack scheme was used for sampling the Brillouin zone (53). The FeN4/GN model was set in a 6 × 6 supercell of graphene (fig. S11A). The vacuum thickness between graphene layers was set as 15 Å to avoid interlayer interactions. The FePc monomer model was set in a 25 × 25 × 16 Å rectangular box (fig. S11B). Spin polarization was considered throughout the calculations. The transition states were searched using the constrained minimization approach (54–56). The free energies (G) of the reactants, surface intermediates, and products were obtained using the equation G = Etotal + ZPE − TS, where Etotal is the total energy of the species, ZPE is the zero point energy, and S is the entropy.
EXAFS analysis
FT EXAFS spectra of the FeN4/GN samples, as well the Fe foil and FePc reference materials, were generated and fitted using WinXAS (57). Scattering paths used in the fitting process were calculated ab initio using FEFF 8.2 (58) and models of the FePc and FeN4/GN structures (fig. S11). The Fe foil spectrum was used to determine an empirical S02 value (0.86), which was then fixed for the fitting of all subsequent samples.
Supplementary Material
Acknowledgments
We thank T. Regier at the CLS for his assistance on XAS measurements and BL14W1 beamline of the SSRF for assistance on XAFS measurements. Funding: This work was supported by the National Natural Science Foundation of China (grant nos. 21321002, 21303191, and 51420105003) and the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA09030100). Author contributions: X.B. and D.D. supervised the work and designed the experiments. D.D. and X. Chen prepared the samples and performed most of the experiments. L.Y. performed the DFT calculations for the catalytic reactions. X.W., T.X., and L.S. performed HRTEM microscopy and simulation. H.Y., H.T., and J.L. performed HAADF-STEM microscopy. Q.L., Y.L., and F.Y. performed STM microscopy. H.L. and J.X. simulated the STM images. Y.H., R.S., P.D., and J.Z. performed XAFS characterization. J.W. performed Mössbauer spectroscopy analysis. P.N.D. and P.Z. performed EXAFS analysis and fitting. X. Cui, J.D., and X.P. helped with the sample preparation and evaluation of catalytic reactions. D.D., X. Chen, and X.B. interpreted the data and wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors at dhdeng@dicp.ac.cn.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/11/e1500462/DC1
Fig. S1. HRTEM images of FeN4/GN-2.7.
Fig. S2. HRTEM image of FeN4/GN-2.7 with the red circles showing some typical single Fe atom positions in the graphene network.
Fig. S3. HAADF-STEM image of FeN4/GN-2.7.
Fig. S4. HRTEM image of FeN4/GN-2.7 with the red circles showing some Fe atoms with different defects in the surroundings.
Fig. S5. XRD patterns of graphite, GN, FeN4/GN-1.5, FeN4/GN-2.7, FeN4/GN-4.0, and FePc.
Fig. S6. Raman spectra of FeN4/GN samples in comparison to their parent materials FePc, GN, and graphite.
Fig. S7. XPS spectra of FePc, FeN4/GN-4.0, FeN4/GN-2.7, and FeN4/GN-1.5.
Fig. S8. Scheme of a proposed mechanism for synthesis of FeN4/GN via a facile ball milling method.
Fig. S9. Low-temperature O2 TPD profiles of FeN4/GN-2.7, GN, and GF.
Fig. S10. The recycling experiments of FeN4/GN-2.7.
Fig. S11. Models of FeN4/GN and the FePc monomer in the DFT calculations.
Fig. S12. Free energy profile of the benzene oxidation reaction intermediates on the iron site of the FePc monomer and FeN4/GN.
Fig. S13. Fe K-edge XANES signal of FeN4/GN samples with H2O2 treatment in comparison to their corresponding original samples.
Fig. S14. The Fe K-edge EXAFS analysis of FeN4/GN samples before and after H2O2 treatment.
Fig. S15. Room-temperature 57Fe Mössbauer spectra of FeN4/GN-2.7, FeN4/GN-2.7-H2O2, and FeN4/GN-2.7-H2O2-Ben.
Table S1. The elemental compositions of FePc, FeN4/GN-4.0, FeN4/GN-2.7, and FeN4/GN-1.5 estimated from XPS and ICP measurements.
