

Significance
Chronic stress alters the hippocampal responses to familiar and novel stressors, behaviorally, physiologically, and epigenetically. In the aftermath of chronic stress in WT mice and in mice with a BDNF loss-of-function allele without any applied stress, there is a window of plasticity that allows familiar and novel experiences to alter anxiety- and depressive-like behaviors, reflected also in electrophysiological changes in the dentate gyrus (DG) in vitro. A consistent biomarker of mood-related behaviors in DG is reduced type 2 metabotropic glutamate (mGlu2), which regulates the release of glutamate. Within this window, familiar stress rapidly and epigenetically up-regulates mGlu2 by a P300-driven histone H3 lysine 27 acetylation and improves mood behaviors. This transient epigenetic plasticity may be useful for treatment of stress-related disorders where dysregulaton of glutamate is involved.
Keywords: H3K27, resilience, mGlu2, NMDA, BDNF
Abstract
Excitatory amino acids play a key role in both adaptive and deleterious effects of stressors on the brain, and dysregulated glutamate homeostasis has been associated with psychiatric and neurological disorders. Here, we elucidate mechanisms of epigenetic plasticity in the hippocampus in the interactions between a history of chronic stress and familiar and novel acute stressors that alter expression of anxiety- and depressive-like behaviors. We demonstrate that acute restraint and acute forced swim stressors induce differential effects on these behaviors in naive mice and in mice with a history of chronic-restraint stress (CRS). They reveal a key role for epigenetic up- and down-regulation of the putative presynaptic type 2 metabotropic glutamate (mGlu2) receptors and the postsynaptic NR1/NMDA receptors in the hippocampus and particularly in the dentate gyrus (DG), a region of active neurogenesis and a target of antidepressant treatment. We show changes in DG long-term potentiation (LTP) that parallel behavioral responses, with habituation to the same acute restraint stressor and sensitization to a novel forced-swim stressor. In WT mice after CRS and in unstressed mice with a BDNF loss-of-function allele (BDNF Val66Met), we show that the epigenetic activator of histone acetylation, P300, plays a pivotal role in the dynamic up- and down-regulation of mGlu2 in hippocampus via histone-3-lysine-27-acetylation (H3K27Ac) when acute stressors are applied. These hippocampal responses reveal a window of epigenetic plasticity that may be useful for treatment of disorders in which glutamatergic transmission is dysregulated.
Stress effects on higher brain regions, such as hippocampus, are known to involve actions of excitatory amino acids to induce structural and functional changes depending upon the type, intensity, and duration of the stressor (1). These differential responses, including determining susceptibility versus resilience to stress, contribute to the pathophysiology of debilitating stress-related disorders (2–5). The hippocampus is a brain region noted for its plasticity in response to stress and sensitivity to adrenal steroid hormones (6). Acute stress enhances synaptic plasticity that is associated with improved cognition and other adaptive functions whereas chronic stress produces opposite effects mediating, in the hippocampus, spine synapse turnover, dendritic shrinkage, impaired long-term potentiation (LTP), and suppression of adult neurogenesis in the dentate gyrus (DG) (1, 7). Importantly, neuroanatomical changes in response to repeated stress recover in young adult animals, based upon the restoration of dendritic length and branching and spine density (8). However, there are underlying changes that can be seen at the level of gene expression and epigenetic regulation that indicate that the brain is continually changing (9, 10). Epigenetic modifications, such as acetylation of histones, have also been involved in the consolidation of contextual memories that allow the brain to respond and adapt to changes in the environment (11).
Among the large number of mediators of brain structural and functional plasticity, the glutamatergic system and BDNF play a key role in mediating the effects of stress on both cognition and psychopathology (1, 12–15). Animal models have shown that chronic stress effects on dendritic remodeling are blocked by blocking NMDA receptors (1, 16) and that adrenalectomy attenuates the acute stress-induced elevations of extracellular glutamate levels in the hippocampus (1, 17). More recently, presynaptic type 2 metabotropic glutamate (mGlu2) receptor, an inhibitor of synaptic glutamate release, has been identified as a marker of stress susceptibility (2) and as a target for novel rapidly acting antidepressants (18–20), such as acetyl-l-carnitine (LAC). LAC rapidly up-regulated mGlu2 in the hippocampus, along with correcting depressive-like behaviors (18–20). LAC also elevated BDNF levels in a genetic animal model of depressive-like behavior (18). From a translational standpoint, BDNF signaling is also relevant. For example, 33% of the human population present a BDNF Val66Met SNP that leads to a valine-to-methionine substitution in the BDNF protein at codon 66, and this SNP has been associated with increased susceptibility to development of stress-related disorders (21, 22).
Because the brain is continually changing its gene expression with experience (10), we wanted to know how a history of stress alters—behaviorally, physiologically, and epigenetically—the responsivity to subsequent life stressful events, conferring a differential susceptibility to psychopathologies. We investigated in depth histone-3-lysine-27-acetylation (H3K27ac), a consistent biomarker of mood-related behaviors and antidepressant action (18–20). We demonstrate that a history of chronic stress alters the response to familiar (known) and unfamiliar (unknown) stressors, such that familiar acute restraint stress (ARS) and a novel acute forced swim stress (AFS) have differential effects upon the putative presynaptic mGlu2 and postsynaptic NR1/NMDA receptors in the hippocampus of naive and chronically stressed mice. These effects are strongest in the dentate gyrus (DG), a region of active neurogenesis and target of antidepressant treatment. Consistent with the responses of these markers to rapid-acting antidepressants (18), these molecular responses are consistently associated with changes in anxiety- and depressive-like behaviors. We also show that DG-LTP is involved in the memory formation associated with habituation to the same stressor (ARS) and sensitization to a novel stressor (AFS). Finally, using both WT BDNF Val66Val and BDNF Val66Met transgenic mice, we demonstrate that the epigenetic regulator of gene transcription, P300, has a pivotal role in the dynamic up- and down-regulation of mGlu2 expression in the hippocampus in the course of the continuously adapting responses of the brain to the environment.
Results
Differential Responses of the Hippocampus to Novel and Familiar Stressors in Chronically Stressed Mice.
Because excessive glutamatergic activity is a key mediator of stress-induced changes in hippocampal morphology and function (1), we evaluated the effects of stress upon key glutamate genes based on the type, intensity, and duration of the stress. We first evaluated the impact of prolonged 21 d of chronic-restraint stress (CRS) on the presynaptic mGlu2 receptor and on the obligatory subunit NR1 of the NMDA receptor, targets for novel rapidly acting antidepressants (18–20, 23). As expected from previous work (24), 21-d CRS, which leads to dendritic remodeling in the CA3 hippocampus (6), resulted in a significant increase in adrenal-to-body weight ratio compared with nonstressed mice (Fig. S1) that is consistent with elevated glucocorticoid production contributing to dendritic remodeling (13). Interestingly, 21-d CRS significantly decreased the transcription of the putative presynaptic mGlu2 receptor (Fig. 1A), an inhibitor of glutamate release that recently has been identified as a marker of mood-related behavior (2, 18). There was also a trend to decrease the mRNA for the NR1 of NMDA receptors in the hippocampus (Fig. 1B). Thus, transcriptional CRS effects in the hippocampus imply a strong increase in glutamatergic activity in line with previous evidence showing that stress-increased extrasynaptic glutamate contributes to structural changes in the CA3 hippocampus (1, 6).