Table S2. Catalytic performance of different samples for the direct oxidation of benzene to phenol.
Table S3. Catalytic performance of FeN4/GN-2.7 for the direct oxidation of benzene to phenol with different reaction times.
Table S4. Catalytic performance of different samples for the direct oxidation of benzene to phenol at 0°C.
Table S5. Fitting parameters for the analysis of the EXAFS spectra of FeN4/GN samples with H2O2 treatment in comparison to their corresponding original samples.
Table S6. Fitting parameters for the 57Fe Mössbauer spectra in fig. S15.
REFERENCES AND NOTES
- 1.Meunier B., de Visser S. P., Shaik S., Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 104, 3947–3980 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Kille S., Zilly F. E., Acevedo J. P., Reetz M. T., Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 3, 738–743 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Burgess B. K., Lowe D. J., Mechanism of molybdenum nitrogenase. Chem. Rev. 96, 2983–3012 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Ambundo E. A., Friesner R. A., Lippard S. J., Reactions of methane monooxygenase intermediate Q with derivatized methanes. J. Am. Chem. Soc. 124, 8770–8771 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Ensing B., Buda F., Gribnau M. C. M., Baerends E. J., Methane-to-methanol oxidation by the hydrated iron(IV) oxo species in aqueous solution: A combined DFT and Car–Parrinello molecular dynamics study. J. Am. Chem. Soc. 126, 4355–4365 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Das T. K., Couture M., Ouellet Y., Guertin M., Rousseau D. L., Simultaneous observation of the O–O and Fe–O2 stretching modes in oxyhemoglobins. Proc. Natl. Acad. Sci. U.S.A. 98, 479–484 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kudrik E. V., Afanasiev P., Alvarez L. X., Dubourdeaux P., Clemancey M., Latour J.-M., Blondin G., Bouchu D., Albrieux F., Nefedov S. E., Sorokin A. B., An N-bridged high-valent diiron–oxo species on a porphyrin platform that can oxidize methane. Nat. Chem. 4, 1024–1029 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Kwak J. H., Hu J., Mei D., Yi C.-W., Kim D. H., Peden C. H. F., Allard L. F., Szanyi J., Coordinatively unsaturated Al3+ centers as binding sites for active catalyst phases of platinum on γ-Al2O3. Science 325, 1670–1673 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Panov G. I., Uriarte A. K., Rodkin M. A., Sobolev V. I., Generation of active oxygen species on solid surfaces. Opportunity for novel oxidation technologies over zeolites. Catal. Today 41, 365–385 (1998). [Google Scholar]
- 10.Zecchina A., Rivallan M., Berlier G., Lamberti C., Ricchiardi G., Structure and nuclearity of active sites in Fe-zeolites: Comparison with iron sites in enzymes and homogeneous catalysts. Phys. Chem. Chem. Phys. 9, 3483–3499 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Thomas J. M., The concept, reality and utility of single-site heterogeneous catalysts (SSHCs). Phys. Chem. Chem. Phys. 16, 7647–7661 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Fu Q., Li W.-X., Yao Y., Liu H., Su H.-Y., Ma D., Gu X.-K., Chen L., Wang Z., Zhang H., Wang B., Bao X., Interface-confined ferrous centers for catalytic oxidation. Science 328, 1141–1144 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Fu Q., Yang F., Bao X., Interface-confined oxide nanostructures for catalytic oxidation reactions. Acc. Chem. Res. 46, 1692–1701 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Novoselov K. S., Geim A. K., Morozov S. V., Jiang D., Zhang Y., Dubonos S. V., Grigorieva I. V., Firsov A. A., Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Allen M. J., Tung V. C., Kaner R. B., Honeycomb carbon: A review of graphene. Chem. Rev. 110, 132–145 