Fig. S1.
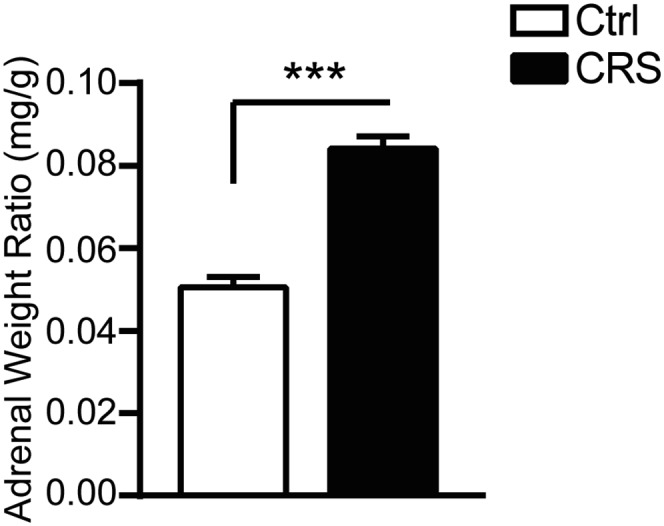
Prolonged 21-d CRS, which we know leads to dendritic remodeling in CA3 hippocampus, results in a significant increase in adrenal-to-body weight ratio compared with nonstressed mice. Bars represent mean + SEM, and * indicates significant comparisons with corresponding controls, ***P < 0.0001.
Fig. 1.

Differential responses of the hippocampus to novel and familiar stressors in chronically stressed mice. (A and B) The 21-d CRS decreased the transcription of presynaptic mGlu2 receptors, inhibitors of synaptic glutamate release, and resulted in a nonsignificant decrease in the transcription of the obligatory subunit NR1 of NMDA receptors in hippocampus. Two hours after AFS in CRS mice, mGlu2 transcripts were still down-regulated whereas hippocampal NR1/NMDA transcripts were significantly up-regulated. Conversely, 2 h after a known ARS in CRS mice, we observed no significant change in NR1/NMDA mRNA levels, but the CRS-induced decrease in mGlu2 in hippocampus was temporarily reversed [mGlu2, F3,20 = 9.75, P < 0.001 (stress); NR1/NMDA, F3,14 = 15.2, P < 0.001 (stress)]. (C) Immunohistochemistry analyses for mGlu2 expression in the subregions of the hippocampal formation (DG, CA3, and CA1) showed a subregional localization of CRS effects within the DG: CRS induced a large decrease in mGlu2 immunoreactivity in the DG, with no change in the CA3 and CA1. However, 2 h after a known ARS in CRS mice, the CRS-induced decrease in mGlu2 expression was temporarily reversed in the DG hippocampus, with no change in the CA3 and CA1 hippocampus. Conversely, 2 h after an unknown AFS in CRS mice, mGlu2 expression was still down-regulated in the DG, with no change in the CA3 and CA1 hippocampus [mGlu2, DG, F3,71 = 5.22, P < 0.01 (stress); CA3, F3,66 = 1.26; CA1, F3,65 = 1.67)]. (D) No significant change was observed, after CRS, in NMDA immunoreactivity within the hippocampal subregions (DG, CA3, and CA1). Two hours after a known ARS in CRS mice, we observed no significant change in NMDA expression within the hippocampal subregions whereas, 2 h after an unknown AFS in CRS mice, NMDA expression was increased in the DG, CA3, and CA1 hippocampus [NMDA, DG, F3,35 = 4.82, P < 0.01 (stress); CA3, F3,35 = 6.03, P < 0.01; CA1, F3,30 = 6.29, P < 0.01]. (E) Representative 10× images of hippocampal sections of NMDA immunoreactivity from unstressed mice (Top Left), CRS mice (Top Right), 2 h after ARS in CRS mice (Bottom Left) and 2 h after AFS in CRS mice (Bottom Right). Bars represent mean + SEM, and * indicates significant comparisons with corresponding controls, *P < 0.05, **P < 0.01.
This plasticity of the glutamate system in the hippocampus that can either increase or decrease the potential for excitoxic damage (6, 25) also affects behaviors, including those related to anxiety and depression. To determine how a chronic stress history alters the brain ability to respond to an acute stress, we measured the levels of mGlu2 and NR1/NMDA transcripts in mice that were chronically stressed for 21 d and then subjected to an episode of either acute novel forced swim stress (AFS) or acute familiar restraint stress (ARS) on day 22 (Fig. S2).
Fig. S2.

Stress challenge. The schematic shows the time course of the stress challenge procedure and behavioral/molecular analyses in chronically stressed mice.
CRS for 21 d altered the response to both AFS and ARS within the hippocampus, but in different ways: 2 h after AFS in CRS mice, mGlu2 transcripts were still down-regulated whereas hippocampal NR1/NMDA transcripts showed a threefold up-regulation (Fig. 1 A and B), pointing to increased glutamatergic responsiveness in CRS mice exposed to AFS, which does not have this effect in naive mice (Fig. S3 A and B). Interestingly, 2 h after the familiar ARS, which, in naive mice, resulted in changes in mGlu2 and NR1/NMDA receptors (Fig. S3 A and B), we found no change in NR1/NMDA mRNA levels, but the CRS-induced decrease in mGlu2 mRNA in the hippocampus was, at least temporarily, reversed (Fig. 1A). Thus, we found opposite changes in mGlu2 in CRS mice after exposure to AFS and ARS, with an up-regulation of mGlu2 in CRS mice after exposure to the familiar ARS, but not after exposure to novel AFS (Fig. 1), thus further elaborating metaplastic changes of the glutamatergic system in stress responses (26).
Fig. S3.

Effects of acute restraint (ARS) and acute forced swim (AFS) stressors in naive mice. ARS, which results in no change in the immobility time at the tail-suspension test [in A, TST, F2,21 = 0.17 (stress)], but in decreased time spent in a light chamber of a light–dark box [in B, LDT, F2,21 = 3.97, P < 0.05 (stress)], decreases mGlu2 mRNA transcripts in hippocampus (C) and has no significant effect in NMDA transcripts (D). Instead, AFS results in no significant change in presynaptic mGlu2 and postsynaptic NMDA transcripts (C and D) and in no significant change at the LDT and TST in naive mice (A and B). [mGlu2, F2,21 = 4.92, P < 0.05 (stress); NMDA, F2,12 = 3.87, P < 0.05 (stress).] Bars represent mean + SEM, and * indicates significant comparisons with corresponding controls, *P < 0.05.
Neuroanatomical Specificity of mGlu2 and NMDA Changes.