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Cretu O., Krasheninnikov A. V., Rodríguez-Manzo J. A., Sun L., Nieminen R. M., Banhart F., Migration and localization of metal atoms on strained graphene. Phys. Rev. Lett. 105, 196102 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Wang H., Wang Q., Cheng Y., Li K., Yao Y., Zhang Q., Dong C., Wang P., Schwingenschlögl U., Yang W., Zhang X. X., Doping monolayer graphene with single atom substitutions. Nano Lett. 12, 141–144 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Zhao J., Deng Q., Bachmatiuk A., Sandeep G., Popov A., Eckert J., Rümmeli M. H., Free-standing single-atom-thick iron membranes suspended in graphene pores. Science 343, 1228–1232 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Claessens C. G., Hahn U., Torres T., Phthalocyanines: From outstanding electronic properties to emerging applications. Chem. Rec. 8, 75–97 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Deng D., Yu L., Pan X., Wang S., Chen X., Hu P., Sun L., Bao X., Size effect of graphene on electrocatalytic activation of oxygen. Chem. Commun. 47, 10016–10018 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Sandoval S., Kumar N., Sundaresan A., Rao C. N. R., Fuertes A., Tobias G., Enhanced thermal oxidation stability of reduced graphene oxide by nitrogen doping. Chem. Eur. J. 20, 11999–12003 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Hentsche M., Hermann H., Gemming T., Wendrock H., Wetzig K., Nanostructured graphite prepared by ball-milling at low temperatures. Carbon 44, 812–814 (2006). [Google Scholar]
- 23.Jeon I.-Y., Shin Y.-R., Sohn G.-J., Choi H.-J., Bae S.-Y., Mahmood J., Jung S.-M., Seo J.-M., Kim M.-J., Chang D. W., Dai L., Baek J.-B., Edge-carboxylated graphene nanosheets via ball milling. Proc. Natl. Acad. Sci. U.S.A. 109, 5588–5593 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Immohr S., Felderhoff M., Weidenthaler C., Schüth F., An orders-of-magnitude increase in the rate of the solid-catalyzed CO oxidation by in situ ball milling. Angew. Chem. Int. Ed. 52, 12688–12691 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Gopakumar T. G., Brumme T., Kröger J., Toher C., Cuniberti G., Berndt R., Coverage-driven electronic decoupling of Fe-phthalocyanine from a Ag(111) substrate. J. Phys. Chem. C 115, 12173–12179 (2011). [Google Scholar]
- 26.Ohta N., Arafune R., Tsukahara N., Kawai M., Takagi N., Enhancement of inelastic electron tunneling conductance caused by electronic decoupling in iron phthalocyanine bilayer on Ag(111). J. Phys. Chem. C 117, 21832–21837 (2013). [Google Scholar]
- 27.Gao L., Ji W., Hu Y. B., Cheng Z. H., Deng Z. T., Liu Q., Jiang N., Lin X., Guo W., Du S. X., Hofer W. A., Xie X. C., Gao H.-J., Site-specific Kondo effect at ambient temperatures in iron-based molecules. Phys. Rev. Lett. 99, 106402 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Pham V. D., Lagoute J., Mouhoub O., Joucken F., Repain V., Chacon C., Bellec A., Girard Y., Rousset S., Electronic interaction between nitrogen-doped graphene and porphyrin molecules. ACS Nano 8, 9403–9409 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Zhao L., He R., Rim K. T., Schiros T., Kim K. S., Zhou H., Gutiérrez C., Chockalingam S. P., Arguello C. J., Pálová L., Nordlund D., Hybertsen M. S., Reichman D. R., Heinz T. F., Kim P., Pinczuk A., Flynn G. W., Pasupathy A. N., Visualizing individual nitrogen dopants in monolayer graphene. Science 333, 999–1003 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Kim S., Ohta T., Kwag G., In situ structural investigation of iron phthalocyanine monolayer adsorbed on electrode surface by X-ray absorption fine structure. Bull. Korean Chem. Soc. 21, 588–594 (2000). [Google Scholar]