To gain insights into the regional localization of the stress-induced changes in presynaptic mGlu2 and postsynaptic NMDA receptors and to explore whether the different responses to acute known or unknown stressors in CRS mice are reflected at the protein level, we measured mGlu2 and NMDA expression in the subregions of the hippocampal formation (DG, CA3, and CA1). Immunohistochemistry analyses showed that CRS induces large decreases in mGlu2 immunoreactivity in the DG, with no change in the CA3 and CA1 (Fig. 1C). No significant change was observed, after CRS, in NMDA expression within the hippocampal formation (Fig. 1 D and E). With regard to the familiar and novel acute stressors in CRS mice, 2 h after ARS in CRS mice, NMDA receptor expression was not altered within the hippocampal subregions (Fig. 1 D and E), and the CRS-induced decrease in mGlu2 expression in the DG hippocampus was, at least temporarily, reversed with no change in the CA3 and CA1 hippocampus (Fig. 1C). Conversely, 2 h after AFS in CRS mice, we observed a significant increase in NMDA expression in the DG, CA3, and CA1 hippocampus (Fig. 1 D and E) whereas mGlu2 is still depressed in the DG hippocampus, showing agreement between mRNA and protein levels and suggesting also that the DG, a zone of active neurogenesis, may play a key role in these response to stressors.
Prior Chronic Stress History Alters the Behavioral Reactivity to Future Stressors.
Because the hippocampus mediates contextual and temporal aspects of stress-related memory, as well as mood-related behaviors, we next investigated how the stress challenges affect behavioral responses. Two hours after AFS, which did not result in depressive-like behavior in naive mice (Fig. S3D), we observed a worsened depressive-like behavior in CRS mice (Fig. 2A). Thus, the chronic stress history (CRS) altered the brain’s ability to respond to a novel acute stressor, and this response was associated with glutamatergic hyperactivity of the hippocampus based on the strong elevation of NR1/NMDA receptors and on the down-regulated mGlu2 receptors (Fig. 1) that inhibit glutamate release. On the other hand, 21-d CRS opens a “window of plasticity” that enables the coping response to a single, familiar ARS on day 22, as shown by decreased immobility time in the tail suspension test (TST) (Fig. 2A) 2 h after ARS in CRS mice. This, at least transient, “reversal effect” was associated with increased mGlu2 expression in the hippocampus and a reduction in glutamatergic activity.
Fig. 2.

Prior chronic stress history alters the behavioral reactivity to future stressors. (A) Immobility time at the tail suspension test (TST) in nonstressed age-matched mice and in mice after chronic restraint stress (21-d CRS) as well as in CRS mice 2 h after either an additional known ARS or an additional unknown AFS. The 21 d of CRS resulted in an increased immobility time at the TST. An unknown AFS, which did not result in depressive-like behavior in naive mice (Fig. S3), worsened, in 21-d CRS mice, the depressive-like behavior (i.e., more depressive-like behavior, thus sensitization). Conversely, 2 h after a known ARS in CRS mice, immobility time at the TST was decreased (i.e., less depressive-like behavior, and thus an habituation and reversal effect) [TST, F3,25 = 21.58, P < 0.0001 (stress)]. (B) The 21-d CRS mice showed different responses to the AFS and ARS at the light–dark test (LDT) because, 2 h after ARS in CRS mice, time spent in the light chamber was increased (i.e., less anxious, and thus a habituation and reversal effect), but time spent in the light chamber was reduced 2 h after AFS in CRS mice (i.e., more anxious, and thus sensitization) [LDT, F3,25 = 18.03, P < 0.0001 (stress)]. Bars represent mean + SEM, and * indicates significant comparisons with corresponding controls, *P < 0.05, **P < 0.01, ***P < 0.0001.
To further explore whether ARS in CRS mice at least temporarily reverses CRS-induced mood abnormalities and to learn how a previous stress history (CRS) alters the brain’s reactivity to unknown stressors, we assessed an anxiety-like behavior in 21-d CRS mice 2 h after an episode of either AFS or ARS using the light–dark test (LDT) (27). The 21-d CRS mice spent more time in the light chamber when subjected to the familiar ARS (i.e., less anxious), but less time in the light chamber (i.e., more anxious) when subjected to a novel AFS (Fig. 2B).
Novel and Familiar Stressors Have Different Effects on Synaptic Plasticity After 21-d CRS.
Because stress hormones with their synergistic interaction with glutamate are suggested to mediate negative and positive influence on memory through induction of long-term potentiation (LTP) (1, 28), we evaluated the effects of either a novel AFS or a familiar ARS on day 22 after 21 d of CRS on basal synaptic transmission and DG-LTP induced by high-frequency stimulation (HFS). We found that hippocampal slices from CRS mice exhibited significantly reduced maximum field excitatory postsynaptic potentials (fEPSPs) compared with nonstressed mice, particularly when the presynaptic afferents were stimulated at higher intensities (Fig. 3A). HFS to the medial perforant pathway produced significantly lower LTP in CRS mice (133.5 ± 5.4%) compared with nonstressed controls (176.3 ± 6.7%) (Fig. 3B). Thus, CRS significantly reduced basal synaptic transmission and DG-LTP (Fig. 3). In hippocampal slices from CRS mice that experienced the novel AFS, DG-LTP continued to be suppressed (128.0 ± 7.2%). In contrast, the familiar ARS increased DG-LTP in hippocampal slices from 21-d CRS mice (176.5 ± 13.8%) (Fig. 3B). Thus, the familiar ARS applied in CRS mice, at least transiently, reversed the CRS-induced decrease in DG-LTP that may represent a habituation memory for familiarity while promoting sensitization to the novel AFS (29). Moreover, the persistence of the different electrophysiological responses to the familiar ARS vs. the novel AFS in vitro in slices of CRS hippocampus indicates a metastable change at the cellular level.
Fig. 3.

Novel and familiar stressors have different effects on synaptic plasticity after 21-d CRS. (A and B) Hippocampal slices from CRS mice exhibited significantly reduced maximum fEPSPs compared with nonstressed mice, particularly when the presynaptic afferents were stimulated at higher intensities. Also, HFS to the medial perforant pathway produced significantly lower DG-LTP in CRS mice compared with nonstressed controls. In hippocampal slices from CRS mice after an unknown AFS, DG-LTP was still reduced whereas DG-LTP was increased in hippocampal slices from CRS mice after a known ARS, suggesting that an additional known ARS, but not a subsequent unknown AFS, applied in CRS mice at least transiently reversed CRS-induced decrease in DG-LTP [LTP, F3,25 = 21.6, P < 0.0001 (stress)].
P300 Regulates the mGlu2-Driven Window of Plasticity Opened by a Familiar Stressor in the Hippocampus After CRS.
To investigate whether the “window of plasticity” (opened by CRS in response to the familiar ARS that is able to reverse, within 2 h, the behavioral and molecular effects of CRS) is a transient or a permanent effect, we used the tail suspension test (TST) in CRS mice to show that, 2 h after ARS, they do not show depressive-like behavior. Indeed, we showed that CRS mice with an additional ARS and 24-h recovery showed a markedly higher immobility time in the TST (depressive-like behavior) (Fig. 4A). Furthermore, this behavioral recurrence of depressive-like behavior is concomitant with a reappearance of down-regulation of mGlu2 mRNA and an immunocytochemical signal in the DG-hippocampus of CRS mice 24 h after the ARS (Fig. 4B and Fig. S4). Thus, the reversal effect at 2 h post-ARS in 21-d CRS mice reveals a temporary “window of plasticity” that might be capitalized for therapeutic purposes.
Fig. 4.

P300 regulates the mGlu2-driven window of plasticity opened by stress in the hippocampus of CRS and BDNF Val66Met mice. (A) At 24 h after an additional ARS in CRS mice, immobility time at the TST was once again significantly increased (reappearance of depressive-like behavior) compared with age-matched controls, suggesting that the reversal effect observed 2 h after ARS in CRS mice was transient [TST, F3,16 = 9.65, P < 0.001 (stress)]. (B) Concomitantly, mGlu2 and P300 transcripts were once again down-regulated 24 h after ARS in CRS mice [mGlu2, F3,15 = 11.74, P < 0.001 (stress); P300, F3,19 = 32.01, P < 0.0001 (stress)]. (C) Chromatin immunoprecipitation assay (ChIP) showed that the CRS-induced decrease in the levels of acetylated H3K27 bound to the Grm2 promoter (which encodes for mGlu2 receptors) was rapidly reversed 2 h after ARS in CRS mice whereas levels of H3K27ac bound to the Grm2 promoter were once again down-regulated 24 h after ARS in CRS mice. (H3K27Ac, F3,20 = 6.54, P < 0.01 (stress)]. (D) P300 regulates acetylation of the lysine K27 on the histone H3 bound to the Grm2 promoter, which regulates expression of the mGlu2, to control stress responses. (E) A subset of susceptible heterozygous BDNF Met mice, identified at the light–dark test as anxiety-prone mice (Fig. S5), showed increased immobility time at the TST compared with age-matched male homozygous BDNF Val mice. The immobility time, in heterozygous BDNF Met mice, measured at the TST, was significantly reduced 2 h after ARS (i.e., less depressive-like behavior) whereas it was once again significantly increased 24 h after ARS (i.e., more depressive-like behavior) [TST, F3,36 = 7.22, P < 0.001 (stress)]. (F) The susceptible heterozygous BDNF Met mice showed reduced hippocampal mGlu2 and P300 levels compared with age-matched male homozygous BDNF Val mice. Two hours after ARS, mGlu2 and P300 levels were rapidly up-regulated in heterozygous BDNF Met mice whereas they were once again down-regulated 24 h after ARS [mGlu2, F3,13 = 36.29, P < 0.0001 (stress); P300, F3,14 = 13.2, P < 0.001 (stress)]. (G) ChIP showed that the susceptible heterozygous BDNF Met mice have reduced hippocampal levels of H3K27ac bound to the Grm2 promoter compared with age-matched male homozygous BDNF Val mice. At 2 h after ARS, levels of H3K27ac bound to the Grm2 promoter were rapidly up-regulated in heterozygous BDNF Met mice whereas they were once again down-regulated 24 h after ARS [H3K27Ac, F3,15 = 18.38, P < 0.0001 (stress)]. Bars represent mean + SEM, and * indicates significant comparisons with corresponding controls, *P < 0.05, **P < 0.01, ***P < 0.0001.
Fig. S4.

Immunohistochemistry analyses for mGlu2 expression in the subregions of the hippocampal formation (DG, CA3, and CA1). At 24 h after ARS in CRS mice, mGlu2 was once again down-regulated in the DG hippocampus and no significant difference was observed in the CA3 and CA1 hippocampal subregions [mGlu2, DG, F3,76 = 6.42, P < 0.001 (stress); CA3, F3,63 = 1.57; CA1, F3,64 = 1.55]. Bars represent mean + SEM, and * indicates significant comparisons with corresponding controls, *P < 0.05, **P < 0.01.
Recently, we discovered that the more anxiety-prone naive C57bl mice, revealed by a light–dark screening method, have higher hippocampal mineralocorticoid (MR) levels and respond to ARS with a robust down-regulation of mGlu2 receptors in the hippocampus (2), the up-regulation of which is mediated by the histone acetyl-transferase (HAT) P300. Chromatin immunoprecipitation (ChIP) assays showed that ARS induced a decrease of P300 that was accompanied by decreased acetylation on the H3K27 bound to Grm2 promoter, which regulates expression of the mGlu2 receptor (18). Here, we determined whether a change in P300 levels is involved in the regulation of the transient effect induced by ARS in CRS. Although 21-d CRS down-regulated P300 as well as mGlu2 mRNA levels, the application of ARS to 21-d CRS mice increased P300 hippocampal transcripts 2 h after ARS (Fig. 4B). As predicted, 24 h after ARS in 21-d CRS mice, mGlu2 mRNA levels were again down-regulated in the hippocampus (Fig. 4B). As noted, the mGlu2 elevation 2 h after ARS in CRS mice is associated with decreased immobility time in the TST (i.e., less depressive-like behavior) and with elevated immobility time 24 h later (Fig. 4A). Consistently, 24 h after ARS in CRS mice, P300 and mGlu2 mRNA levels were once again lower in the hippocampus, showing, again, the tight association with the biphasic changes observed in mGlu2 transcripts and the epigenetic mechanism for its regulation in the hippocampus.
Because P300 is a regulator of acetylation of histone-3-lysine-27-acetylation (H3K27ac), an epigenetic marker that has been correlated with transcriptional activation (30) and a consistent marker of antidepressant action (18–20), we performed ChIP for the H3K27ac bound to the promoter of the Grm2 gene, which encodes for mGlu2 receptors. In keeping with the finding that many epigenetic markers are rapidly regulated and transient (30), we found that the CRS-induced decrease in H3K27ac in the Grm2 promoter was rapidly reversed 2 h after ARS in CRS mice and was once again down-regulated 24 h after ARS in CRS mice (Fig. 4C). This dynamic epigenetic regulation of the mGlu2 promoter driven by H3K27ac in our mouse model is consistent with previous findings showing a key role of the same marker H3K27ac in rat studies in the control of mGlu2 transcription (18).
Further Support for the P300 Role in the Control of the mGlu2-Driven “Window of Epigenetic Plasticity” Opened by Stress in Heterozygous BDNF Val66Met Mice.