- 31.Choi H. J., Kwag G., Kim S., Electrochemical and XAFS investigation of nitrite reduction by heat-treated μ-oxo derivative of iron phthalocyanine supported on high area carbon. J. Electroanal. Chem. 508, 105–114 (2001). [Google Scholar]
- 32.Betti M. G., Gargiani P., Frisenda R., Biagi R., Cossaro A., Verdini A., Floreano L., Mariani C., Localized and dispersive electronic states at ordered FePc and CoPc chains on Au(110). J. Phys. Chem. C 114, 21638–21644 (2010). [Google Scholar]
- 33.Calabrese A., Floreano L., Verdini A., Mariani C., Betti M. G., Filling empty states in a CuPc single layer on the Au(110) surface via electron injection. Phys. Rev. B 79, 115446 (2009). [Google Scholar]
- 34.Koch E. E., Jugnet Y., Himpsel F. J., High-resolution soft x-ray excitation spectra of 3d-metal phthalocyanines. Chem. Phys. Lett. 116, 7–11 (1985). [Google Scholar]
- 35.Guo X., Fang G., Li G., Ma H., Fan H., Yu L., Ma C., Wu X., Deng D., Wei M., Tan D., Si R., Zhang S., Li J., Sun L., Tang Z., Pan X., Bao X., Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen. Science 344, 616–619 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Niwa S.-i., Eswaramoorthy M., Nair J., Raj A., Itoh N., Shoji H., Namba T., Mizukami F., A one-step conversion of benzene to phenol with a palladium membrane. Science 295, 105–107 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Tanev P. T., Chibwe M., Pinnavaia T. J., Titanium-containing mesoporous molecular sieves for catalytic oxidation of aromatic compounds. Nature 368, 321–323 (1994). [DOI] [PubMed] [Google Scholar]
- 38.K. Weissermel, H.-J. Arpe, Industrielle Organische Chemie: Bedeutende Vor- und Zwischenprodukte (Wiley-VCH, Weinheim, Germany, 1988). [Google Scholar]
- 39.B. Elvers, S. Hawkins, G. Schulz, Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH, Weinheim, Germany, 2004). [Google Scholar]
- 40.Ding G., Wang W., Jiang T., Han B., Fan H., Yang G., Highly selective synthesis of phenol from benzene over a vanadium-doped graphitic carbon nitride catalyst. ChemCatChem 5, 192–200 (2013). [Google Scholar]
- 41.Yang J.-H., Sun G., Gao Y., Zhao H., Tang P., Tan J., Lu A.-H., Ma D., Direct catalytic oxidation of benzene to phenol over metal-free graphene-based catalyst. Energy Environ. Sci. 6, 793–798 (2013). [Google Scholar]
- 42.Zhang H., Pan X., Han X., Liu X., Wang X., Shen W., Bao X., Enhancing chemical reactions in a confined hydrophobic environment: An NMR study of benzene hydroxylation in carbon nanotubes. Chem. Sci. 4, 1075–1078 (2013). [Google Scholar]
- 43.Parton R. F., Neys P. E., Jacobs P. A., Sosa R. C., Rouxhet P. G., Iron–phthalocyanine immobilized on activated carbon black: A selective catalyst for alkane oxidation. J. Catal. 164, 341–346 (1996). [Google Scholar]
- 44.Dahlberg S. C., Musser M. E., Electron acceptor surface states due to oxygen adsorption on metal phthalocyanine films. J. Chem. Phys. 72, 6706–6711 (1980). [Google Scholar]
- 45.Westre T. E., Kennepohl P., DeWitt J. G., Hedman B., Hodgson K. O., Solomon E. I., A multiplet analysis of Fe K-edge 1s → 3d pre-edge features of iron complexes. J. Am. Chem. Soc. 119, 6297–6314 (1997). [Google Scholar]
- 46.Kresse G., Hafner J., Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993). [DOI] [PubMed] [Google Scholar]
- 47.Kresse G., Hafner J., Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994). [DOI] [PubMed] [Google Scholar]
- 48.Kresse G., Furthmüller J., Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996). [DOI] [PubMed] [Google Scholar]
- 49.Blöchl P. E., Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Kresse G., Joubert D., From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999). [Google Scholar]
- 51.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Perdew J. P., Burke K., Ernzerhof M., Generalized gradient approximation made simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396–1396 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Monkhorst H. J., Pack J. D., Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976). [Google Scholar]