Next, we evaluated the occurrence of this window of epigenetic plasticity also in the humanized mouse model of BDNF loss-of-function (BDNFVal66Met). The Val66Met SNP is carried by 33% of the human population and has been associated with the development of neuropsychiatric disorders (21, 22). Here, we found that a subset of naive heterozygous BDNFVal66Met mice, identified at the light–dark screening method as particularly anxiety-prone (2) (Fig. S5), showed increased immobility time in the TST (Fig. 4E) along with reduced hippocampal P300 levels (Fig. 4F) that led to a decrease in the acetylation of H3K27 bound to the Grm2 promoter as shown by ChIP (Fig. 4G). In turn, CRS-decreased acetylation of H3K27 led to a decreased transcription for mGlu2 in the hippocampus (Fig. 4F). However, 2 h after ARS, without any prior history of CRS, heterozygous BDNFmet mice showed reduced immobility time at the TST (i.e., less depressive-like behavior) (Fig. 4E). As was the case for WT mice given CRS, this enhanced plasticity 2 h after ARS was accompanied by a rapid up-regulation of P300 transcript levels in the hippocampus (Fig. 4F) that increases transcription of mGlu2 receptors through acetylation of H3K27 (Fig. 4 F and G). Moreover, 24 h after ARS in heterozygous BDNFVal66Met mice, the transient increase was reversed, with increased immobility time in the TST, along with reduced P300 transcripts and H3K27ac levels in the hippocampus and down-regulation of mGlu2 transcripts (Fig. 4 E–G). Thus, ARS opens the window of plasticity in BDNFVal66Met mice, as well as in CRS-WT mice.
Fig. S5.

Individual differences in heterozygous BDNF Met mice. The use of the light–dark test as a screening method (A and B) allowed identification of clusters of animals with a different baseline anxiety profile. The susceptible and anxiety-prone heterozygous BDNF Met mice (about 70%) that are characterized by a lower time spent in a light chamber (A) of a light–dark box (B) compared with the subgroup of low susceptible heterozygous BDNF Met mice (about 30%), which spent higher time in the light chamber.
Discussion
We have uncovered mechanisms of epigenetic plasticity of the hippocampus in response to acute and chronic stressors, revealing that the putative presynaptic mGlu2 receptors and the postsynaptic NR1/NMDA receptors are dynamically regulated in the hippocampus in relation to anxiety- and depressive-like behaviors. Our findings demonstrate that a history of chronic stress determines the responses to future familiar or novel acute stressors. Furthermore, we showed that stress-induced changes in DG-LTP reveal memory-like plasticity, with habituation to the same stressor (ARS) and sensitization to the novel stressor (AFS). Using both WT mice and a humanized mouse model of reduced BDNF function (Val66Met SNP), we found that the epigenetic activator of histone acetylation, P300, plays a pivotal role in the dynamic up- and down-regulation of mGlu2 expression in the hippocampus in response to chronic and acute novel and familiar stressors. This dynamic reaction of the hippocampus reveals a window of epigenetic plasticity, a temporary timeframe of dynamic neuroplasticity, in response to stress that could allow interventions to rapidly promote resilience through regulation of acetylation of histones.
Glutamatergic Regulation.
CRS for 21 d in naive mice causes potentially maladaptive neural responses with no significant change in NR1/NMDA receptors and a decrease in mGlu2 expression in the DG hippocampus, both in terms of mRNA levels and immunocytochemistry for the receptors. Therefore, a paradigm of 21-d CRS, which results in increased immobility time in the TST (depressive-like behavior) and decreased time in the light chamber of a light–dark box (anxiety-like behavior), alters glutamate homeostasis in the DG hippocampus. The strength of changes of mGlu2 expression in the DG points to a region of active neurogenesis and a target of antidepressant treatment (31). Indeed, reduced activity of mGlu2, an inhibitor of glutamate release, would lead to increased glutamatergic activity, which inhibits neurogenesis (32). It is noteworthy that neither ARS nor AFS in naive animals resulted in increased immobility time in the TST whereas 21-d CRS was needed for this effect. In the present study, AFS in naive animals resulted in no significant behavioral changes at the LDT (Fig. S3C) whereas, as reported recently, ARS results in anxiety-like behavior in susceptible mice characterized by an MR-driven down-regulation of mGlu2 receptors in the hippocampus, suggesting that higher MR in susceptible mice may be the result of an early life history of stress (2).
History of Chronic Stress.
Does a history of chronic stress alter the brain’s ability to respond appropriately to an acute stress challenge? Our findings demonstrate the importance of a stress history in determining the response to future familiar or novel stressors. Here, we show that a history of stress (21-d CRS) alters the reactivity to future stressors that may be, at least temporarily, adaptive for known stressors (ARS) or may sensitize to unknown stressors (AFS). CRS mice exposed to a novel AFS were sensitized and showed more depressive- and anxiety-like behaviors, which were not observed in naive animals after AFS. These behavioral outcomes were accompanied by a glutamatergic hyperactivity of the hippocampus based on the strong elevation of NR1/NMDA receptors and on the continued down-regulation of mGlu2 receptors, which normally inhibit glutamate release. We detected these changes in mGlu2 and NR1 receptors at both the mRNA and immunocytochemical levels.
On the other hand, 21-d CRS altered the ability to respond to an additional familiar ARS, as shown by the transiently decreased immobility time at the TST and increased time spent in the light chamber of a light–dark box. This “reversal effect” is transient and is associated with an increase in hippocampal mGlu2 expression, which would reduce glutamate overflow.
Recently, mGlu2 receptors have been identified as markers of mood-related behavior and individual responsiveness to stress (2, 18); thus, it is noteworthy that we found an opposite trend in mGlu2 expression in CRS mice after exposure to AFS and ARS, with an up-regulation of mGlu2 in CRS mice after exposure to the familiar ARS, but not after exposure to the novel AFS. Previous studies showed that prior daily exposure to the same stressor confers protection from some effects of acute superimposed stressors (29). In the present study, our stress challenge paradigm may have induced a form of metaplasticity in the responses of the glutamate mGlu2 and NMDA receptors through mechanisms of adaptation that promote increased DG-LTP that we found in the response of CRS mice to the familiar ARS (33). The metaplasticity we describe is similar to that reported to enhance the neuronal environment for learning and memory through increased late-phase LTP in animals trained on memory tasks (34).
Conversely, after the novel AFS, CRS mice showed decreased neuronal responsiveness and lower DG-LTP. How these responses may be related to altered learning, and learning ability remains to be determined. Future studies should address whether the large up-regulation of the essential subunit NR1 of NMDA receptors after AFS in CRS mice is a metaplastic effect induced by stress that helps to keep synaptic efficacy in the appropriate state for learning and memory processes to adapt to future stressful life episodes or whether the huge up-regulation in NR1/NMDA receptors is a maladaptive response that could lead to permanent damage if not appropriately treated.
Role of P300 in a Window of Epigenetic Plasticity Unveiled by Stress.
Recently, we discovered that, in susceptible, naive individuals with higher MR levels, ARS induces anxiety-like behavior through a down-regulation of mGlu2 receptors in the hippocampus mediated by the HAT P300 (2). Chromatin immunoprecipitation assays showed that the decrease of P300 expression decreases acetylation of the lysine K27 on the histone H3 bound to the Grm2 promoter, which regulates expression of the mGlu2 receptor (18). In the present study, we found that an additional known ARS applied to CRS mice at least transiently reverses the CRS effects, showing that the brain and, more specifically, the hippocampus are continuously adapting to the environment. Here, we report that the transient reversal effect induced by ARS in CRS animals is driven by a dynamic and transient up-regulation of P300 leading to increased acetylation of histone H3K27 and increased expression of mGlu2 in the hippocampus (Fig. 4D). Interestingly, heterozygous BDNFVal66Met mice without any chronic stress history react similarly to WT BDNFVal66Val mice with a history of CRS, showing the same transient up-regulation of mGlu2 mediated by the same mechanism accompanied by reduced anxiety- and depressive-like behavior. Therefore, ARS in CRS mice and ARS applied to heterozygous BDNF Val66Met mice open a window of epigenetic plasticity in the hippocampus in terms of regulation of mGlu2 expression driven by P300, revealing epigenetic mechanisms through which the brain is continuously adapting to external influences.
Translational Implications.
The window of epigenetic plasticity offers opportunity for behavioral and pharmacological interventions that can increase resilience by correcting imbalances of excitatory transmission through regulation of gene transcription via histone modifications and related epigenetic alterations. Thereby, this window of epigenetic plasticity can be capitalized by behavioral and pharmacological interventions to reestablish balanced neural circuitry in the hippocampus, prefrontal cortex, and amygdala that become unbalanced in stress-related disorders (3, 35–38). Pharmacological interventions may include acetyl-l-carnitine (LAC), a donor of acetyl groups that has been shown to rapidly counteract depressive-like behavior and rectify glutamate dysregulation (18). Behavioral and pharmacological interventions that are rapid may be useful in relation to suicide prevention because they can quickly alleviate depression, counteract impulsiveness, and improve self-regulatory ability (39, 40). Furthering our understanding of how stress affects glutamate homeostasis, altering glutamate pre- and postsynaptic receptors, will ultimately contribute to improved therapeutics for more rapidly promoting resilience through more balanced cognitive function and decision making.
Methods
All procedures were carried out in accordance with the National Institutes of Health, The Rockefeller University Institutional Animal Care and Use Committee guidelines, and the European (86/609/EEC) and Italian (D.Lgs 116/92) guidelines of animal care. See SI Methods for details of stress procedure, behavioral tests (tail suspension, light–dark screening method), gene expression protocol, ChIP procedure and promoters’ primers, immunohistochemistry protocol, densitometric analysis, and electrophysiology.
SI Methods
All procedures were carried out in accordance with the National Institutes of Health, The Rockefeller University Institutional Animal Care and Use Committee guidelines and the European (86/609/EEC) and Italian (D.Lgs 116/92) guidelines of animal care. All efforts were made to minimize animal suffering.
Animals.
Male C57black (7-wk-old, 20–25 g; Charles River) and BDNF Val66Met mice were housed five per cage under controlled conditions (12-h light/dark cycle, 22 °C, food and water ad libitum) and individually gently handled daily for 1 wk before the beginning of the stress procedures. The 21-d CRS resulted in a significant increase in adrenal-to-body weight ratio compared with nonstressed mice (Fig. S1) that is consistent with elevated glucocorticoid production as a contributor to dendrite remodeling. An n of 4–8 animals per group was used for the molecular analysis. An n of 6–9 animals per group was used for the behavioral experiments.
Stress Challenge Procedure.
We devised an acute stress challenge (Fig. S2) in chronically stressed mice. Naive mice were stressed chronically for 21 consecutive days and then subjected to an episode of either acute novel forced swim stress (AFS) or acute familiar restraint stress (ARS) on day 22 (Fig. S2). Effects of chronic stress were assessed 24 h after the last episode of stress (CRS); effects of acute stressors in CRS mice were assessed either 2 h after the acute stressors (CRS+ARS, CRS+AFS) or 24 h after the acute stressors (CRS+ARS+Rec). The stress challenge paradigm allowed us to evaluate the acute and chronic stress effects and whether the brain response to acute stressors is modified by a previous period of chronic stress. The whole stress procedure is schematized in Fig. S2. The restraint device contained two 0.4-cm air holes and allowed mice to stretch their legs but not to move within the tube. Naive age-matched not-stressed animals were used as controls. For the chronic stress procedure, mice were placed in the restrainers for 2 h per day for 21 consecutive days. Successively, additional groups of chronically restraint-stressed mice were subjected to either an acute novel stress in the form of a forced swim stressor or an acute known stressor in the form of restraint stress on day 22 after 21 d of chronic restraint stress. The swim stress was performed placing individually each mouse into a vertical glass cylinder (height, 25 cm; diameter, 12 cm) filled with 12-cm-deep water (23–24 °C) for 6 min, followed by 2 h or 24 h recovery in the home cage before behavioral and molecular assessment and killing (15 min after behavioral/molecular assessment).
Chromatin Immunoprecipitation.
ChIP was performed as previously described (2, 18) using the Ez-Magna ChIP kit (Millipore) with a few modifications to adapt the protocol to mouse brain tissue. Hippocampal tissue was isolated from one hemisphere of a mouse brain. To fix the complexes’ DNA proteins, hippocampal chopped tissues from each animal were incubated with 500 μL of PBS containing 1% formaldehyde in a rotator. After 10 min, the fixation reaction was stopped by adding 2 M glycine. After cross-linking with formaldehyde, chromatin was sheared using the Covaris Ultrasonicator, and the fragments’ size was confirmed in a 2% (wt/vol) agarose gel. The ChIP validated H3K27ac antibody (3 μg, overnight incubation, cat. no. 4729; Abcam) was used to immunoprecipitate the material of interest. Input genomic DNA and H3K27ac-genomic DNA were reverse cross-linked at 65 °C. RT-PCR was used to quantify differential binding on the genomic DNA using custom-designed primers with the following sequences: Grm2 (mGlu2) promoter: Forward, GCCACTGTCTCATCTGTTCC; Reverse, ATCCCGCTCTTGACAGGT; Probe, ATTCAGCACCACACTGTGGACAGC; β-actin promoter: Forward, GAGACATTGAATGGGGCAGT; Reverse, ATGAAGAGTTTTGGCGATGG; Probe, ACAAGGGCGGAGGCTATTCCTGTA.
Gene Expression Analysis.
After killing by rapid decapitation, hippocampal tissues were dissected, flash frozen, and stored at −80 °C until processing. Tissue samples were weighed before homogenization, and about 60–90 mg of hippocampal tissue was lysed. Total RNA was extracted using QIAzol reagent and an RNeasy mini Kit (Qiagen), according to the manufacturer’s instructions. RNA quality was checked using the 2100 Bioanalyzer (Agilent Technologies) and quantified spectrophotometrically. The acceptance criteria for RNA quality was a 260/280 ratio of ≥1.80 and ≤2.20. Two micrograms of total RNA was then used for cDNA synthesis according to the manufacturer’s instructions of the high capacity cDNA reverse transcription kit (Life Technologies). The reverse transcribed reaction was carried out according to the following condition steps: 25 °C/10 min, 37 °C/120 min, and 85 °C/5 min. Quantitative PCR was performed according to the protocols of the manufacturer (Life Technologies), using TaqMan Universal PCR Master Mix and gene-specific primers, synthetized by the Applied Biosystem Company (Life Technologies). GAPDH was used as a reference control. The following PCR conditions were used: an initial incubation of 50 °C for 2 min and a denaturation step of 95 °C for 10 min, followed by 40 cycles of 95 °C/15 s and 60 °C/1 min. All reactions were performed in triplicate. The threshold cycle (CT), which correlates inversely with the levels of target mRNA, was measured as the number of cycles at which the reporter fluorescence emission exceeded the preset threshold level. The amplified transcripts were quantified using the comparative CT method, with the formula for relative fold change equal to 2ΔΔCT (41).