- 54.Alavi A., Hu P., Deutsch T., Silvestrelli P. L., Hutter J., CO oxidation on Pt(111): An ab initio density functional theory study. Phys. Rev. Lett. 80, 3650–3653 (1998). [Google Scholar]
- 55.Michaelides A., Hu P., Insight into microscopic reaction pathways in heterogeneous catalysis. J. Am. Chem. Soc. 122, 9866–9867 (2000). [Google Scholar]
- 56.Liu Z.-P., Hu P., General rules for predicting where a catalytic reaction should occur on metal surfaces: A density functional theory study of C–H and C–O bond breaking/making on flat, stepped, and kinked metal surfaces. J. Am. Chem. Soc. 125, 1958–1967 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Ressler T., WinXAS: A program for x-ray absorption spectroscopy data analysis under MS-Windows. J. Synchrotron Radiat. 5, 118–122 (1998). [DOI] [PubMed] [Google Scholar]
- 58.Ankudinov A. L., Ravel B., Rehr J. J., Conradson S. D., Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B 58, 7565–7576 (1998). [Google Scholar]
- 59.Cançado L. G., Takai K., Enoki T., Endo M., Kim Y. A., Mizusaki H., Jorio A., Coelho L. N., Magalhães-Paniago R., Pimenta M. A., General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 88, 163106 (2006). [Google Scholar]
- 60.Malard L. M., Pimenta M. A., Dresselhaus G., Dresselhaus M. S., Raman spectroscopy in graphene. Phys. Rep. 473, 51–87 (2009). [Google Scholar]
- 61.Deng D., Pan X., Zhang H., Fu Q., Tan D., Bao X., Freestanding graphene by thermal splitting of silicon carbide granules. Adv. Mater. 22, 2168–2171 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/11/e1500462/DC1
Fig. S1. HRTEM images of FeN4/GN-2.7.
Fig. S2. HRTEM image of FeN4/GN-2.7 with the red circles showing some typical single Fe atom positions in the graphene network.
Fig. S3. HAADF-STEM image of FeN4/GN-2.7.
Fig. S4. HRTEM image of FeN4/GN-2.7 with the red circles showing some Fe atoms with different defects in the surroundings.
Fig. S5. XRD patterns of graphite, GN, FeN4/GN-1.5, FeN4/GN-2.7, FeN4/GN-4.0, and FePc.
Fig. S6. Raman spectra of FeN4/GN samples in comparison to their parent materials FePc, GN, and graphite.
Fig. S7. XPS spectra of FePc, FeN4/GN-4.0, FeN4/GN-2.7, and FeN4/GN-1.5.
Fig. S8. Scheme of a proposed mechanism for synthesis of FeN4/GN via a facile ball milling method.
Fig. S9. Low-temperature O2 TPD profiles of FeN4/GN-2.7, GN, and GF.
Fig. S10. The recycling experiments of FeN4/GN-2.7.
Fig. S11. Models of FeN4/GN and the FePc monomer in the DFT calculations.
Fig. S12. Free energy profile of the benzene oxidation reaction intermediates on the iron site of the FePc monomer and FeN4/GN.
Fig. S13. Fe K-edge XANES signal of FeN4/GN samples with H2O2 treatment in comparison to their corresponding original samples.
Fig. S14. The Fe K-edge EXAFS analysis of FeN4/GN samples before and after H2O2 treatment.
Fig. S15. Room-temperature 57Fe Mössbauer spectra of FeN4/GN-2.7, FeN4/GN-2.7-H2O2, and FeN4/GN-2.7-H2O2-Ben.
Table S1. The elemental compositions of FePc, FeN4/GN-4.0, FeN4/GN-2.7, and FeN4/GN-1.5 estimated from XPS and ICP measurements.
Table S2. Catalytic performance of different samples for the direct oxidation of benzene to phenol.
Table S3. Catalytic performance of FeN4/GN-2.7 for the direct oxidation of benzene to phenol with different reaction times.
Table S4. Catalytic performance of different samples for the direct oxidation of benzene to phenol at 0°C.
Table S5. Fitting parameters for the analysis of the EXAFS spectra of FeN4/GN samples with H2O2 treatment in comparison to their corresponding original samples.
Table S6. Fitting parameters for the 57Fe Mössbauer spectra in fig. S15.