Immunohistochemistry.
For immunohistochemistry, animals were killed by deep anesthesia with pentobarbital (100 mg/kg), followed by transcardial perfusion with heparinized saline and then 4% (wt/vol) paraformaldehyde (PFA) using a peristaltic perfusion pump (Fisher Scientific). Brains were postfixed for 4 h in 4% PFA before sinking in 30% (wt/vol) sucrose for 3 d. Finally, brains were flash-frozen on dry ice and stored at −80 °C until they were cut using a microtome into 40-mm-thick sections, which were then stored in a cryoprotectant solution at −20 °C before being processed for immunohistochemistry.
Immunohistochemical staining was carried out on 3–4 serial 40-mm coronal sections from the rostrocaudal extent of the hippocampus (selected from −1.58 to −2.06 mm caudal to bregma) as follows: sections were washed twice (2 × 15 min) in 0.1 M PBS solution (pH 7.4), blocked in 0.5% BSA in 0.1 M PBS with 0.25% Triton X-100 for 30 min, and, successively, incubated in the following primary antibodies: mGlu2 (1:500; Abcam) or NMDA (1:500; Abcam) overnight at 4 °C in the blocking solution. On the following day, sections were rinsed in 0.1 M PBS (3 × 10 min) before being incubated with secondary antibody (biotinylated anti-rabbit) diluted 1:400 in the blocking solution for 30 min. After three washes in 0.1 M PBS (3 × 10 min), tissues were incubated in the avidin–biotin peroxidase complex (Vector ABC Labs) for 30 min at room temperature, followed by three more 10-min PBS washes. Subsequently, brain sections were randomly slide-mounted to avoid any difference among control and stressed groups in the peroxidase reaction, which was developed for 4 min using a diaminobenzidine (DAB) substrate (Sigma), followed by three brief PBS washes. Finally, the immunoreactivity for each cover-slipped section was assessed by a Nikon ECLIPSE 90i microscope, equipped with the digital camera DS-Fi1.
All antibodies were tested by blocking with their respective immunizing peptides to confirm their specificity. Immunoreactivity was also confirmed by control staining, which was performed without the primary and/or secondary antibodies.
Densitometry.
Densitometric analyses were performed by two independent blind observers. All groups were represented on each slide to avoid any difference in the DAB reaction. The image analysis was assessed on three to four sections per mouse by two independent observers in the following subregions: CA1, CA3, and dentate gyrus (DG) regions of the hippocampus. Densitometric analysis was accomplished using MCID software and a light box equipped with a CCD camera with a 60-mm Nikon lens. Three to four sections per animal were examined, and each region was analyzed across its full length (excluding transition zones between regions: e.g., CA2) and after subtracting background (e.g., corpus callosum). The values for each region for each animal were averaged to provide a single data point for that region and animal in relative optical density (arbitrary units). We report that mGlu2 immunoreactivity was higher in the DG compared with the CA3 and CA1 and that, within the DG, the higher immunoreactivity was observed into the granule cell layers of the hippocampal formation.
Behavioral Tests.
The tail suspension test (TST) and light–dark test (LDT) were performed as previously described (2, 18, 27, 42, 43).
TST.
Two independent blind observers scored the total duration of immobility at the tail suspension during a 6-min test. Each mouse was visually isolated from the other mice and was suspended 50 cm above the floor by adhesive tape placed ∼1 cm from the tip of the tail. Mice were considered immobile only when they hung passively and completely motionless.
LDT.
The LDT was performed as previously described (27). The apparatus consisted of a rectangular Plexiglas box (20 × 50 × 20 cm) with a black chamber constituting one-third of the total volume. The two chambers were separated by a Plexiglas septum with an open door (12 × 5 cm) that permitted the passage from the illuminated chamber (300 luminex) to the enclosed dark chamber (4 luminex). Mice were videotaped, and the time spent by each mouse in the light chamber was measured and scored by two independent blind observers. Mice were considered to have entered a chamber when all four paws were positioned into the chamber. No pretest (habituation session) was carried out before the test session.
Electrophysiology.
Electrophysiological experiments were assessed at the same time point of the behavioral and molecular analyses (Fig. S2). Mice were anesthetized with halothane, and their brains were removed rapidly and placed in an ice-cold artificial cerebrospinal fluid (ACSF) solution containing 124 mM NaCl, 2.5 mM KCl, 1.3 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, 2.4 mM CaCl2, and 10 mM glucose. The hippocampus was dissected out, and slices were cut at a thickness of 350 μm, using a Vibroslice (VT 1000S; Leica), and placed in a storage container containing oxygenated medium at room temperature (20–22 °C) for at least 1 h before recordings. A single slice was then placed on a nylon mesh, completely submerged in a small chamber (0.5 mL) and superfused with oxygenated ACSF (∼30 °C) at a constant flow rate of 3 mL⋅min−1. Presynaptic stimulation was applied to the medial perforant pathway by using a bipolar insulated tungsten wire electrode, and field excitatory postsynaptic potentials (fEPSPs) were recorded at a control test frequency of 0.033 Hz from the middle one-third of the molecular layer of the DG with a glass microelectrode. In each experiment, an input–output curve was plotted at the test frequency, and the amplitude of the test EPSP was adjusted to one-third of maximum. LTP was evoked by HFS consisting of eight trains, each of eight stimuli at 200 Hz, and an intertrain interval of 2 s, with the stimulation voltage increased during the HFS so as to elicit an initial EPSP of the train of double the normal test EPSP amplitude. All solutions contained 50 μM picrotoxin to block GABAA-mediated transmission. Statistical analysis was performed using one-way ANOVA. Dunnett’s test was used for multiple comparisons. The values were considered to be significantly different when P < 0.05.
Statistics.
Statistical analyses were performed using the two-tailed unpaired Student’s t test (for comparison of two groups) and one-way analysis of variance (ANOVA) followed by Tukey’s test for the post hoc analysis (for three and four groups).
Acknowledgments
We thank Timothy Lau and Kathleen Jedruszczuk for their commitment in performing blind behavioral and molecular analyses. This work was supported by the American Foundation for Suicide Prevention, the Hope for Depression Research Foundation, NIH Grant R01 MH41256, and by Grant UL1 TR000043 from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) Clinical and Translational Science Award (CTSA) program.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1516016112/-/DCSupplemental.
References
- 1.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13(1):22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nasca C, Bigio B, Zelli D, Nicoletti F, McEwen BS. Mind the gap: Glucocorticoids modulate hippocampal glutamate tone underlying individual differences in stress susceptibility. Mol Psychiatry. 2015;20(6):755–763. doi: 10.1038/mp.2014.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McEwen BS, Gray J, Nasca C. Recognizing resilience: Learning from the effects of stress on the brain. Neurobiol Stress. 2015;1(1):1–11. doi: 10.1016/j.ynstr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Southwick SM, Charney DS. The science of resilience: Implications for the prevention and treatment of depression. Science. 2012;338(6103):79–82. doi: 10.1126/science.1222942. [DOI] [PubMed] [Google Scholar]
- 5.Wood SK, Walker HE, Valentino RJ, Bhatnagar S. Individual differences in reactivity to social stress predict susceptibility and resilience to a depressive phenotype: Role of corticotropin-releasing factor. Endocrinology. 2010;151(4):1795–1805. doi: 10.1210/en.2009-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 7.McEwen BS. Stress, sex, and neural adaptation to a changing environment: Mechanisms of neuronal remodeling. Ann N Y Acad Sci. 2010;1204(Suppl):E38–E59. doi: 10.1111/j.1749-6632.2010.05568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McEwen BS. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol Rev. 2007;87(3):873–904. doi: 10.1152/physrev.00041.2006. [DOI] [PubMed] [Google Scholar]
- 9.Datson NA, et al. Previous history of chronic stress changes the transcriptional response to glucocorticoid challenge in the dentate gyrus region of the male rat hippocampus. Endocrinology. 2013;154(9):3261–3272. doi: 10.1210/en.2012-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gray JD, Rubin TG, Hunter RG, McEwen BS. Hippocampal gene expression changes underlying stress sensitization and recovery. Mol Psychiatry. 2014;19(11):1171–1178. doi: 10.1038/mp.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reul JM, Chandramohan Y. Epigenetic mechanisms in stress-related memory formation. Psychoneuroendocrinology. 2007;32(Suppl 1):S21–S25. doi: 10.1016/j.psyneuen.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 12.McEwen BS, Nasca C, Gray JD. Stress effects on neuronal structure: Hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology. 2015 doi: 10.1038/npp.2015.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McEwen BS, Gray JD, Nasca C. 60 years of neuroendocrinology: Redefining neuroendocrinology: Stress, sex and cognitive and emotional regulation. J Endocrinol. 2015;226(2):T67–T83. doi: 10.1530/JOE-15-0121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10(8):561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 15.Nicoletti F, et al. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology. 2011;60(7-8):1017–1041. doi: 10.1016/j.neuropharm.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: Involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69(1):89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- 17.Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61(5):1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- 18.Nasca C, et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA. 2013;110(12):4804–4809. doi: 10.1073/pnas.1216100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo SJ, Charney DS. Next generation antidepressants. Proc Natl Acad Sci USA. 2013;110(12):4441–4442. doi: 10.1073/pnas.1301593110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flight MH. Antidepressant epigenetic action. Nat Rev Neurosci. 2013;14(4):226. doi: 10.1038/nrn3466. [DOI] [PubMed] [Google Scholar]
- 21.Chen ZY, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314(5796):140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dincheva I, Glatt CE, Lee FS. Impact of the BDNF Val66Met polymorphism on cognition: Implications for behavioral genetics. Neuroscientist. 2012;18(5):439–451. doi: 10.1177/1073858411431646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murrough JW, Charney DS. Is there anything really novel on the antidepressant horizon? Curr Psychiatry Rep. 2012;14(6):643–649. doi: 10.1007/s11920-012-0321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magariños AM, et al. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus. 2011;21(3):253–264. doi: 10.1002/hipo.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapolski RM. Stress, the Aging Brain, and the Mechanisms of Neuron Death. MIT Press; Cambridge, MA: 1992. [Google Scholar]
- 26.Karst H, Joëls M. Effect of chronic stress on synaptic currents in rat hippocampal dentate gyrus neurons. J Neurophysiol. 2003;89(1):625–633. doi: 10.1152/jn.00691.2002. [DOI] [PubMed] [Google Scholar]
- 27.Nasca C, et al. Exposure to predator odor and resulting anxiety enhances the expression of the α2 δ subunit of voltage-sensitive calcium channels in the amygdala. J Neurochem. 2013;125(5):649–656. doi: 10.1111/j.1471-4159.2012.07895.x. [DOI] [PubMed] [Google Scholar]
- 28.Joëls M, Krugers HJ. LTP after stress: Up or down? Neural Plast. 2007;2007:93202. doi: 10.1155/2007/93202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastor-Ciurana J, et al. Prior exposure to repeated immobilization or chronic unpredictable stress protects from some negative sequels of an acute immobilization. Behav Brain Res. 2014;265(265):155–162. doi: 10.1016/j.bbr.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 30.Nestler EJ. Epigenetic mechanisms of depression. JAMA Psychiatry. 2014;71(4):454–456. doi: 10.1001/jamapsychiatry.2013.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cameron HA, Gould E. Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience. 1994;61(2):203–209. doi: 10.1016/0306-4522(94)90224-0. [DOI] [PubMed] [Google Scholar]
- 32.Schlett K. Glutamate as a modulator of embryonic and adult neurogenesis. Curr Top Med Chem. 2006;6(10):949–960. doi: 10.2174/156802606777323665. [DOI] [PubMed] [Google Scholar]
- 33.Abraham WC. Metaplasticity: Tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9(5):387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- 34.Swanson CJ, et al. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4(2):131–144. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 35.McEwen BS, Morrison JH. The brain on stress: Vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16–29. doi: 10.1016/j.neuron.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: Implications for dopamine-associated psychiatric disorders. Biol Psychiatry. 2002;51(10):775–787. doi: 10.1016/s0006-3223(01)01362-2. [DOI] [PubMed] [Google Scholar]
- 37.McEwen BS, et al. Mechanisms of stress in the brain. Nat Neurosci. 2015;18(10):1353–1363. doi: 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matrisciano F, et al. Enhanced expression of the neuronal K+/Cl- cotransporter, KCC2, in spontaneously depressed Flinders Sensitive Line rats. Brain Res. 2010;1325:112–120. doi: 10.1016/j.brainres.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 39.Kinnally EL, Mann JJ. Early life stress programming and suicide risk. Psych Ann. 2012;42(3):95–100. [Google Scholar]
- 40.Oquendo M, et al. Posttraumatic stress disorder comorbid with major depression: Factors mediating the association with suicidal behavior. Am J Psychiatry. 2005;162(3):560–566. doi: 10.1176/appi.ajp.162.3.560. [DOI] [PubMed] [Google Scholar]
- 41.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 42.Nestler EJ, et al. Neurobiology of depression. Neuron. 2002;34(1):13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 43.Cryan JF, Mombereau C, Vassout A. The tail suspension test as a model for assessing antidepressant activity: Review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev. 2005;29(4-5):571–625. doi: 10.1016/j.neubiorev.2005.03.009. [DOI] [PubMed] [Google Scholar]